
Abstract
HAPE susceptible (HAPE-S, had HAPE episode in past) subjects may have subclinical cardio-pulmonary dysfunction. We compared the results of pulmonary function tests in 25 healthy HAPE-S non-mountaineers and 19 matched HAPE resistant (HAPE-R, no HAPE episode in past). Acute normobaric hypoxia (FIo2 0.12) was administered at sea level to confirm hypoxia intolerance in HAPE-S. Unlike HAPE-R, HAPE-S subjects had elevated baseline and post-hypoxia systolic pulmonary arterial pressures (20.9 ± 3 vs 27.3 ± 5 mm Hg during normoxia and 26.2 ± 6 vs 45.44 ± 10 mm Hg during hypoxia, HAPE-R vs HAPE-S). Forced vital capacity (FVC) and single breath alveolar volume (SBVA) were significantly lower in HAPE-S compared to HAPE-R (FVC: 4.33 ± 0.5 vs 4.6 ± 0.4; SBVA: 5.17 ± 1 vs 5.6 ± 1 Lt; HAPE-S vs HAPE-R). Two subgroups with abnormal pulmonary function could be identified within HAPE-S; HAPE-S1 (n = 4) showed DLCO>140% of predicted, suggestive of asthma and HAPE-S2 (n = 12) showed restrictive pattern. Each of these patterns have previously been linked to early small airway disease and may additionally represent a lower cross-sectional area of the pulmonary vascular bed, related to lower lung volumes. HAPE susceptibility in healthy non-mountaineers may be related to sub-clinical pulmonary pathology that limits compensatory rise in ventilation and pulmonary circulation during hypoxic stress.
Similar content being viewed by others

Introduction
High Altitude Pulmonary Edema (HAPE) is a hydrostatic oedema which occurs at altitude more than 2500 m in susceptible individuals1,2,3. The risk is believed to be multifactorial with prior work from our and other labs implicating genetic abnormalities in the hypoxic response pathway as well as chronically up regulated hypoxic response4,5,6,7. Subclinical cardiopulmonary dysfunction may have a synergistic role in this context and it has been previously reported that HAPE resistant (HAPE-R) subjects have a significantly greater forced vital capacity (FVC), on spirometry, than HAPE susceptible (HAPE-S)8,9. Low FVC volumes have been reported to be one of the strongest non-invasive predictors of cardiopulmonary risk10,11,12. While the mechanism for this remains unknown, it is thought that the underlying chronic low-grade inflammation have a significant role in causing reduced lung function13,14. To determine the possible role of pulmonary function testing, especially FVC, for predicting HAPE-risk, we conducted this study in a non mountaineer population that was physically fit and carefully followed a prescribed acclimatisation schedule during ascent to high altitude. We measured pulmonary functions in both HAPE-S and HAPE-R followed by acute normobaric hypoxia stress given to all individuals at sea level and pulmonary vascular responses were recorded to confirm hypoxia intolerance in HAPE-S.
Methods
We studied 44 male Army soldiers whose susceptibility and resistance for HAPE was known from their previous stay at high altitude. All participants were non smokers, lowlanders, free of airway infection and receiving no medication at the time of study. None of the subject has resided above 2000 m within last six months before the baseline measurements were carried out in Delhi, India at an altitude 293 m above sea level. The HAPE susceptible subjects (HAPE-S, n = 25) suffered the illness which was confirmed radiologically in spite of observing acclimatisation schedule. HAPE resistant (HAPE-R) subjects were drawn from colleagues who were deployed similarly but did not have any adverse event and showed normal pulmonary vascular response15 at sea level (sPpa <38 mm Hg at Fio2 = 0.12). Two groups were screened out of twenty-five HAPE-S subjects based on abnormal pulmonary functions: a) DLCO>140% predicted16 for inclusion under HAPE-S1, n = 4. b) FVC and TLC < average normal level of % predicted for inclusion under HAPE-S2, n = 12. We selected 140% of the predicted DLCO as the cutoff for the selection of subjects under HAPE-S1 group for the following reasons: 1) this is well outside the common range of inter subject variability and (2) this increase in DLCO would be unlikely to be secondary to technical or physiological variations during testing. All experimental protocols were approved by Defence Institute of Physiology and Allied Sciences Ethics Committee for scientific experiments. Informed written consent was obtained from all participants before enrolment in the study. All methods were carried out in accordance with the approved guidelines and regulations.
General Procedures
The subjects were investigated in the supine position while breathing synthetic gas mixtures consisting of 21 or 12% oxygen (FiO2 = 0.12) mixed in nitrogen. The hypoxic gas mixture corresponded to an altitude of 4500 m. Inhalation was performed via a tight fit face mask. Systolic pulmonary artery pressure (sPpa) was recorded before and at the end of 30 min. of hypoxic stress.
Determination of pulmonary artery systolic pressure
Pulmonary17 artery hemodynamics was measured non invasively using echocardiography. Echocardiography studies were performed with My Lab 30 Gold Line ultrasonograph (Esaote India). Standard parasternal and apical two dimensional views were obtained, and color flow directed pulse wave Doppler measurements of transvalvular flows and continuous wave Doppler measurements of tricuspid regurgitant flow were obtained. A single lead electrocardiogram was recorded on the ultrasonograph. Measures obtained using this noninvasive technique correlates closely with those obtained using cardiac catheterisation.
Pulmonary artery systolic pressure (sPpa) was calculated as follows
where TRvel is tricuspid regurgitation jet velocity and RAP is the estimated right atrial pressure based on respiration variation in inferior venacava size.
Pulmonary function measurement
Spirometry and DLCO measurements were performed in compliance with American Thoracic Society (ATS) guidelines18,19,20. Basal pulmonary function data was measured using dry, rolling seal spirometer (P. K. Morgan, Kent, UK). In addition to lung volumes and flow measurements, functional residual capacity (FRC) was measured by the closed circuit Helium dilution test. Diffusion capacity of the lung (DLCO) was determined by single-breath, breath-holding technique. The DLCO was routinely adjusted for hemoglobin if value was outside the normal range. Reference values for predicted DLCO were from North Indian population21,22. The best value from three attempts were recorded as both absolute values and as percentage of the predicted values, based on age and body weight.
Quality control
DLCO tests were performed by trained technician using equipment that was calibrated using a 3 litre syringe. Subjects performed at least one repeat DLCO test after the initial manure. If DLCO and SBVA values were not within 10%, the test was repeated a third time, and the values that were the closest match were used.
Statistics
Difference between two groups was assessed by t-test after confirming normal distribution. Since the values are highly auto-correlated, no further correction was performed. Analysis was done using R statistical programming language. All data are presented as means ± SD. P < 0.05 is considered significant.
Results
HAPE-S showed abnormal pulmonary vascular response to hypoxia. Table 1 shows anthropometric data, pulmonary functions and pulmonary vascular response to hypoxic challenge for HAPE-S and HAPE-R subjects. The two groups were similar in terms of age, height and weight. Hb was normal in both groups but slightly higher in HAPE-R. Baseline systolic pulmonary artery pressure (sPpa) was high and showed exaggerated response to acute hypoxia in HAPE-S.
HAPE-S showed low lung volumes. Pulmonary function data (Table 1) shows significantly lower FVC, FEV1 and SBVA in HAPE-S compared to HAPE-R. To determine how many of the otherwise healthy HAPE-S subjects would be defined as clinically abnormal using standard PFT definitions, we used predicted values for each subject and classified each subject, using standard clinical guidelines (see methods). All HAPE-R subjects and nine of twenty-five HAPE-S subjects had normal PFT. In the sixteen HAPE-S subjects with at least one abnormal PFT result, two types of abnormal patterns emerged —abnormally high DLCO (HAPE-S1) or a restrictive pattern in (HAPE-S2), as shown in Tables 2 and 3.
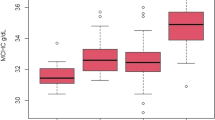
Pulmonary function testing has modest utility to discriminate HAPE susceptibility. To determine the usefulness of PFT parameters in predicting abnormal hypoxia-induced sPpa increase, we looked at the degree of correlation between the pulmonary function parameters found to be different in HAPE-S subjects and sPpa response to hypoxia (Fig. 1). Negative correlations were observed between SBVA or FVC, with baseline sPpa as well as hypoxia induced sPpa elevation (Table 4). Further, to determine their suitability as predictive markers, we also plotted receiver-operating characteristics (ROC) for these parameters (Fig. 2). None had a ROC area under curve greater than 0.8, when used individually. Even using a random forest technique based multi-parametric classification system, error rates remained close to 40%, comparable to the number of HAPE-S subjects with normal PFT.

Scatter plots showing individual data of different HAPE-S subgroups: HAPE-S1, HAPE-S2, HAPE-N (HAPE-S with normal PFT) and HAPE-R individuals. Plots between Hypoxic Response with single breath alveolar volume (SBVA) (panel A) and forced vital capacity (FVC) (panel C) and Systolic pulmonary artery pressure (sPpa) in hypoxic condition with SBVA (panel B) and FVC (panel D) showing negative correlation.

Receiver operator curve to predict HAPE susceptibility indicating the AUC for forced vital capacity (FVC) as panel A and single breath alveolar volume (SBVA) as panel B.
Discussion
Identification of otherwise fit subjects at risk of developing HAPE when taken to high altitude is an important problem. Others and we have previously reported a multitude of potential genetic or biochemical biomarkers4,5,6,7. However, there continues to be a need for simple nonspecialised physiological tests that can be done in resource-limited settings. While assessment of hemodynamic response during hypoxic challenge is a gold standard physiological test, it is neither simple, nor can it be performed outside specialised centres like ours. Receiver operator curve (ROC) curves represents excellent, good, and worthless tests plotted on the same graph. The accuracy of the test depends on how well the test separates the group being tested into those with and without the disease in question. Accuracy is measured by the area under the ROC curve (AUC).Here we show that low FVC is a potential marker of HAPE risk, but has poor sensitivity at acceptable levels of specificity (AUC = 0.66, sensitivity of about 40% at specificity of 80%), probably due to multiple etiological factors contributing to HAPE susceptibility like abnormal haemoglobin fractions as discussed in our previous study7. Additional PFT parameters beyond spirometry improve the discriminatory capacity somewhat, but about 40% of subjects who are HAPE susceptible may remain unidentified. Nevertheless, even modest predictive capacity of spirometry to identify HAPE-S subjects is potentially important since the test is easy to perform in any setting and is a good health screening tool in general. Using strict cutoffs that correctly classified all the HAPE-R subjects, about 20% of HAPE-S subjects could have been identified by FVC, which can be increased by simultaneous measurement of brain natriuretic peptide (BNP) showing AUC value 0.85, HIF1alpha and haemoglobin fractions6,7.Our findings are generally comparable to other reports, but with some important differences. HAPE-resistant subjects have previously been reported to have about 10% greater FVC compared to susceptible subjects in a small study9. Another study showed reduced FVC and TLC with no reduction in resting DLCO, but rebreathing DLCO was low at all times while doing normobaric and hypoxic exercise in HAPE-S8. We found no difference in FEV1/FVC and FRC in HAPE-S in contrast to previous studies23 however a sub group (HAPE-S2) showed low FVC, TLC and DLCO compared to control. Some of these differences may simply relate to different populations under study. Previous studies on susceptibility to HAPE were done on mountaineers and experimental protocols involved rapid ascent to high altitude. It is well known that physical exertion associated with rapid ascent is an independent factor for occurrence of HAPE. In contrast, our study is on Indian Army personnel who were low landers and deployed at high altitude on tenure basis. Strict instructions were followed to prevent high altitude maladies. Yet, despite these precautions during deployment, some of these personnel develop HAPE, presumably reflecting a greater susceptibility to HAPE than the mountaineers who developed HAPE during rapid ascent. This may explain why prior studies show comparable baseline sPpa between HAPE-S and HAPE-R subjects from a mountaineer population24,25 while we find higher baseline sPpa in HAPE-S subjects of this and previous studies6,7. Baseline difference in sPpa between two groups is partly related to inclusion criteria where subjects with normal pulmonary vascular response (normal PVR: sPpa < 38 mm Hg at Fio2 = i.e.0.12), apart from uneventful stay at high altitude, were included in HAPE-R group in this study15. However, HAPE-S was clinically defined since diagnosis was made on chest radiography and showed baseline raised BNP levels6 which in turn is inversely correlated with arterial oxygen levels thus the physiological differences remain meaningful.
The mechanistic basis of these observations remains unclear but there is strong clinical evidence linking FVC to vascular as well as pulmonary disease. John Hutchison, the inventor of spirometry, coined the term “vital capacity” i.e., capacity for life, as this seemed predictive for premature mortality26. This was subsequently confirmed by the Framingham Study where it was found to predict cardiovascular and all-cause mortality. It is worth mentioning here that FVC < 85% predicted has the highest hazard ratios for all cause mortality within framingham risk score groups27 which is consistent with present study showing FVC 84.83 ± 7.15% predicted in HAPE-S2 group. Since then additional studies have also confirmed associations between reduced FVC and a variety of non-pulmonary diseases including stroke, diabetes, hypertension, amongst others28. The chronic systemic inflammation29 and chronic hypoxia mediated vascular remodeling6,7 observed in our previous studies on non mountaineer HAPE-S can lead to a combination of small airway disease and reduced pulmonary vascular bed capacity, which may not manifest at sea level but prevent adequate compensatory increase in ventilation and perfusion, leading to HAPE. In support, the restrictive pattern shown by HAPE-S2 and asthma pattern16 shown by HAPE-S1 in our study has been strongly associated with presence of small airway or vascular disease in a large observational cross-sectional and longitudinal study30. Interestingly, interstitial lung diseases are an uncommon basis for such a pattern in otherwise healthy subjects from the general population. Surprisingly, raised baseline and increased endothelia-1(ET-1) levels at high altitude were reported in HAPE-S mountaineers31. ET-1 has been linked to lung fibrosis although ET-1 antagonist are ineffective in true fibrotic lung diseases like idiopathic pulmonary fibrosis, suggesting that ET-1 is part of a general lung remodeling pathway.
The major limitations of our study are that the subjects were all male and were studied after the development of HAPE. The incidence of HAPE is very low in army troops so prospective study was not feasible and the troops were all male. The possibility that the observed differences are a consequence of HAPE, rather than risk marker appears unlikely since it is well known that HAPE is non-inflammatory in nature and resolves quickly and completely with descent and/or oxygen32. Similar results have been shown in larger number of subjects studied earlier33. Thus it appears unlikely that the differences seen between HAPE-S and HAPE-R groups are a consequence of HAPE. Based on the total evidence available, it can be reasonably concluded that a restrictive pattern or an abnormally low vital capacity are predictive of increased HAPE risk. While the test characteristics are modest for FVC (AUC = 0.66) in the present study as expected due to multifactorial reasons for HAPE susceptibility however, its relevance is increased as it can be applied to both mountaineer and non mountaineer populations to predict HAPE susceptibility. Keeping in mind the ease and low-cost of performing spirometry, the potentially fatal course of HAPE and the huge logistical challenges of emergency transport to low altitude, this is a useful step towards personalised risk assessment.
References
Swenson, E. R. & Bärtsch, P. High Altitude Pulmonary Edema. Compr Physiol 2, 2753–2773 (2012).
Hultgren, H. N., Lopez, C. E., Lundberg, E. & Miller, H. Physiological studies of pulmonary edema at high altitude. Circulation 29, 393–408 (1964).
Roy, S. B. et al. Haemodynamic studies in high altitude pulmonary edema. Br. Heart J 31, 52–58 (1969).
Mishra, A., Mohammad, G., Thinlas, T. & Pasha, M. A. EGLN1 variants influence expression and SaO2 levels to associate with high-altitude pulmonary oedema and adaptation. Clinical Sciences 124, 479–489 (2013).
Aggarwal S. et al. Indian Genome Variation Consortium, Prasher B, Mukerji M. EGLN1 involvement in high-altitude adaptation revealed through genetic analysis of extreme constitution types defined in Ayurveda. Proc Natl Acad Sci USA 107(44), 18961–6 https://doi.org/10.1073/pnas.1006108107 (Nov 2 2010).
Gupta, R. K. et al. Elevated pulmonary artery pressure and brain natriuretic peptide in high altitude pulmonary edema susceptible non-mountaineers. Sci. Rep. 6, 21357, https://doi.org/10.1038/srep21357 (2016).
Soree, P. et al. Raised HIF1αduring normoxia in high altitude pulmonary edema susceptible non-mountaineers. Sci. Rep. 6, 26468, https://doi.org/10.1038/srep26468 (2016).
Steinacker, J. M. et al. Lung diffusion capacity and exercise in subjects with previous high altitude pulmonary edema. EurRespir J 11, 643–650 (1998).
Eldridge, M. W. et al. Pulmonary hemodynamic response to exercise in subjects with prior high-altitude pulmonary edema. Journal of Applied Physiolog 81(2), 911–921 (1996).
Lee, H. M., Truong, S. T. & Wong, N. D. Evidence of Lung Function for Stratification of Cardiovascular Disease Risk. Korean Circ J 41, 171–174 (2011).
Friedman, G. D., Klatsky, A. L. & Siegelaub, A. B. Lung function and risk of myocardial infarction and sudden cardiac death. N Engl J Med 294, 1071–1075 (1976).
Lee, H. M., Le, H. & Lee, B. T. et al. Forced vital capacity paired with Framingham Risk Score for prediction of all-cause mortality. Eur Respir J. 36(5), 1002–6, https://doi.org/10.1183/09031936.00042410 (Nov, 2010).
Hancox R. J. et al. Systemic inflammation and lung function in young adults. Thorax 62(12), 1064–1068, https://doi.org/10.1136/thx.2006.076877 (2007 Dec).
Shaabana, R. et al. Change in C-reactive protein levels and FEV1 decline: A longitudinal population-based study. Respiratory Medicine 100(12), 2112–2120 (2006).
Dehnert, C. et al. Exaggerated hypoxic pulmonary vasoconstriction without susceptibility to high altitude pulmonary edema. High Alt Med Biol 16, 11–17 (2015).
Saydain, G., Beck, K. C., Decker, P. A., Cowl, C. T. & Scanlon, P. D. Clinical Significance of Elevated Diffusing Capacity. CHEST 125, 446–452 (2004).
Yock, P. G. & Popp, R. L. Non invasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation 70, 657–62 (1984).
American Thoracic Society. Single breath carbon monoxide diffusing capacity (transfer factor): recommendations for a standard technique; 1995 update. Am J RespirCrit Care Med 152, 2185–2198 (1995).
American Thoracic Society. Standardization of spirometry: 1994 update. Am J RespirCrit Care Medm 152, 1107–1136 (1995).
American Thoracic Society. Lung function testing: selection of reference values and interpretive strategies. Am Rev Respir Dis 144, 1202–1218 (1991).
Jain, S. K. & Gupta, C. K. Age, height and body weight as determinants of ventilator norms in healthy men above forty years of age. Indian J Med Res 55, 599–607 (1967).
Jain, S. K. & Ramiah, T. J. Spirometric studies in healthy men and women 15–40 years age. Indian J Chest Dis 9, 1–8 (1967).
Viswanathan, R. et al. Pulmonary edema of high altitude II: clinical, aerohemodynamic, and studies in a group with history of pulmonary edema of high altitude. Am Rev RespirDis. 100, 334–341 (1969).
Busch, T. et al. Swenson ER.Hypoxia decreases exhaled nitric oxide in mountaineer susceptible to high altitude pulmonary edema. Am J RespirCrit care Med 163, 368–373 (2001).
Berger, M. M. et al. Hypoxia impairs systemic endothelial function in individuals prone to high altitude pulmonary edema. Am J RespirCrit Care Med 172, 763–767 (2005).
Petty, T. L. John Hutchinson’s Mysterious Machine Revisited. Chest 121(5), 219S–223S (2002).
Bang, K. M., Gergen, P. J., Kramer, R. & Cohen, B. The effect of pulmonary impairment on all-cause mortality in a national cohort. Chest 103, 536–540 (1993).
Hickson, D. A. et al. Diabetes, Impaired Glucose Tolerance, and Metabolic Biomarkers in Individuals with Normal Glucose Tolerance are Inversely Associated with Lung Function: The Jackson Heart Study. Lung 189(4), 311–321, https://doi.org/10.1007/s00408-011-9296-1 (2011).
Mishra, K. P. et al. Hypoxia induced inflammatory chemokines in subjects with history of high altitude pulmonary oedema. Indian J. Clin Biochem 31(1), 81–6, https://doi.org/10.1007/s12291-015-0491-3. (2016).
Bidaud, B. C. et al. Non specific pattern of lung function in a respiratory physiology unit: causes and prevalence: results of an observational cross-sectional and longitudinal study. BMC Pulmonary Medicine 14, 148, https://doi.org/10.1186/1471-2466-14-148. (2014).
Sartori, C. et al. Exaggerated Endothelin Release in High-Altitude Pulmonary Edema. Circulation 99, 2665–2668 (1999).
Swenson, E. R. et al. Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. JAMA 287(17), 2228–35 (2002).
Hohenhaus, E., Paul, A., McCullough, R. E., Kucherer, H. & Bartsch, P. Ventilatory and pulmonary vascular response to hypoxia and susceptibility to high altitude pulmonary edema. EurRespir J 8, 1825–1833 (1995).
Acknowledgements
The authors are grateful to the commanding officers and the troops of different units for volunteering to participate in the study. We also express our gratitude to Army Medical Corps authorities including DGAFMS, AFMC Pune and SD branch for providing nominal roll of HAPE patients. Due thanks are also expressed to Mr. CGC Sekharan, V. P. Chest Institute for conducting lung functions and diffusion studies. DRDO, Ministry of Defence, Govt. of India.
Author information
Authors and Affiliations
Contributions
R.K.G. has set up the study concept and design, data acquisition, interpretation and drafting of manuscript. P.S. performed data acquisition and preparation of manuscript. K.D. and A.A. were responsible for drafting of manuscript, statistical analysis and preparation of figures. S.B.S. was responsible for drafting of manuscript, supervision and coordination of study process. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gupta, R.K., Soree, P., Desiraju, K. et al. Subclinical pulmonary dysfunction contributes to high altitude pulmonary edema susceptibility in healthy non-mountaineers. Sci Rep 7, 14892 (2017). https://doi.org/10.1038/s41598-017-14947-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14947-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
