
Abstract
Next-generation sequencing (NGS)-based circulating tumor DNA (ctDNA) assays have provided a new method of identifying tumor-driving genes in patients with advanced non-small cell lung carcinoma (NSCLC), especially in those whose cancer tissues are unavailable or in those that have acquired treatment resistance. Here, we describe a total of 119 patients with advanced EGFR-TKI-naive NSCLC and 15 EGFR-TKI-resistant patients to identify somatic SNVs, small indels, CNVs and gene fusions in 508 tumor-related genes. Somatic ctDNA mutations were detected in 82.8% (111/134) of patients in the total cohort. Of the 119 patients with advanced NSCLC, 27.7% (33/119) were suitable for treatment with National Comprehensive Cancer Network (NCCN) guideline-approved targeted drugs. Actionable genetic alterations included 25 EGFR mutations, 5 BRAF mutations, and 1 MET mutation, as well as 1 EML4-ALK gene fusion and 1 KIF5B-RET gene fusion. In 19.3% (23/119) of the patients, we also identified genomic alterations with that could be targeted by agents that are in clinical trials, such as mTOR inhibitors, PARP inhibitors, and CDK4/6 inhibitors. Additionally, the EGFR T790M mutation was found in 46.7% (7/15) of the patients with EGFR-TKI-resistant NSCLC, suggesting that the NGS-based ctDNA assay might be an optional method to monitor EGFR-TKI resistance and to discover mechanisms of drug resistance.
Similar content being viewed by others


Introduction
Most tumors are discovered to be locally advanced or metastatic, as is the case for lung cancer, which is a prevailing cause of death worldwide1. With advances in molecular diagnosis and targeted therapies, molecular genotyping is now routinely used to guide the clinical treatment of patients with non-small cell lung carcinoma (NSCLC). The efficacy of targeted kinase inhibitors was demonstrated to be superior to that of standard chemotherapy for patients with EGFR mutations or ALK/Ros1 fusions2. In addition, NSCLC frequently harbors genomic alterations in KRAS, BRAF, ERBB2, RET and MET. Potential targeted agents for these genomic mutations are available from an ongoing trial or are being used off protocol2.
Currently, ARMS PCR, Sanger sequencing and FISH are commonly used to detect a few targetable oncogenes and hotspot mutations3. However, such assays are insufficient since most of these genes are not altered in a large proportion of patients. In terms of the complex genomic alterations in NSCLC, there is an urgent need to screen potentially actionable targets simultaneously. Next-generation sequencing (NGS) has revolutionized molecular diagnostics, and enabled the simultaneous detection of multiple alterations in a single test. NGS-based hybrid capture assays not only allow the identification of hotspot mutations but also allow the assessment of unknown alterations, all from a single formalin-fixed, paraffin-embedded (FFPE) specimen or serum sample4.
Circulating tumor DNAs (ctDNAs), which carry tumor-specific sequence alterations, represent a variable and generally small fraction of the total circulating DNA5. Studies have shown that ctDNA is an informative, inherently specific and highly sensitive biomarker of metastatic breast cancer6. Analysis of ctDNA is particularly attractive for those patients without enough tissue samples or those who cannot be repeatedly sampled with invasive procedures after disease progression. In NSCLC, both non-NGS-based and NGS-based assays with variable sensitivity have been used to detect genomic alterations in serum samples7,8. The detection of ctDNA in NSCLC could be used to guide targeted therapy, identify resistance mechanisms, and monitor clinical prognosis9,10,11. Most of the ctDNA detection approaches are limited to hotspot mutations in a few genes. By comparison, NGS-based ctDNA assays offer the ability to profile a much broader range of genetic alterations in a single test. To date, studies have tried to apply ctDNA NGS panels that screen from 70 to 252 genes for the detection of clinically actionable variants and resistance mutations in patients with lung cancer12,13.
In this study, a broad hybrid capture-based 508-gene panel NGS assay (Oseq-NT) was used to screen targetable genomic alterations of ctDNA from patients with NSCLC. We intended to confirm the potential benefits of this ctDNA detection method in guiding personalized therapy in patients with NSCLC.
Materials and Methods
Patients and Samples
We analyzed 119 patients with stage IIB-IV NSCLC and 15 patients who had developed drug-resistance to EGFR-TKIs. Patient characteristics were shown in Table 1. The diagnosis was verified by fine-needle aspirations or cell pathology of pleural effusion before any therapy. Peripheral blood sample collections (10 ml) were approved by the Ethics Committee of the Affiliated Hospital of Qingdao University, and all patients signed informed consent. All the experiments were carried out in accordance with the guideline released by the National Health and Family Planning Commission of the PRC.
NGS-based ctDNA assay
Circulating DNA was isolated from 2 mL plasma with the QIAamp Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer’s instructions. Genomic DNA from peripheral blood was purified using QIAamp DNA Blood Mini Kit (Qiagen). DNA purity and concentration were examined by the NanoDrop2000 spectrophotometer and Qubit 2.0 Fluorometer with Quant-IT dsDNA HS Assay Kit (Thermo Fisher Scientific), respectively. The quality of genomic DNA from peripheral blood was assessed by agarose gel electrophoresis and the size distribution of circulating DNA was evaluated on a 2100 Bioanalyzer using the DNA 1000 Kit (Agilent).
Library construction with peripheral blood DNA was performed as previously described using 1mg of DNA sheared by an ultrasonoscope to generate fragments with a peak of 250 bps, followed by end repair, A-tailing and ligation to the Illumina-indexed adapters according to the standard library construction protocol. Target enrichment was performed on a custom sequence capture-probe (Nimblegen, USA) which targeted 7,708 exons of 508 cancer-related genes and 78 introns from 19 genes recurrently rearranged in solid tumor representing totaling ~1.7Mb of the human genome (Supplement Table S1). Library for circulating DNA was constructed by KAPA LTP Library Preparation Kit for Illumina Platform (Kapa Biosystems) following the manufacturer’s instructions without modification. Sequencing was performed with 2 × 101 bp paired-end reads and 8-bp index read on an Illumina Hiseq. 2500 plateform (Illumina, San Diego, USA).
Raw reads were first processed by removing adaptors and filtering low-quality ones using SOAPnuke (http://soap.genomics.org.cn/). Clean reads were then aligned to the human reference GRCh37 using BWA aligner (v0.6.2-r126)14. PCR duplication were removed by PICARD (v1.98). Local realignment and base quality score recalibration were performed using GATK (v2.3–9)15, based on which we removed poorly mapped reads. Then we identified SNVs using Mutect and SOMATK-SNV (developed by BGI, manuscript in preparation), and InDels were detected using GATK and SOMATK-INDEL (developed by BGI, manuscript in preparation). CNV calling was applied by CONTRA (v2.0.4)16. The CNV analysis was performed based on off-target sequencing data, which is used as low-depth whole genome sequencing data described in research by Bellos E et al.17. We split the whole genome into bins with 500kb length and count the read depth (RD) in each bin, followed by GC normalization method described by Yu Z et al.18. Bic-seq19 was used for segmentation of off-target sequencing data.
Statistical analysis
The experimental data are presented as the mean ± SEM and were analyzed with the two-tailed Student’s t test. The threshold of P < 0.05 was considered as statistically significant.
Results
Use of the NGS-based ctDNA assay to screen 119 patients with advanced NSCLC
The clinical and pathological features of the patients are summarized in Table 1. The median age at diagnosis was 59.7 years with a range of 31–94, and 57.1% of the patients were smokers (68 of 119). Screening of the patients’ ctDNA identified somatic mutations in a total of 189 genes (with a mean of 5.5 ± 5.4 mutations per patient). Of all the samples, 81.5% (97/119) exhibited at least one genetic alteration and 18.5% (22/119) of samples exhibited no detectable alterations. The median average sequencing depth was 950 × for cell-free DNA from each sample and the basic summary of the genomic sequencing can be found in Supplementary Table S2.
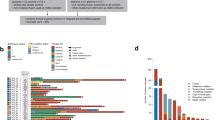
In total, 37.0% (44/119) of the patients with NSCLC had at least one targetable alteration (mean 1.38 ± 0.53) (Fig. 1), and the frequency of each specific alteration ranged from 0.5% to 56.9% (Supplementary Table S3). In terms of pathology type, targetable alterations were found in 37.1% (39/105) of the patients with adenocarcinoma and in 35.7% (5/14) of the patients with squamous cell carcinoma. Genomic alterations with corresponding targeted agents approved by the NCCN guidelines for NSCLC were identified in 27.7% (33/119) of the patients (Table 2). These actionable alterations included EGFR (n = 25), BRAF (n = 5), EML4-ALK (n = 1), KIF5B-RET (n = 1), and MET (n = 1). In addition, actionable genomic alterations corresponded to targeted therapy options that were available from ongoing trials or are being used off protocol were identified in another 19.3% (23/119) of the patients (Table 2). The most common alterations identified in our analysis included those in K-RAS (n = 4), TP53 (n = 5), N-ras (n = 1), ATM (n = 1) and CDKN2A (n = 1). Notably, we also identified 6 mutated genes that encode proteins involved in the mTOR pathway: namely, FBXW7 (n = 5), PIK3CA (n = 2), PTEN (n = 1), NF1 (n = 1), NF2 (n = 1) and STK11 (n = 1).

Comprehensive annotation of 44 NSCLC patients harboring targetable genomic alterations.
Numerous other alterations with no available targeted drugs were also observed in our cohort. Mutations in the following ten genes were observed in more than 5 samples: TP53, EGFR, FBXW7, BRAF, MLL2, MLL3, NAV3, K-RAS, FAT3 and TRRAP. Of these, no targeted agents have been reported for MLL2, MLL3, NAV3, FAT3, and TRRAP. The most frequent mutant genes were TP53 (n = 49, 41.2%) and EGFR (n = 30, 25.2%).
The EGFR mutation spectrum and concomitant actionable genetic alterations
Of the 119 patients with NSCLC, 25 (21.0%) were found to harbor actionable EGFR mutations. It should be noted that EGFR mutations were detected in 21.9% (23/105) of the patients with adenocarcinoma and 14.3% (2/14) of the patients with squamous cell carcinoma. The EGFR mutations in these 25 patients with NSCLC were located in the following exons: exon 18 (n = 2), exon 19 (n = 11), exon 20 (n = 2), exon 21 (n = 9), and exons 20 and 21 (n = 1) (Fig. 2). Notably, 7 of the 25 patients were found to harbor alternative actionable genetic alterations in addition to the EGFR mutation. Concomitant BRAF or NRAS mutations were found in two patients. In 4 patients, an EGFR mutation was accompanied with a mutation in an mTOR pathway-related gene (2FBXW7, 1NF1, and 1PTEN) (Table 2).

EGFR mutation spectrum in this 25 NSCLC patients.
An NGS-based ctDNA assay at the time of acquired EGFR-TKI resistance
Fifteen patients with NSCLC who had developed resistance to EGFR-TKIs underwent blood sampling and NGS-ctDNA assays (Table 3). Tissue sample analysis confirmed that all these patients harbored EGFR-activating mutations before EGFR-TKI treatment. Of the 15 plasma samples obtained from patients post-EGFR-TKI treatment, EGFR-sensitive mutations were detected in 10 patients, which was consistent with prior tissue-based assays. Noticeably, the EGFR T790M mutation was also identified in 46.7% (7/15) of the patients. The allele frequency of the T790M mutation in this study varied from 0.8 to 28.3%. More importantly, the following mutations, which might be involved in EGFR-TKI resistance, were each identified in 1 patient: ERBB2 L755S, NRAS Q61K and EGFR amplification.
Discussion
The identification of specific molecular targets in NSCLC has led to the development of oncogene-directed targeted therapies. At present, several methods have been used to identify targetable mutations, and these methods have mainly analyzed cancer tissue samples20,21. However, it is often difficult to obtain sufficient tumor samples for genetic analysis in clinical practice. For patients who develop resistance to targeted therapy, molecular analysis is urgently required to identify the prevalence of both known and new resistance mutations that occur during therapy22. Under these circumstances, a noninvasive and real-time genomic detection assay is particularly valuable. In this study, we intended to discover actionable genomic alterations using an NGS-based ctDNA assay in patients with advanced NSCLC so that approved drugs or investigational agents in clinical trials could be selected. Through the study of 119 patients, ctDNA screening identified somatic mutations in a total of 189 genes across all patients, and somatic mutations were present in 81.5% of the patients. More importantly, 37.0% (44/119) of patients with NSCLC harbored at least one targetable alteration.
The detection and monitoring of tumors via noninvasive methods is a major challenge in oncology. ctDNA is composed of small fragments of nucleic acid that could be released into the circulation from CTCs and/or cancer tissues. Through the use of digital PCR technologies, Bettegowda et al. detected ctDNA in most patients with advanced cancers23. ctDNA might be a broadly applicable, sensitive, and specific biomarker that could be used for clinical purposes in patients with multiple different types of cancer24,25. In NSCLC, Newman et al. detected ctDNA in 100% of patients with stage II-IV NSCLC and in 50% of patients with stage I NSCLC using deep sequencing (CAPP-seq)26. In this study, somatic mutations were detected in the ctDNA of 97 of the 119 (81.5%) patients with advanced NSCLC using a broad, hybrid capture-based NGS assay (Oseq-NT) that has sufficient sensitivity and clinical applicability.
In advanced NSCLC, patients who have genomic alterations that are matched a targeted therapy have been found to live substantially longer than do those without such alterations27. However, largely due to limited tissue resources, not all the patients in this previous study could be analyzed by a genomic assay. Through the use of an NGS-based ctDNA assay in the present study, genomic alterations were identified in 27.7% of patients, and included alterations in EGFR, ALK, RET, BRAF, and MET. Consequently, these patients could receive corresponding targeted agents approved by the NCCN guidelines. In plasma DNA samples of Chinese patients with advanced adenocarcinoma, Qin et al. compared three methods for detecting EGFR mutations. EGFR mutations were detected in 6.9% samples by direct DNA sequencing, in 30.1% by denaturing high-performance liquid chromatography, and in 38.4% by Scorpions ARMS-PCR28. In contrast, EGFR mutations were detected in 21.9% (23/105) of adenocarcinoma samples by the NGS-based ctDNA assay in this study, which is a much higher proportion than detected by direct DNA sequencing yet a lower proportion than identified by Scorpions ARMS-PCR in the previous study. In addition, EGFR mutations were detected in 40.0% of non-paired tissue samples by the same NGS method (data not shown). Consequently, the approximate sensitivity of the NGS-based EGFR ctDNA assay reached 54.8%, and this needs to be improved by increasing the sequencing depth in the future.
Notably, our research discovered an additional 19.3% actionable genomic alterations for which targeted agents are available from ongoing clinical trials or are being used off protocol. The NCCN NSCLC Guidelines Panel strongly endorses broader molecular profiling with the goals of identifying rare driver mutations for which effective drugs may already be available and appropriately counselling patients regarding the availability of clinical trials. A study by Drilon et al. found that broad, hybrid capture-based NGS approaches could identify actionable genomic alterations in 65% of tumors from lung cancers that had been shown to lack targetable genomic alterations by earlier extensive non-NGS testing4. First-line profiling of lung adenocarcinomas using broad, hybrid capture-based NGS might be a more comprehensive and efficient strategy than is non-NGS testing. Therefore, NGS combined with a Scorpions ARMS-PCR ctDNA assay is a very promising diagnostic regimen for patients with NSCLC for whom limited tissue samples are available. Numerous other alterations with no available targeted drugs were also observed. The frequencies of mutations in several genes, including TP53, EGFR, FBXW7, BRAF and K-RAS, were consistent with previous massively parallel sequencing studies in lung adenocarcinoma29.
Although the application of TKI therapy to patients with EGFR-mutant NSCLC has led to a dramatic lengthening of both progression-free survival (PFS) and overall survival (OS), nearly all patients ultimately progress while receiving EGFR-TKI treatment. EGFR T790M has been identified as the most common mechanism of acquired resistance, and other, less frequent mechanisms have also been reported, including MET amplification, HER2 amplification, mutation of PIK3CA and BRAF, transformation to small-cell lung cancer and epithelial-to-mesenchymal transition30. The mechanism of EGFR-TKI resistance will determine the next treatment required, which emphasizes the need to repeat the biopsy and carry out molecular characterization of tumor samples at clinical progression. However, it is challenging to obtain serial tumor rebiopsies following disease progression in clinical practice owing to the invasiveness of the procedure and tumor heterogeneity. Several studies have assessed the ability of different technology platforms to detect EGFR mutations, including T790M7,9,26, in ctDNA. Detection rates of T790M ctDNA in plasma from patients post-EGFR-TKI treatment ranged from 43 to 47%, as determined by digital PCR21,31. In this study, EGFR T790M was detected in 46.7% of the patients with EGFR-TKI-resistant NSCLC by our NGS assay. Our results indicated that NGS was not inferior to digital PCR at detecting T790M in ctDNA. Interestingly, 3 EGFR-TKI resistance-related genetic alterations aside from EGFR-790M were identified in this study by the NGS assay: namely, ERBB2 L755S, NRAS Q61K and EGFR amplification. All of these EGFR-TKI-resistant genetic alterations might be missed by low-throughout non-NGS tests, which suggests the NGS ctDNA assay might be a more sensitive and comprehensive approach for drug resistance surveillance.
Recently, the most commonly used methods to detect EGFR mutations in ctDNA from patients with NSCLC depends on PCR-based techniques, and there has been a recent emergence of digital PCR and NGS. Representative studies are displayed in Table 4. Compared with these studies, the NGS-based ctDNA assay used in this study could identify more targeted alterations in addition to EGFR and could disclose EGFR-TKI resistance-related mutations aside from EGFR T790M. NGS-based ctDNA assays, especially with large gene panels, have a great advantage in guiding personalized treatment considering the growing number of therapeutic targets and known genetic alterations that confer resistance in NSCLC.
In summary, actionable genetic alterations were identified in 37.0% of patients with advanced NSCLC by an NGS-based ctDNA assay. Noninvasive NGS-based ctDNA assays might be a potential method for monitoring EGFR-TKI resistance and discovering drug-resistance mechanisms. The NGS-based ctDNA assay might be a comprehensive and efficient strategy that complements tissue analysis and expands the scope of personalized targeted therapies for advanced NSCLC.
Change history
15 January 2018
A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
References
Torre, L. A. et al. Global cancer statistics, 2012. CA Cancer J Clin. 65, 87–108 (2015).
Tsao, A. S. et al. Scientific Advances in Lung Cancer 2015. J Thorac Oncol. 11, 613–638 (2016).
Liu, X. et al. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol. 66, 1065–1069 (2013).
Drilon, A. et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin Cancer Res. 21, 3631–3639 (2015).
Mouliere, F. & Rosenfeld, N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. Proc Natl Acad Sci USA 112, 3178–3179 (2015).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 368, 1199–1209 (2013).
Jiang, T., Ren, S. & Zhou, C. Role of circulating-tumor DNA analysis in non-small cell lung cancer. Lung Cancer. 90, 128–134 (2015).
Qiu, M. et al. Circulating tumor DNA is effective for the detection of EGFR mutation in non-small cell lung cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 24, 206–212 (2015).
Thress, K. S. et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 90, 509–515 (2015).
Que, D. et al. EGFR mutation status in plasma and tumor tissues in non-small cell lung cancer serves as a predictor of response to EGFR-TKI treatment. Cancer Biol Ther. 17, 320–327 (2016).
Guo, N. et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci Rep. 6, 33519 (2016).
Thompson, J. C. et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin Cancer Res. 22, 5772–5782 (2016).
Chabon, J. J. et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 7, 11815 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, J. et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 28, 1307–1313 (2012).
Bellos, E. & Coin, L. J. cnvOffSeq: detecting intergenic copy number variation using off-target exome sequencing data. Bioinformatics. 30, i639–645 (2014).
Yu, Z., Liu, Y., Shen, Y., Wang, M. & Li, A. CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data. Bioinformatics. 30, 2576–2583 (2014).
Xi, R. et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA 108, E1128–1136 (2011).
Lopez-Rios, F. et al. Comparison of molecular testing methods for the detection of EGFR mutations in formalin-fixed paraffin-embedded tissue specimens of non-small cell lung cancer. J Clin Pathol. 66, 381–385 (2013).
Shao, D. et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sci Rep. 6, 22338 (2016).
Lin, Y., Wang, X. & Jin, H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 4, 411–435 (2014).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 6, 224 (2014).
Lipson, E. J. et al. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother. Cancer. 2, 42 (2014).
Thierry, A. R. et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 20, 430–435 (2014).
Newman, A. M. et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 20, 548–554 (2014).
Nguyen, K. S., Neal, J. W. & Wakelee, H. Review of the current targeted therapies for non-small-cell lung cancer. World J Clin Oncol. 5, 576–587 (2014).
Qin, L., Zhong, W., Zhang, L., Li, L. Y. & Wang, M. Z. Comparison of three methods for detecting epidermal growth factor receptor mutations in plasma DNA samples of Chinese patients with advanced non-small cell lung cancer. Chin Med J (Engl). 124, 887–891 (2011).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 150, 1107–1120 (2012).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 19, 2240–2247 (2013).
Wang, Z. et al. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS One 9, e110780 (2014).
Bai, H. et al. Epidermal growth factor receptor mutations in plasma DNA samples predict tumor response in Chinese patients with stages IIIB to IV non-small-cell lung cancer. J Clin Oncol. 27, 2653–2659 (2009).
Jian, G. et al. Prediction of epidermal growth factor receptor mutations in the plasma/pleural effusion to efficacy of gefitinib treatment in advanced non-small cell lung cancer. J Cancer Res Clin Oncol. 136, 1341–1347 (2010).
Jiang, B. et al. Serum detection of epidermal growth factor receptor gene mutations using mutant-enriched sequencing in Chinese patients with advanced non-small cell lung cancer. J Int Med Res. 39, 1392–1401 (2011).
Goto, K. et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol. 7, 115–121 (2012).
Bai, H. et al. Influence of chemotherapy on EGFR mutation status among patients with non-small-cell lung cancer. J Clin Oncol. 30, 3077–3083 (2012).
Zhang, L. et al. Detection of EGFR somatic mutations in non-small cell lung cancer (NSCLC) using a novel mutant-enriched liquidchip (MEL) technology. Curr Drug Metab. 13, 1007–1011 (2012).
Zhao, X. et al. Comparison of epidermal growth factor receptor mutation statuses in tissue and plasma in stage I-IV non-small cell lung cancer patients. Respiration. 85, 119–125 (2013).
Kim, S. T. et al. Can serum be used for analyzing the EGFR mutation status in patients with advanced non-small cell lung cancer? Am J Clin Oncol. 36, 57–63 (2013).
Duan, H. et al. Comparison of EGFR mutation status between plasma and tumor tissue in non-small cell lung cancer using the Scorpion ARMS method and the possible prognostic significance of plasma EGFR mutation status. Int J Clin Exp Pathol. 8, 13136–13145 (2015).
Zheng, D. et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci Rep. 6, 20913 (2016).
Acknowledgements
This work was supported by the Taishan Scholar foundation (No.tshw201502061), the Qingdao Key Discipline foundation and the Qingdao Leading Innovation Team foundation.
Author information
Authors and Affiliations
Contributions
Helei Hou, Xiaonan Yang, Jinping Zhang, Xiaochun Zhang, Zhuokun Li designed the project. Helei Hou and Zhaoyang Qian wrote the paper. Helei Hou, Zhe Zhang, Xiaomei Xu, Chuantao Zhang, Dong Liu and Na Zhou conducted experiments. Zhaoyang Qian and Helei Hou did the data analysis. Hongmei Zhu and Xiaoping Zhang directed the NGS test. Weihua Yan performed the pathological diagnosis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Change History: A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A correction to this article is available online at https://doi.org/10.1038/s41598-018-19648-9.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hou, H., Yang, X., Zhang, J. et al. Discovery of targetable genetic alterations in advanced non-small cell lung cancer using a next-generation sequencing-based circulating tumor DNA assay. Sci Rep 7, 14605 (2017). https://doi.org/10.1038/s41598-017-14962-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14962-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
