
Abstract
Fragile-X syndrome (FXS) patients display intellectual disability and autism spectrum disorder due to silencing of the X-linked, fragile-X mental retardation-1 (FMR1) gene. Dysregulation of cAMP metabolism is a consistent finding in patients and in the mouse and fly FXS models. We therefore explored if BPN14770, a prototypic phosphodiesterase-4D negative allosteric modulator (PDE4D-NAM) in early human clinical trials, might provide therapeutic benefit in the mouse FXS model. Daily treatment of adult male fmr1 C57Bl6 knock-out mice with BPN14770 for 14 days reduced hyperarousal, improved social interaction, and improved natural behaviors such as nesting and marble burying as well as dendritic spine morphology. There was no decrement in behavioral scores in control C57Bl6 treated with BPN14770. The behavioral benefit of BPN14770 persisted two weeks after washout of the drug. Thus, BPN14770 may be useful for the treatment of fragile-X syndrome and other disorders with decreased cAMP signaling.
Similar content being viewed by others


Introduction
Mutational inactivation of the gene encoding the Fragile X Mental Retardation protein (FMRP) causes a spectrum of symptoms including seizures, sleep disorders, anxiety, irritability, autism, mild to severe cognitive impairment and intellectual disability1,2. The constellation of symptoms is known as Fragile-X syndrome (FXS). The syndrome in humans is caused by expansion of an unstable, CGG triplet repeat with subsequent silencing of the fragile-X mental retardation-1 (FMR1) gene that encodes FMRP. FXS is a common monogenic disorder that affects 1 in 3,600 males and 1 in 4,000–6,000 females2. FXS is severely debilitating in males3. Females generally are less affected than males due to mosaicism resulting from X-chromosome inactivation which occurs randomly early in embryogenesis.
Present both pre- and post-synaptically, FMRP associates with and suppresses the translation of mRNA important for synaptic function4,5. Consequently, genetic ablation of FMRP allows the over expression of proteins that normally would be tightly regulated components of the synapse such as channels, signaling and structural components. The syndrome can be modeled in mice and fruit flies by deletion of the fmr1 gene in mice and the highly conserved Dfmr1 gene in Drosophila 6,7.
Initial findings supporting phosphodiesterase-4 (PDE4) as a therapeutic target in FXS come from work in Drosophila 8,9. Interest in PDE4 derived from the observations by Berry-Kravis and coworkers of reduced cAMP levels in FXS patient cells10,11. Drosophila has a single, FMR1 gene otholog that when deleted produces deficits in neuronal development and biochemical and behavioral changes reminiscent of human FXS7,12,13. Dfmr1 null flies have impaired associative memory in an olfactory conditioning paradigm and structural alterations in mushroom body neurons, a neural center important for associative learning, accompanied by decreased cAMP in tissues of the head8,9,14,15. Using a Drosophila model in which the flies are heterozygous for the Dmfr1 3 null allele, Kanellopoulos and coworkers reported that treatment with rolipram, a PDE4 inhibitor, rescued the olfactory learning deficit9. The learning deficit also was rescued by crossing the Dmfr1 3 allele onto a genetic background lacking the fly dunce gene (Dnc), the single Drosophila PDE4 gene. These findings were extended by Choi and coworkers to a Drosophila model in which Dmfr1 was completely absent8. Two PDE4 inhibitors, rolipram and RO201724, were shown to reverse the behavioral deficits in Dmfr1 null flies. A low dose of rolipram did not rescue the structural abnormalities in the mushroom body neurons, while a high dose rescued both behavioral and structural phenotypes. Choi and coworkers also showed genetic rescue of the Dmfr1 null behavioral and structural phenotypes on the dunce background. Thus, reducing PDE4 activity in the Drosophila Dmfr1 models rescues multiple aspects of the Fragile-X phenotype.
While the Drosophila genome contains a single PDE4 gene, this has been expanded to a small gene family in higher organisms. The genomes of humans and other mammals contain four PDE4 genes (PDE4A-D)16. The gene family contains two upstream conserved regions (UCR1 & UCR2) important for regulation of PDE4 enzymatic activity that distinguish the PDE4 enzymes from other PDE. UCR1 and UCR2 are ancestral domains that are conserved in Drosophila and Caenorhabditis elegans but not in Dictyostelium or yeast17. Each gene expresses multiple proteins that differ in N-terminal targeting sequences, their assembly into dimeric or monomeric forms of the PDE4 enzyme, and their post-translation regulation through protein kinase A (PKA) phosphorylation18,19. The importance of PDE4D for human cognition is shown by ultra-rare, autosomal dominant mutations in PDE4D that cause acrodysostosis without hormone resistance (ACRDYS2), a neurodevelopmental syndrome causing short stature, brachydactyly (short fingers and toes), nasal hypoplasia and intellectual disability with speech and psychomotor retardation20,21. All of the ACRDYS2 mutations described to date are missense mutations that alter amino acids on the surface of the protein such as the contact residues between the PDE4D catalytic domain and the UCR2 regulatory domain20,22,23,24,25,26,27. One mutation (serine129 to alanine) removes the PKA phosphorylation site on the UCR1 regulatory domain, and therefore prevents activation of PDE4D enzymatic activity in response to cAMP signaling. The implication is that dysregulation of the spatial and temporal patterning of cAMP signaling by reducing cAMP hydrolysis, as in Drosophila dunce mutant flies, impairs associative memory28. PDE4D negative allosteric modulators (PDE4D-NAM) such as BPN14770 inhibit the enzyme by closing the UCR2 regulatory domain across the active site, thereby limiting access of cAMP29. Unlike rolipram and RO201724, which inhibit all subtypes of PDE4, BPN14770 is selective for the PDE4D subtype. We therefore sought to assess the therapeutic benefit of BPN14770 in adult, male fmr1 gene deleted mice in order to extend previous studies in the Drosophila FXS model.
FXS patients display a range of neuropsychiatric symptoms including intellectual disability, delayed language acquisition, poor social interaction, hyperarousal, hypersensitivity, repetitive behaviors, disrupted sleep, attention deficit hyperactivity disorder and autism2. These behavioral changes are modeled in adult male fmr1 KO mice which display a spectrum of behavioral phenotypes due to the fmr1 gene deletion6. The mutant mice show hyperarousal in the open field test, have impaired social interaction, are less likely to build nests when provided cotton batting and are less likely to bury marbles in the cage bedding. Adult male mice were used for all studies as male FXS patients typically suffer more severe symptoms than do female patients due to the single X chromosome2. In both FXS patients and the fmr1 KO mice, there are alterations in the density, size, shape and maturity of dendritic spines, the principle recipients of excitatory inputs from other neurons30,31.
When treated daily with the PDE4D inhibitor BPN14770 for 14 days, fmr1 KO mice showed significantly reduced hyperarousal in the open field, significantly increased frequency of social interaction, and significantly improved natural behaviors (nesting and marble burying) in comparison to fmr1 KO mice treated with vehicle. Indeed, measures of activity, social interaction, nesting and marble burying were not significantly different from wild-type C57Bl6 mice. There was no decrement in behavioral scores in control C57Bl6 treated with BPN14770. The benefit of BPN14770 persisted two weeks after washout of the drug. The present study supports PDE4D as a therapeutic target for the treatment of FXS. BPN14770 has completed human Phase 1 clinical testing with evidence of safety and tolerability. Our results suggest that BPN14770 may be useful for the treatment of fragile-X syndrome with potential expansion to the treatment of autism spectrum disorder.
Results
BPN14770 Improves Behavioral Phenotypes of fmr1 KO Mice
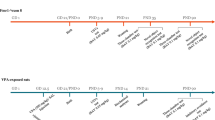
The design of the study is shown in Fig. 1. Four groups of mice were dosed in each experiment. The groups were wild-type C57Bl6 mice dosed orally with vehicle (WT-Veh), wild-type mice dosed with BPN14770 (WT-BPN14770), fmr1 gene deleted or knock-out (KO) mice dosed with vehicle (fmr1 KO-Veh), and fmr1 KO mice dosed with BPN14770 (fmr1 KO-BPN14770). Mice were dosed orally with vehicle or BPN14770 once daily for 14 days. BPN14770 was dosed at 0.3 mg/kg PO in 0.5% methylcellulose. This was 3 times the minimum effective oral dose (0.1 mg/kg) found in separate studies to improve novel object recognition after a 24 hr delay in ICR mice (unpublished data). Three separate experiments with fmr1 KO mice were performed sequentially. In the first experiment, the groups of mice were dosed for 14 days and behavioral assessments were performed 2 hours after the last dose on day 14. In the second experiment, mice were dosed for 14 days, the open field test was performed 2 hour after the last dose on day 14, and then the mice were killed and the brains were prepared for spine morphometry. In the third experiment, mice were dosed for 14 days, the drug was withdrawn, and then behavioral assessments were performed 14 days after the last dose of drug or vehicle.

Schematic Diagram Depicting Treatment Groups and Schedule of Events. Treatment groups were wild-type C57Bl6 mice exposed to vehicle (WT-Vehicle), wild-type C57Bl6 mice exposed to BPN14770 (WT-BPN14770), fmr1 KO mice exposed to vehicle, or fmr1 KO mice exposed to BPN14770. Three experiments were performed. In each experiment, mice were treated daily for 14 days by oral gavage with either vehicle or BPN14770. In the first experiment (Expt 1), mice were profiled for open field activity, social interaction, and natural behaviors (nesting & marble burying) on the 14th day of treatment. In the second experiment (Expt 2), mice were treated daily for 14 days, profiled for open field activity on the 14th day of treatment, then killed and prepared for spine morphometry. In the third experiment (Expt 3), mice were treated daily for 14 days, treatment was then stopped, and 14 days after cessation of treatment the mice were profiled for open field activity, social interaction, nesting & marble burying. Each treatment group contained 10 adult male mice.
BPN14770 improved a spectrum of behavioral deficits in adult male fmr1 KO mice (Fig. 2). For behavioral assessment, mice were randomized based on genotype to groups treated with vehicle or BPN14770. Based on two-way ANOVA, there was a highly significant interaction between treatment and genotype (F (1,36) = 41.78, p < 0.001). Fmr1 C57Bl6 knock-out mice treated with vehicle were significantly more active in the open field than wild type C57Bl6 mice treated with vehicle (p < 0.001). Fmr1 C57Bl6 knock-out mice treated with BPN14770 for 14 days at 0.3 mg/kg showed significantly reduced open field activity in comparison to vehicle treated fmr1 knock-out mice (p < 0.001). There was no effect of BPN14770 on open field activity of wild-type mice, and indeed, after treatment with BPN14770, the open field activity of fmr1 knock-out mice was not significantly different from wild-type mice treated with either BPN14770 or vehicle. Safety studies have shown no effect of BPN14770 on locomotion in C57Bl6 mice up to 30 mg/kg. Toxicological studies in rats identify the No Observed Effect Level (NOEL) of BPN14770 as greater than 60 mg/kg based on inappetence and weight loss. Thus, the reduction in open field activity of fmr1 knock-out mice caused by BPN14770 at the low dose explored is unlikely to be due to an adverse effect of the drug.

BPN14770 Improved Behavioral Phenotypes of fmr1 KO Mice (A) Wild-type (WT) or fmr1 KO (KO) adult male mice were treated with vehicle or BPN14770 daily for 14 days (Expt 1) and then profiled for behavior. From left-to-right, data are shown for hyperarousal (Number of Squares crossed in the open field test), social interaction (Duration of Sniffing), nesting (Nesting Score), and marble burying (Marbles Buried). (B) Mice were treated daily for 14 days with vehicle or BPN14770 and then drug was withdrawn for 14 days (Expt 3). Mice were profiled for hyperarousal, social interaction, nesting and marble burying on the 14th day after drug withdrawal. Data were analyzed by Two-way ANOVA followed by Tukey’s post-test corrected for multiple comparisons. P-values shown are: n.s. not significant, *p < 0.05, **p < 0.01, ***p < 0.001 (N = 10 mice per group).
Social interaction was assessed as the duration of sniffing when the fmr1 KO mouse was tested with a juvenile mouse. There was a highly significant interaction between treatment and genotype (F (1,36) = 17.05, p < 0.001). The fmr1 KO mice treated with vehicle showed a significantly reduced duration of sniffing of the juvenile mouse (p < 0.001) compared to wild type C57Bl6 mice treated with vehicle. These social interaction deficits are consistent with those reported by other researchers32. Fmr1 KO mice treated with BPN14770 showed increased duration of sniffing (p < 0.001) that was not significantly different from wild-type mice. There was no effect of BPN14770 on the behavior of wild-type C57Bl6 mice.
Both male and female mice will build nests for thermoregulation when given access to nesting material in their home cage. This test has been used as an indicator of hippocampal lesion and dysfunction33. To assess nesting, mice were placed individually into the nesting cages about one hour before the dark phase, and the results were assessed the next morning. As with burrowing, and for similar reasons, timing was not critical. Nest building was scored on a 5-point scale and the amount of unturned nestlets was also weighed. As a reference, most C57BL/6 mice score 4–5 on nest construction, but when the hippocampus is lesioned the median score would be around 1–2 and a score of 3 is unlikely to be exceeded33. There was a highly significant interaction between treatment and genotype (F (1,36) = 21.76, p < 0.001). Nesting was significantly impaired in vehicle-treated fmr1 C57Bl6 KO as compared to wild type C57Bl6 mice treated with vehicle (p < 0.001). Fmr1 C57Bl6 KO mice treated with BPN14770 showed significant improvement in nest building in comparison to vehicle treated fmr1 KO mice (p < 0.001) and indeed, were not significantly different from control C57Bl6 mice treated with vehicle. There was no effect of BPN14770 on nest building by wild-type C57Bl6 mice.
Mice spontaneously dig in many substrates in the laboratory. This behavior comes from their ancestry in the wild, where they forage for seeds, grain, insects, and other food to be found buried in the soil or leaf litter in their natural habitat. It exploits a common natural rodent behavior and provides quantitative data under controlled laboratory conditions. There was a highly significant interaction between treatment and genotype (F (1,36) = 69.82, p < 0.001). Marble burying was significantly impaired in vehicle- treated fmr1 C57Bl6 KO as compared to wild-type C57Bl6 mice treated with vehicle (p < 0.001). Fmr1 C57Bl6 KO mice treated with BPN14770 showed significant improvement in marble burying in comparison to vehicle treated fmr1 KO mice (p < 0.001) and were not significantly different from control C57Bl6 mice. There was no effect of BPN14770 on marble burying by wild-type C57Bl6 mice.
Thus, treatment with BPN14770 reversed a range of behavioral phenotypes in the fmr1 KO mutant mice.
BPN14770 Improves Dendritic Spine Morphology in fmr1 KO Mice
Dendritic spine morphology is altered in human FXS and in the fmr1 KO mouse model34,35. There is an abundance of spines with a thin, elongated morphology rather than a mature, squat, bulbous morphology. To assess if BPN14770 affected dendritic spine morphology, adult male fmr1 KO mice were dosed daily with BPN14770 or vehicle for 14 days. When tested in the open field 2hrs after the last dose, mice treated with BPN14770 showed reduced hyperarousal (p < 0.001) consistent with the behavioral findings in the first experiment (Supplementry Fig. 1). The mice were killed and prepared for spine morphometry. The spine morphometry analysis focused on medial prefrontal cortex, a region of cortex in which spine morphology is altered by the fmr1 mutation (Fig. 3)31,34,35. Layer 3 and layer 5 pyramidal cells were chosen for morphometry. Immunohistochemical studies in mice indicate that PDE4D is mostly present in neurons of cortical layers 2 and 336. Spines were sampled on the apical dendrite of layer 3 pyramidal cells and at two sites on layer 5 pyramidal neurons, on the apical dendrite and on the apical tufts.

Representative Laser Scanning Confocal Micrographs Of Dendritic Segments And Dendritic Spines For Pyramidal Neurons In Medial Prefrontal Cortex. Dendritic spine morphometry was performed on dendritic segments of layer 3 and layer 5 pyramidal cells in medial prefrontal cortex (schematic). The sampling region for layer 3 pyramidal cells (L3P) was a segment of the primary apical dendrite extending from the soma to a distance of 100–150 µM. Layer 5 pyramidal cells were sampled from the soma to a distance of 200–250 µM of the primary apical dendrite and from the branch point of the apical tuft for a distance of 50 µM. High magnification insets for each segment sampled show the presence of thin-necked, immature spines (arrow) as well as mushroom (arrowhead only) and stubby spine (double arrowhead) types.
There was a highly significant effect of BPN14770 on the spine length of layer 3 pyramidal cells (p < 0.0001) which is consistent with an effect of BPN14770 on spine maturation (Figs 4 and 5). Treatment with BPN14770 causes reduction in layer 3 pyramidal cell spine length of about 10%. As an internal control, there was no effect of BPN14770 on spine length on layer 5 pyramidal cells on either the apical dendrite or the apical tufts.

Dendritic Spine Multi-Dimensional Feature Distribution Plots. Multi-dimensional spine length x head diameter plots are shown for (A) the apical dendrite of Layer 3 pyramidal cells (N = 3,502 spines, 70 cells vehicle; N = 3,322 spines, 69 cells treated), (B) the apical dendrite of Layer 5 pyramidal cells (N = 3,792 spines, 70 cells vehicle; N = 3,443 spines, 69 cells treated), and (C) the apical dendrite tufts of Layer 5 pyramidal cells (N = 2,626 spines, 65 cells vehicle; N = 3,026 spines, 68 cells treated). The first two panels compare spines from fmr1 KO mice exposed to vehicle or BPN14770. The third panel shows the difference plot. The difference map (vehicle subtracted from BPN14770) shows maturation (shortening) of spines on the apical dendrites of layer 3 pyramidal neurons. There was no change in dendritic spine maturation on layer 5 pyramidal neurons.

Effect of BPN14770 on Dendritic Spine Length. The distribution of spine length is shown for the apical dendrite of Layer 3 pyramidal cells (L3P apical), the apical dendrite of Layer 5 pyramidal cells (L5P apical), and the apical dendrite tufts of Layer 5 pyramidal cells (L5P tufts). Dendritic spines were sampled for length, head dimeter and neck width. Data are plotted for vehicle (red circles) and BPN14770-treated (blue squares) fmr1 KO mice. Data were analyzed by the 2-sample Komolgorov-Smirnov test. P-values shown are ***p < 0.0001.
The spine data also showed a trend by animal (N = 10 mice per group; p < 0.1) for an increase in mature spines on layer 3 pyramidal cells after treatment with BPN14770, while there was no change in spine morphometry on layer 5 pyramidal neurons (Sup Fig. 2). When the data were analyzed by dendrite (N = 70 dendrites from mice treated with BPN14770, N = 69 dendrites from vehicle treated mice), there was a significant decrease in intermediate spines on layer 3 pyramidal cells (p < 0.05) and a trend for an increase in mature spines (p < 0.1) (Sup Fig. 3). There was no effect of BPN14770 on the spines of layer 5 pyramidal neurons on either the apical dendrite or the apical tufts.
Behavioral Benefit of BPN14770 Endures After Washout of the Drug
The third experiment explored whether or not the behavioral benefit of BPN14770 persisted after washout of the drug. Mice were treated daily with BPN14770 or vehicle for 14 days and then the drug was withdrawn for 14 days (Fig. 1). Even 14 days after withdrawal of the drug, fmr1 KO mice previously treated with BPN14770 showed decreased hyperarousal in the open field (p < 0.001), increased social interaction (p < 0.005), improved nesting behavior (p < 0.005) and increased marble burying (p < 0.001). The activity of fmr1 KO mice previously treated with BPN14770 was significantly greater than wild-type C57Bl6 mice treated with vehicle or BPN14770 (p < 0.05) indicating that the improvement in behavior waned somewhat after washout of the drug.
Discussion
Multiple drug classes have shown evidence of therapeutic efficacy in fragile X animal models; however, many have been validated via acute dosing in limited model systems. BPN14770 shows a broad range of therapeutic effects in fmr1 KO mice with no loss of efficacy over an extended dosing period. The pro-cognitive effects of PDE4 inhibitors are potentially beneficial to humans with mosaic fragile X, a common presentation in males with the fragile X mutation and a universal condition in females with fragile X. The benefit of rolipram, an older PDE4 inhibitor, also strongly suggests class-wide antidepressant effects which could be useful in fragile X37,38. Other drug classes, such as GABA(B) agonists and mGluR5 NAMs are known to cause significant cognitive impairment and adverse psychiatric effects in human subjects39,40, suggesting that some agents with disease-specific effects in fragile X may be harmful to normal cells in a mosaic brain.
The cAMP dysregulation in FXS affects multiple tissues. In the original study by Berry-Kravis and Huttenlocher41, platelets from FXS patients were found to have lower basal and stimulated cAMP levels than controls. Basal, prostaglandin E1 and forskolin-stimulated cAMP levels were 50–60% of controls. In their experiment, a non-selective PDE inhibitor (1-isobutyl-3-methylxanthine) was used to inhibit all platelet PDE activity. This indicates that cAMP dysregulation is likely at the level of adenylate cyclase rather than due to dysregulation of a PDE. The dominant PDE in platelets is PDE3, the target of cilostazol, a selective PDE3 inhibitor indicated for the treatment of intermittent claudication in individuals with peripheral vascular disease42. PDE4 isoenzymes are absent from platelets43. Thus, it is unlikely that dysregulation of PDE4D could account for the alteration in cAMP levels seen in multiple tissues in FXS, nor is treatment with BPN14770 likely to normalize cAMP levels in all tissues. In brain, PDE4D is only one of three PDE4 subtypes expressed, the others being PDE4A and PDE4B, thus PDE4A, PDE4B and PDE4D all contribute to cAMP homeostasis36,44,45. At the low dose of BPN14770 used in this study (0.3 mg/kg), inhibition is still relatively selective for PDE4D, although the effect of BPN14770 on basal cAMP levels in fmr1 KO mouse brain should be addressed in future studies.
Chronic dosing of 14 days duration with BPN14770 had broad efficacy across multiple behavioral measures in the fmr1 mouse model of fragile X. In contrast to CTEP, an mGluR5 antagonist, BPN14770 improves social interaction and natural behaviors such as building nests and burying marbles46. Also in contrast to the mGluR5 antagonists, BPN14770 maintains efficacy with chronic dosing while the benefit of the mGluR5 antagonists may wane with repeat dosing47. Similarly, GABA(B) agonists have demonstrated acute efficacy in fragile X models, but chronic dosing leads to rapid development of tolerance48. In addition, unlike mGluR5 signaling, cAMP signaling has been shown to be altered in human fragile X tissues10,11,49 as well as multiple fragile X model systems8,9, providing an added level of validation for PDE4 as a therapeutic target in fragile X.
Chronic dosing with BPN14770 also improved spine morphology on layer 3 pyramidal cells in medial prefrontal cortex. As an internal control, there was no effect on the dendritic spines of layer 5 pyramidal cells. Layer 3 pyramidal cells are the principle input neurons for intracortical association while layer 5 pyramidal cells are the principle output cells. The selective effect of BPN14770 on layer 3 pyramidal neurons may be due to the preferential expression of PDE4D in this cortical cell type36. Such cells may be more responsive to PDE4D inhibition. Cruz-Martin et al. also has provided intriguing data showing that dendritic spine dynamics of layer 2/3 pyramidal neurons is altered in the barrel cortex of Fragile X knock-out mice31. Although pyramidal neurons were not probed in deeper layers of cortex, intracortical association neurons may be more plastic, and in adults, maturation of their dendritic spines may be more responsive to signaling through the PKA-CREB pathway and thereby to inhibition of PDE4D. Consistent with the effect of BPN14770 on cortical neuron structure, the behavioral benefit of BPN14770 endures well after washout of the drug. The half-life of BPN14770 in mice is 11 hr (unpublished data), therefore the enduring behavioral benefit is not due to slow washout of the drug.
These studies were conducted in adult male fmr1 gene-deleted mice suggesting that adolescent and adult FXS patients may benefit from treatment with a PDE4D-NAM. One of the challenges in the design of clinical efficacy studies in FXS patients is the number of concomitant medications used to reduce symptoms of FXS, their impact on clinical aspects of the disorder that the novel neurotherapeutic may address, and the potential for pharmacokinetic as well as pharmacodynamic drug-drug interactions. FXS patients are prescribed multiple classes of psychotropic drugs including, but not limited to, antipsychotics to reduce irritability and aggression, serotonin reuptake inhibitors (SSRI) to reduce anxiety, and dopaminergic stimulants to reduce inattention and hyperactivity3. Younger patients are at higher risk for seizure and may be prescribed anticonvulsants. In contrast, when potential therapeutic agents are evaluated in animal models, either Dfmr1 mutant flies or fmr1 mutant mice, the agents are evaluated on a “clean” background in the absence of other psychotropic drugs. SSRIs are known to increase brain cAMP and PDE4D levels in preclinical models50, and indeed, upregulate PDE4 binding in patients with major depression51. Although SSRIs provide some control of anxiety in FXS patients, concomitant administration of an SSRI and a second drug that elevates cAMP may reduce the likelihood of discerning clinical benefit of the new agent on measures of anxiety or on clinical global impression scales incorporating anxiety in a composite measure.
In addition to FXS, there is a clear link between other monogenic disorders associated with intellectual disability and aberrant cAMP signaling through activation of PKA and consequent phosphorylation of the cAMP response element binding factor (CREB). CREB phosphorylation mediates the synaptic changes needed for the formation of various forms of memory52,53,54. Many of these disorders are amenable to study in model systems such as mice or Drosophila. Neurofibromatosis is linked to the cAMP pathway through the regulation by neurofibromin of adenylate cyclase production of cAMP with neuronal apoptosis phenotypes in an NF1 mouse model shown to be rescued by a PDE4 inhibitor55. Cognitive benefit was not examined in that study. CREB requires the cofactor CREB binding protein (CBP), a mutation in which causes Rubinstein-Taybi syndrome. Despite the mutation of CBP, PDE4 inhibitors are able to rescue memory phenotypes in a mouse model56. Finally, Rett syndrome, which is due to mutation of the MeCP2 gene, also affects CREB signaling by reducing CREB expression and phosphorylation by PKA, while activation of CREB signaling by a PDE4 inhibitor ameliorates multiple cellular and behavioral phenotypes57. Drugs in clinical development such as BPN14770 that target a specific molecular signaling pathway, as in the case of the cAMP-PKA-CREB pathway, may facilitate the new model of experimental medicine in which proof of concept studies in preclinical models and their translation to patients are used to interrogate specific biological processes and neurocognitive outcomes related to the drug mechanism of action.
Methods
Compounds
BPN14770 was synthesized as described17. The fifty percent inhibitory (IC50) potency of BPN14770 against mouse PDE4D is 133 ± 18 nM (mean + SEM) as compared to mouse PDE4B (IC50 = 2,124 ± 527 nM). BPN14770 does not inhibit other PDE (BPS Biosciences panel), GPCR, ion channels or transporters (CEREP panel) at 10 µM. BPN14770 has 100% bioavailability in mice when dosed by perioral gavage as a suspension in 0.5% methylcellulose.
Animals
Fmr1 KO2 C57Bl6 knock-out mice (The Dutch-Belgium Fragile X Consortium 1994)6 and wild-type (WT) littermates were generated on a C57BL/6J background and repeatedly backcrossed onto a C57BL/6J background for more than eight generations. Initial stocks of mice were obtained from the Jackson Laboratory. The fmr1 KO2 is a null allele at fmr1 generated by deletion of the promoter and first exon. It is both protein and mRNA null. This model is widely used by the FXS research community. The fmr1 KO2 C57Bl6 KO mice were housed in groups of the same genotype in a temperature- and humidity-controlled room with a 12-hr light–dark cycle (lights on 7 a.m.–7 p.m.). Testing was conducted during the light phase. Food and water were available ad libitum. Testing was conducted on fmr1 KO mice and their WT littermates. Ten male mice per treatment group, 14 weeks of age, were used for behavioral experiments. Mice were housed in commercial plastic cages, and experiments were conducted in accordance with the requirements of the UK Animals (Scientific Procedures) Act, 1986. Protocols were reviewed and approved by the Fraunhofer Chile Institute review board.
Randomization and Blinding
All experiments were conducted with the Fraunhoeffer Institute staff blinded to genotype and drug treatment. Separate investigators prepared and coded the dosing solutions, allocated the mice to the study treatment groups, dosed the animals, and collected the behavioral data.
Treatment Groups and Dosing
There were four treatment groups in the study with 10 male mice used per treatment group:
Group 1: fmr1 C57Bl6 knock-out mice treated with vehicle,
Group 2: wild-type C57Bl6 mice treated with vehicle,
Group 3: fmr1 C57Bl6 knock-out mice treated with BPN14770,
Group 4: wild-type C57Bl6 mice treated with BPN14770.
Mice were dosed by perioral gavage using a gavage tube suitable for mice. BPN14770 was dosed at 0.3 mg/kg once daily for 14 days as a suspension in 0.5% methylcellulose in sterile water. The suspension was stirred or shaken to prevent settling and maintain homogeneity. The dosing solution was prepared fresh daily. Vehicle-treated mice were dosed orally with 0.5% methylcellulose in sterile water. Behavioral assessment was after 14 days of dosing (Expt 1 and Expt 2) or after 14 days of dosing followed by 14 days of drug withdrawal (Expt 3).
Behavioral Testing
For experiments, all mice were tested once in the same apparatus. Prior to testing, mice not on study were placed in the apparatus for some minutes before the experiment. The apparatus was cleaned with moist and dry tissues before testing each mouse. The aim was to create a low but constant background mouse odor for all experimental subjects. Testers were blind to the genotype and treatment during all testing and data analysis
Open Field
The open-field apparatus was used to test multiple processes including anxiety/hyperactivity and habituation to a novel environment. The apparatus was a gray PVC-enclosed arena 50 × 9 × 30 cm divided into a 10 × 10 cm grid. Mice were brought to the experimental room 5–20 min before testing. A mouse was placed into a corner square facing the corner and observed for 3 min. The number of squares entered by the whole body (locomotor activity) and rears (both front paws off the ground, but not as part of grooming) were counted. The latency to the first rear was noted. The movement of the mouse around the field was recorded with a video tracking device for 3 min (version NT4.0, Viewpoint). The latency for the mouse to enter the brightest, central part of the field, total time spent in this central region, and total activity (as path length in cm) were recorded.
Social Interaction
The apparatus was a test arena/cage roughly the same size as the home cage. Typically, this was a 40 × 23 × 12 cm cage, with a Perspex lid to facilitate viewing the mice. The arena had fresh wood chips on the floor. Non-experimental mice were introduced to the cage and then removed before starting the test to provide a background mouse odor. Mice were transferred to the experimental room 10–15 min prior to test to wake them up. Simultaneously, both a test subject and a juvenile were placed in the test cage. The total duration and number of bouts of social investigation, defined as sniffing and close following (<2 cm from the tail) of the stimulus juvenile, were measured for 3 min.
Nesting
The test was performed in the same individual cages as above. Normal bedding covered the floor to a depth of 0.5 cm. Each cage was supplied with a “Nestlet,” a 5 cm square of pressed cotton batting (Ancare). Mice were placed individually into the nesting cages 1 hr before the dark phase, and the results were assessed the next morning. The nests were assessed on a 5-point scale, and the amount of un-torn Nestlet was weighed. Nest building was scored on a 5 point scale.
Score 1: The Nestlet was largely untouched (>90% intact).
Score 2: The Nestlet was partially torn up (50–90% remaining intact).
Score 3: The Nestlet was mostly shredded but often there was no identifiable nest site: <50% of the Nestlet remained intact but <90% was within a quarter of the cage floor area, i.e. the cotton was not gathered into a nest but spread around the cage. Note: the material may sometimes be in a broadly defined nest area but the critical definition was that 50–90% had been shredded.
Score 4: An identifiable, but flat nest: >90% of the Nestlet was torn up, the material was gathered into a nest within a quarter of the cage floor area, but the nest was flat, with walls higher than mouse body height (curled up on its side) on less than 50% of its circumference.
Score 5: A (near) perfect nest: >90% of the Nestlet was torn up, the nest was a crater, with walls higher than mouse body height on more than 50% of its circumference.
Marble Burying
Transparent plastic cages were filled with a 10-cm deep layer of sawdust on top of which 10 glass marbles were placed in two rows. Each animal was left undisturbed in such a cage for 30 min, after which the number of marbles that were buried to at least two-thirds of their depth was recorded.
Statistical Analysis of Behavioral Data
Data were analyzed by two-way analysis of variance (ANOVA) followed by post-test comparisons where appropriate using Tukey’s Multiple Comparison Test. Data are represented as the mean and standard error of the mean (SEM). Statistical analyses were performed in GraphPad Prism 7.03.
Ballistic dye labeling and microscopy
Ballistic dye labeling (DiI and DiO) was performed according to protocols developed by Afraxis to label target neurons (Afraxis, San Diego, CA). Sections were slide mounted and cover slipped. Laser-scanning confocal microscopy (Olympus FV1000) was performed using a 63X objective (1.42 NA) to scan individually labeled neurons at high resolution (0.098 × 0.098 × 0.33 μm voxels). Target neurons were identified in the brain region of interest by anatomical location and cell morphology. Microscopy was performed blind to experimental conditions. 10 mice were tested in each experimental condition. A minimum of 5 cells were sampled from each mouse.
Dendritic spine analysis, database lock and unblinding
Blind deconvolution (AutoQuant) was applied to raw three-dimensional digital images which were then analyzed for spine density and morphology by trained analysts. Individual spines were measured manually for (a) head diameter, (b) length, and (c) neck thickness from image Z-stacks using custom-built Afraxis ESP software. Each dendrite was analyzed by (on average) 2–3 independent analysts. Automated image assignment software distributed images to analysts in a randomized manner and ensured that each analyst performed measurements of near equal numbers of dendrites per group. Analysts were blinded to all experimental conditions (including treatment, brain region, and cell type). Statistical analysis of interanalyst variability for each dendrite was examined on-line and used to eliminate dendrites that did not meet interanalyst reliability criteria: For spine density and spine morphological classification, data across analysts were averaged to report data for each dendrite. Population distributions of each measure were compiled for each dendritic sample and pooled by group. Quantitative comparisons using the non-parametric Kolmolgorov-Smirnov test were used to assess group differences (Fig. 5). Considering the sensitivity of pooled-population non-parametric statistics, a conservative hypothesis test was applied (α = 0.0001). Population distributions for combinations of measures (spine length x head diameter or spine length x head diameter / neck width) were compiled for each dendritic sample and pooled by group. Difference maps were constructed by subtraction of raw distributions (control subtracted from target) as in Fig. 4. A 12-category classification scheme that describes highly granulated dendritic spine phenotypes was used to categorize every spine (Sup Fig. 8 & 9). These categories were collapsed into three categories representing immature, intermediate, and mature spine morphologies (Sup Fig. 8 & 9). Finally, an assessment independent from the 12-point scheme was used to describe classic spine phenotypes (e.g. mushroom, stubby, etc.).
All Afraxis experimenters were fully blinded to treatment conditions during the collection, assembly, and interpretation of the data. The database was locked after all spine data was collected prior to unblinding by treatment for statistical analysis.
Statistical Analysis of Spine Morphometry
Values are reported in tables and plots as group means ± standard errors of the mean (SEMs) where each data value (individual mouse) represents the mean value of all segments sampled from a given mouse. For all group comparisons of parametric values, statistical significance was determined using the analysis of variance test (ANOVA; SPSS). Post-hoc comparisons were assessed using the Student’s t-test (2-tailed). For the spine analysis, non-parametric comparisons of individual measure population distributions were conducted using the 2-sample Kolmogorov-Smirnov test (α = 0.0001).
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Verkerk, A. J. et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991).
Hagerman, R. J. et al. Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390 (2009).
Lozano, R., Azarang, A., Wilaisakditipakorn, T. & Hagerman, R. J. Fragile X syndrome: A review of clinical management. Intractable Rare Dis Res 5, 145–157 (2016).
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A. & Fischer, U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet 10, 329–338 (2001).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Fmr1. knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33 (1994).
Morales, J. et al. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972 (2002).
Choi, C. H. et al. PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J Neurosci 35, 396–408 (2015).
Kanellopoulos, A. K., Semelidou, O., Kotini, A. G., Anezaki, M. & Skoulakis, E. M. Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci 32, 13111–13124 (2012).
Berry-Kravis, E. & Sklena, P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. American Journal of Medical Genetics 45, 81–87 (1993).
Berry-Kravis, E., Hicar, M. & Ciurlionis, R. Reduced cyclic AMP production in fragile X syndrome: cytogenetic and molecular correlations. Pediatric Research 38, 638–643 (1995).
Zhang, Y. Q. et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603 (2001).
Dockendorff, T. C. et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984 (2002).
McBride, S. M. et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764 (2005).
Bolduc, F. V., Bell, K., Cox, H., Broadie, K. S. & Tully, T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nature Neuroscience 11, 1143–1145 (2008).
Houslay, M. D., Schafer, P. & Zhang, K. Y. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today 10, 1503–1519 (2005).
Gurney, M. E. et al. Heteroaryl Inhibitors of PDE4. United States Patent US 9,221,843 B2 (2015).
Hoffmann, R., Wilkinson, I. R., McCallum, J. F., Engels, P. & Houslay, M. D. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: generation of a molecular model. Biochem J 333(Pt 1), 139–149 (1998).
Sette, C. & Conti, M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem 271, 16526–16534 (1996).
Linglart, A. et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. The Journal of Clinical Endocrinology and Metabolism 97, E2328–2338 (2012).
Butler, M. G., Rames, L. J. & Wadlington, W. B. Acrodysostosis: report of a 13-year-old boy with review of literature and metacarpophalangeal pattern profile analysis. American Journal of Medical Genetics 30, 971–980 (1988).
Lee, H. et al. Exome sequencing identifies PDE4D mutations in acrodysostosis. American Journal of Human Genetics 90, 746–751 (2012).
Michot, C. et al. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. American Journal of Human Genetics 90, 740–745 (2012).
Lynch, D. C. et al. Identification of novel mutations confirms Pde4d as a major gene causing acrodysostosis. Human Mutation 34, 97–102 (2013).
Lindstrand, A. et al. Different mutations in PDE4D associated with developmental disorders with mirror phenotypes. Journal of Medical Genetics 51, 45–54 (2014).
Kaname, T. et al. Heterozygous mutations in cyclic AMP phosphodiesterase-4D (PDE4D) and protein kinase A (PKA) provide new insights into the molecular pathology of acrodysostosis. Cellular Signalling 26, 2446–2459 (2014).
Gurney, M. E., D’Amato, E. C. & Burgin, A. B. Phosphodiesterase-4 (PDE4) Molecular Pharmacology and Alzheimer’s Disease. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics 12, 49–56 (2015).
Gervasi, N., Tchenio, P. & Preat, T. PKA dynamics in a Drosophila learning center: coincidence detection by rutabaga adenylyl cyclase and spatial regulation by dunce phosphodiesterase. Neuron 65, 516–529 (2010).
Burgin, A. B. et al. Design of Phosphodiesterase Type 4D (PDE4D) Allosteric Modulators for Cognition with Improved Safety. Nature Biotechnology 28, 63–70 (2010).
Comery, T. A. et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci USA 94, 5401–5404 (1997).
Cruz-Martin, A., Crespo, M. & Portera-Cailliau, C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30, 7793–7803 (2010).
Thomas, A. M. et al. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res 223, 310–321 (2011).
Deacon, R. M., Croucher, A. & Rawlins, J. N. Hippocampal cytotoxic lesion effects on species-typical behaviours in mice. Behav Brain Res 132, 203–213 (2002).
He, C. X. & Portera-Cailliau, C. The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128 (2013).
Irwin, S. A., Galvez, R. & Greenough, W. T. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10, 1038–1044 (2000).
Cherry, J. A. & Davis, R. L. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. The Journal of Comparative Neurology 407, 287–301 (1999).
Bobon, D. et al. Is phosphodiesterase inhibition a new mechanism of antidepressant action? A double blind double-dummy study between rolipram and desipramine in hospitalized major and/or endogenous depressives. Eur Arch Psychiatry Neurol Sci 238, 2–6 (1988).
Bertolino, A. et al. Rolipram versus imipramine in inpatients with major, “minor” or atypical depressive disorder: a double-blind double-dummy study aimed at testing a novel therapeutic approach. Int Clin Psychopharmacol 3, 245–253 (1988).
Pecknold, J. C., McClure, D. J., Appeltauer, L., Wrzesinski, L. & Allan, T. Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. Journal of Clinical Psychopharmacology 2, 129–133 (1982).
Berry-Kravis, E. M. et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Science Translational Medicine 4, 152ra127 (2012).
Berry-Kravis, E. & Huttenlocher, P. R. Cyclic AMP metabolism in fragile X syndrome. Annals of Neurology 31, 22–26 (1992).
Gresele, P., Momi, S. & Falcinelli, E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol 72, 634–646 (2011).
Ito, M. et al. Characterization of the isoenzymes of cyclic nucleotide phosphodiesterase in human platelets and the effects of E4021. Cellular Signalling 8, 575–581 (1996).
Richter, W., Jin, S. L. & Conti, M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J 388, 803–811 (2005).
Kelly, M. P. et al. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cellular Signalling 26, 383–397 (2014).
Michalon, A. et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 (2012).
Yan, Q. J., Rammal, M., Tranfaglia, M. & Bauchwitz, R. P. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066 (2005).
Pacey, L. K., Tharmalingam, S. & Hampson, D. R. Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J Pharmacol Exp Ther 338, 897–905 (2011).
Kelley, D. J. et al. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS One 2, e931 (2007).
Dlaboga, D., Hajjhussein, H. & O’Donnell, J. M. Regulation of phosphodiesterase-4 (PDE4) expression in mouse brain by repeated antidepressant treatment: comparison with rolipram. Brain Res 1096, 104–112 (2006).
Fujita, M. et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Molecular Psychiatry (2016).
Bailey, C. H., Bartsch, D. & Kandel, E. R. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA 93, 13445–13452 (1996).
Kandel, E. R. The biology of memory: a forty-year perspective. J Neurosci 29, 12748–12756 (2009).
Tully, T., Bourtchouladze, R., Scott, R. & Tallman, J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov 2, 267–277 (2003).
Brown, J. A., Gianino, S. M. & Gutmann, D. H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci 30, 5579–5589 (2010).
Bourtchouladze, R. et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA 100, 10518–10522 (2003).
Bu, Q. et al. CREB Signaling Is Involved in Rett Syndrome Pathogenesis. J Neurosci 37, 3671–3685 (2017).
Acknowledgements
This work was supported by research grants from the FRAXA Foundation to P.C, the National Institute of Neurological Disorders and Stroke to M.G. (NS078034), and by Tetra Discovery Partners, Inc.
Author information
Authors and Affiliations
Contributions
M.G., M.T. and P.C. designed the experiments, P.C. conducted the behavioral tests and performed the statistical analysis with oversight by R.D., C.R. conducted the spine morphometry analysis. M.G. and M.T. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
M.E.G is an employee of Tetra. Tetra owns BPN14770. All other authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gurney, M.E., Cogram, P., Deacon, R.M. et al. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci Rep 7, 14653 (2017). https://doi.org/10.1038/s41598-017-15028-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15028-x
This article is cited by
-
mGluR7 allosteric modulator AMN082 corrects protein synthesis and pathological phenotypes in FXS
EMBO Molecular Medicine (2024)
-
An Update on Psychopharmacological Treatment of Autism Spectrum Disorder
Neurotherapeutics (2022)
-
Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders
Molecular Psychiatry (2021)
-
Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial
Nature Medicine (2021)
-
A genetic basis for friendship? Homophily for membrane-associated PDE11A-cAMP-CREB signaling in CA1 of hippocampus dictates mutual social preference in male and female mice
Molecular Psychiatry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
