
Abstract
Endolysins are peptidoglycan-degrading enzymes utilized by bacteriophages to release the progeny from bacterial cells. The lytic properties of phage endolysins make them potential antibacterial agents for medical and industrial applications. Here, we present a comprehensive characterization of phage AP3 modular endolysin (AP3gp15) containing cell wall binding domain and an enzymatic domain (DUF3380 by BLASTP), both widespread and conservative. Our structural analysis demonstrates the low similarity of an enzymatic domain to known lysozymes and an unusual catalytic centre characterized by only a single glutamic acid residue and no aspartic acid. Thus, our findings suggest distinguishing a novel class of muralytic enzymes having the activity and catalytic centre organization of DUF3380. The lack of amino acid sequence homology between AP3gp15 and other known muralytic enzymes may reflect the evolutionary convergence of analogous glycosidases. Moreover, the broad antibacterial spectrum, lack of cytotoxic effect on human cells and the stability characteristics of AP3 endolysin advocate for its future application development.
Similar content being viewed by others

Introduction
Muralytic enzymes are peptidoglycan (PG) degrading proteins widely represented within bacteriophages (bacterial viruses), bacteria, archaea, and eukaryotes. Among bacteriophages, muralytic enzymes may occur as virion-associated lysins serving for PG degradation at the first step of host infection, or as endolysins required at the end of a viral lytic cycle to allow progeny release from the infected cell1,2,3. Intensive research has been conducted in the recent years on antibacterial properties of phage endolysins. In vitro studies and in in vivo investigation in animal models, recently reviewed by Nelson and co-workers4, demonstrated that these enzymes are highly effective in the eradication of Gram-positive pathogens such as Streptococcus pneumoniae, Staphylococcus aureus, Bacillus anthracis and Enterococcus faecalis. In contrast, endolysins have a limited activity against Gram-negatives because of their impermeable outer membrane (OM) which protects the bacterial cell wall from exogenous PG-degrading enzymes. Only a few endolysins were proved to be exogenously effective against Gram-negative bacteria5,6,7. Nevertheless, recent studies have reported the effectiveness of modified endolysins or enzymes combined with membrane-permeabilizing agents to overcome OM barrier and destroy bacterial cells8,9,10,11.
The vast majority of known endolysins derived from Gram-negatives infecting phages are 15–20 kDa proteins containing a single enzymatic domain of simple globular structure. Rare examples of modular endolysins are usually composed of a single N-terminal cell wall-binding domain (CBD) responsible for high-affinity tying to the cell wall receptor and a C-terminal enzymatically active domain (EAD) responsible for breaking specific bonds within the PG structure4. Modular architecture has been demonstrated for several endolysins produced by Gram-negatives infecting phages: KZ144, EL188, OBPgp279 and 201φ2-1gp229 encoded by Pseudomonas phages12,13 and PVP-SE1gp146, SPN1S_0028, Lys68 and Gp110 encoded by Salmonella phages10,13,14,15.
The cleavage specificity of muralytic enzymes (including endolysins) falls into three major categories: amidases, endopeptidases, and glycosidases, depending on the type of the chemical bond that is cleaved within PG. Amidases hydrolyse the amide bond between the sugar and the peptide moieties and endopeptidases cleave the PG within the peptides4. The major group, however, consists of glycosidases, which cleave one of the two glycosidic bonds in the glycan chain, and are subdivided into glucosaminidases, lysozymes (muramidases) and lytic transglycosylases3. Glucosaminidases hydrolase β-1,4–glycosidic bond between N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) on the reducing side of GlcNAc. Lysozymes and lytic transglycosylases cleave the same bond, but on the reducing side of MurNAc, wherein lysozymes simply hydrolase the β-1,4–glycosidic bond and lytic transglycosylase release a product containing 1,6-anhydro ring at the MurNAc residue16. Lysozymes are the most common muralytic enzymes. Based on the fold similarity, four general families of lysozymes with demonstrated PG-hydrolytic activity have been described so far: (i) hen egg-white lysozyme (HEWL), (ii) goose egg-white lysozyme (GEWL), (iii) bacteriophage T4 lysozyme (T4L) and (iv) Chalaropsis lysozyme17. A characteristic feature of lysozymes is their great diversity in terms of the amino-acid sequence. HEWL, GEWL and T4L lysozymes share closest similarities in three-dimensional structure and catalytic centre organization. All share the same characteristic set of hydrogen bonds between the backbone of the enzyme and the 2-acetamido group of the saccharide in subsite C. HEWL catalytic centre is located in a crevice between two sub-domains, which are connected by a long α-helix. In a vast majority of lysozymes, two residues: a general-acid catalyst residue - glutamic acid (Glu, E) and a general-base catalyst residue - aspartic acid (Asp, D) or cysteine (Cys, C) are involved in catalysis. Besides the Glu and Asp/Cys, the third accessory and catalytically important residue – threonine (Thr) or serine (Ser) may take part in the catalytic reaction18. Thr/Ser as a catalytic water positioning residue (in the sequence of Glu-8aa-Asp/Cys-5aa-Thr catalytic triad) was previously demonstrated for lysozymes of coliphages T4 and P21 (Glu11-Asp20-Thr26 and Glu35-Asp44-Thr50 respectively)18,19. GEWL lysozymes are an exception having only a single catalytic residue – Glu20. Despite the differences in the cleavage products, the single catalytic residue (Glu) is also characteristic for lytic transglycosylases including phage lambda endolysin17.
The initial classification of the activity type of new enzymes is often based on in silico homology. It is therefore hampered if a large sequence diversity is encountered in a single class, particularly with a limited number of well-defined canonical members21. Currently, large sets of uncharacterized protein families are still found in databases. Indeed, ca. 1,600 domains of unknown function (DUFs) exist in Pfam protein database. A wide distribution of certain DUFs among different organisms suggests their evolutionary importance. Based on HMMER reference proteome database the number of DUF3380 domain homologues is currently estimated at 233 (209 bacteria, 23 viruses, and 1 eukaryote). Therefore, it is crucial to determine the structure and catalytic activity of this family representative to guide future development in the field.
In this study, we present a comprehensive characterization of recombinant AP3gp15 endolysin encoded by the recently described phage AP3 (vB_BceM_AP3), specific to multidrug-resistant Burkholderia cenocepacia 22. This modular endolysin is composed of CBD domain and EAD domain classified by BLAST into DUF3380 family. In this study, we describe its crystal structure, cleavage specificity, antibacterial activity and spectrum, as well as stability and cytotoxicity to human cells. It is the first report describing the three-dimensional structure of DUF3380 domain combined with the correlation analysis of enzymatic function. Thus, our study provides a general structural and functional description of over two hundred DUF3380 homologues.
Results
In silico analysis of AP3 endolysin
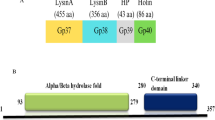
Phage AP3, a representative of Gram-negatives infecting phages, utilizes a complex of four proteins which ensures efficient progeny release from an infected host cell. Its endolysin (AP3gp15), together with antiholin (AP3gp13), holin (AP3gp14) and bimolecular spanin (AP3gp16 and AP3gp17) form the lysis cassette22. AP3gp15 is a 266-amino acid protein with molecular mass of 28.9 kDa and theoretical pI of 8.82. The structure of endolysin, predicted in silico in BLASTP, is modular, consisting of CBD (10-65 aa; PG_binding_1 domain; pfam01471) at the N-terminus, and DUF3380 domain (pfam11860) at the C-terminus (90-262 aa) (Fig. 1A). There are at least 73 known significant homologues to modularly organized AP3gp15 and 37 homologues with reversely arranged domains. There are also 2428 and 233 homologues (E-value < 0.0001) of N-terminal CBD and C-terminal EAD, respectively, if compared separately. Domains similar to AP3gp15 CBD have been already well characterized in terms of the structure and function23,24, however, structural information concerning DUF3380 is non-existent. Recently, the gp110 endolysin derived from Salmonella phage 10, possessing DUF3380 has been analysed regarding its cleavage specificity and stability features15. However, the amino acid identity between phage 10 gp110 and AP3gp15 is less than 50% (46% for the whole protein sequence and 49% for CBD and 45% for EAD, if compared separately) (Fig. 1A). Moreover, both proteins differ significantly in terms of activity strength and stability features. Referring to above differences, the determination of the cleavage specificity and stability of AP3gp15 had to be examined (see below).

The modular structure of AP3gp15: blue, grey and red colours represent the CBD, hinge region and EAD (DUF3380), respectively. (A) The amino acid sequence comparison of AP3gp15 and 10gp110: the active residue Glu (E) at the position 101 is in black and framed; the Asp (D) at the position 110 in AP3gp15 (black) and its equivalent Thr (T) in 10gp110 (red) are framed. (B) Two asymmetrical domains are visible, CBD (N-terminal bundle of three helices representing the putative PG binding domain) encompassing residues Lys3 – Ser66; the catalytic domain EAD (residues Asp77 – Ala264); the hinge region (residues Ala67 to Thr76) connects both subunits.
The modular structure of AP3 endolysin
To provide the structural basis of the catalytic activity of DUF3380, the X-ray crystal structure of AP3gp15 has been evaluated (Fig. 1B; Table 1). The overall structure comprises of two clearly distinguished domains, a smaller N-terminal CBD domain (Lys3 – Ser66), and a larger EAD domain (Asp77 – Ala264) located towards the C-terminus (Fig. 1B). CBD domain is composed of a bundle of three antiparallel helices (α1-α3) and is joined with EAD via a hinge region spanning amino acids (Ala67 – Thr76). The EAD domain is organized into two sub-domains: first encompassing helices α4-α5 (Asp77 – Glu101) and helices α10 to α15 (Gly185 – Ala266), and the second containing β1 (Ile115 – Tyr117), β2-β3 (Thr176 – Met184) sheet intervened by helices α6 to α9 (Arg119 – Ala175). The structure of the EAD domain of AP3gp15 is characterized by a deep concave cleft at the subdomain interface. In order to identify the likely functional role of domains within AP3gp15 with particular emphasis on EAD, we compared them with all known structures within PDB using algorithms implemented in the Dali server25. The N-terminal domain demonstrated significant structural homology to PG binding domains (CBDs) found in other endolysins and multiple functionally distant proteins. The closest structural homologues, include respective domains of Zn-dependent amidase from Clostridium acetobutylicum (PDB ID 4XXT; RMSD = 1.1 for 65 Cα atoms (95%))26 and Pseudomonas aeruginosa phage phiKZ gp144 endolysin (PDB ID 3BKH; RMSD = 1.1 for 65 Cα atoms (94%))24 (Fig. 2). Despite a relatively low homology of amino acid sequence (31% and 39% identity, respectively), a high structural homology of the entire AP3gp15 N-terminal part to the domains of clearly identified function strongly suggests its role in PG interaction. The surface residue analysis of CBD, similarly to homologues, did not reveal any distinct negatively or positively charged patches. However, surface amino acids (Arg6, Arg10, Asp13, Asp31, and Asp54) were spatially homologous to Arg5, Arg9, Asp11, Asp31 and Asp 54 of Zn-dependent amidase, respectively. Also, the residues of AP3gp15: Arg6, Asp13, Asp31, Asp40, Asp54 have their equivalents in phage phiKZ endolysin residues: Lys58, Asp65, Asp83, Asp91, and Asp106. The most common fingerprint between those three proteins comprises of Arg6, Asp31, and Asp54 of AP3gp15 CBD. The conservative character of residues likely suggests their role in PG recognition and binding.

Structural alignment of CBD domains of AP3gp15 (blue), phage phiKZ endolysin (orange) and Zn-dependent amidase from C. acetobutylicum (grey) highlights a strong conservancy of three antiparallel helices bundle motif.
The large enzymatic C-terminal domain of AP3 endolysin shows structural homology to proteins of lysozyme family. HEWL, GEWL, bacteriophage T4 lysozyme and phage lambda lysozyme have been used for further comparison due to the detailed structure-function relationship data available for those proteins. The structural similarity of AP3gp15 to HEWL (PDB ID 4HPI)27 (rmsd~3.3 Å over 128 residues: 12% identity, Z-factor of 5.1), GEWL (PDB ID 153 L)20 (rmsd~3.7 Å over 185 residues: 8% identity, Z-factor of 5.1), T4L (PDB ID 1LYD)28 (rmsd~3.8 Å over 164 residues: 10% identity, Z-factor of 2.3), and lambda endolysin (PDB ID 1D9U)29 (rmsd~3.8 Å over 154 residues: 15% identity, Z-factor of 5.7) has been established by Dali server algorithm. Despite the low similarities, AP3gp15 reveals several common structural components with HEWL, GEWL, T4L, lambda endolysin (Fig. 3A,C,E,G). The comparison to HEWL, the closest structural homologue among lysozymes, allowed to define the core structure of AP3 endolysin, where the entire fold of HEWL is encompassed within α5, α10, α12, α14 and α15. This core is decorated with large protrusions (α6-α9) characteristic only for AP3 phage endolysin and not present in classical lysozymes (Fig. 3A).

Structural alignment of AP3gp15 DUF3380 domain (red) and: (A) HEWL (yellow); (C) GEWL (green); (E) T4 lysozyme (purple) and (G) phage lambda lytic transglycosylase (silver). Structural alignment of the catalytic center of the AP3 endolysin DUF3380 domain (red) and: (B) HEWL (yellow); (D) GEWL (green); (F) T4 lysozyme (purple) and (H) phage lambda lytic transglycosylase (silver).
Structural homology suggests an unusual catalytic mechanism of the AP3gp15 enzymatic domain
The structure alignment of AP3 endolysin to HEWL, GEWL, T4 lysozyme and lambda lytic transglycosylase suggests the probable catalytic center elements interacting with the substrate (Fig. 3B,D,G,H). Glu35 and Asp52 of HEWL, Glu73 of GEWL, Glu11 and Asp20 of T4L, and Glu19 of lambda endolysin have been identified as the key residues involved in the catalytic process29,30. AP3gp15 contains only Glu equivalent at the presumed catalytic centre (position 101). No Asp equivalent, as the second catalytic residue, could have been identified in the active center of AP3gp15.The substrate is most likely recognized by residues located in α5, α10, a12, α14 helices and a loop region (Ser102 – Val114) of AP3gp15. In particular, Asp221 and His187 as homologues of Asp101 and Trp62-63 (involved in recognition and binding of PG sugar residues by HEWL27) were identified as taking part in the binding of the oligosaccharide substrate (Fig. 4).

Structural alignment of AP3gp15 and HEWL loop region responsible for substrate recognition. Homologies between HEWL and AP3gp15 residues that are possibly involved in the substrate recognition and other interactions. Only the homologous catalytic Glu35 of HEWL is highlighted as it is represented by its counterpart residue of Glu101 in AP3gp15. Asp101 of HEWL plays a role in substrate positioning and its putative homologue is Asp221 of AP3gp15. The closest homologue of similarly functioning Trp62 and Trp63 of HEWL is represented by His187 that positions between these two amino acids in the structural alignment.
AP3 endolysin has lysozyme specificity
The enzymatic specificity of AP3 endolysin was examined in vitro using purified PG as a substrate with previously described methodology31. The chromatograms (Fig. 5A) demonstrated AP3gp15 muropeptide profiles similar to those obtained after lysozyme treatment suggesting similar specificity of both enzymes. Further confirmation by MS analysis of all major peaks present in both samples (Fig. 5B) identified identical products. Thus, comparable to lysozyme, AP3gp15 cleaves β-1,4-glycosidic bond of PG and releases GlcNAc and MurNAc. These results confirm that AP3 endolysin is, in fact, a lysozyme, not a lytic transglycosylase.

Cleavage specificity of AP3gp15. (A) UPLC chromatogram of in vitro digestion of E. coli sacculus either with muramidase, AP3gp15 or without enzyme (non-digested). (B) Mass spectrometry muropeptide analysis. M2, N-acetylglucosamine-(GlcNAc)-N-acetylmuramic acid (MurNAc)-l-Ala-d-Glu; M3, GlcNAc-MurNAc-l-Ala-d-Glu-meso-DAP; M4, GlcNac-MurNAc-l-Ala-d-Glu-meso-DAP-d-Ala; M3L, GlcNAc-MurNAc-l-Ala-d-Glu-meso-DAP-Arg-Lys, oligopeptide from Braun’s lipoprotein which remains bound to the muropeptide after cleavage by Pronase E; D33, dimer muropeptide of a M3 cross-linked with a M3; D34, dimer muropeptide of a M3 l,d-cross-linked with a M4; D34 G, dimer muropeptide of a M3 cross-linked with a M3G (GlcNAc-MurNac-l-Ala-d-Glu-meso-DAP-Gly); D43, dimer muropeptide of a M4 d,d-cross-linked to a M3; D44, dimer muropeptide of a M4 cross-linked with a M4; D44N, dimer muropeptide of a M4 cross-linked with a M4N (anhydrous in the MurNac).
Activity and cytotoxicity analysis of recombinant AP3 endolysin
The muralytic activity of AP3 endolysin was assessed on permeabilized Gram-negative strains according to a standardized assay methodology32. The AP3gp15 turned out to be two times stronger than the commercially available lysozyme (Table 2). This was consistent for all tested strains, including E. coli, K. pneumoniae, P. aeruginosa, B. cenocepacia and S. enterica Typhimurium. No lytic activity was detected against Gram-positive Staphylococcus aureus and S. epidermidis strains. To verify that Glu101 is a key residue of AP3gp15 catalytic centre, the site-directed mutagenesis was performed and the Glu101 has been converted to an alanine (Glu101Ala). The muralytic activity of modified enzyme tested on P. aeruginosa PAO1 permeabilized cells, dropped down from 14,710 U/mg for wild-type version to 90 U/mg for protein mutant, reducing the lytic activity of 99.4%.
The endolysin showed to be relatively stable at low temperatures but heat-sensitive. It retained > 95% activity after 1 week storage at −20 °C or 4 °C, at the concentration of 0.5 mg/ml in PBS buffer (pH 7.4). After 1 week at RT, the protein started to precipitate but still retained 70% residual activity. Ten minutes incubation at higher temperatures: 30 °C, 40 °C, 50 °C, 60 °C resulted in a decrease of around 2%, 25%, 80% and 92% of its hydrolytic activity, respectively. The incubation for 10 minutes at 80 °C caused a complete inactivation. The pH optimum was in the range of 7–9. The activity dropped at pH 6 and 10 by 10% and 25%, respectively. Low pH (pH = 5) reduced the activity to 58%. The cytotoxicity of AP3gp15 on mammalian cells was also evaluated and 50 µg/ml of the enzyme exhibited no adverse effect on A549 and THP-1 cells. After 48 h of incubation, no changes in cell viability and number were observed in comparison to endolysin free buffer control (data not shown).
Discussion
Phage endolysins have a large, but yet unexplored potential as antimicrobials, due to poor structural and biochemical characterization. The annotation of endolysin encoding genes and their qualification to a specific enzymatic group are generally based on the amino acid sequence similarity to known proteins. Lack of a large reference library results, among others, in a false classification of newly characterized enzymes. One of the examples is the phage lambda endolysin which operates in the databases and in numerous publications as lysozyme, but in fact, has lytic transglycosylase activity29,33. The experimental verification of structural features and enzymatic specificity is a crucial step for prospective enzyme modification and further application. The thorough characterization of representative members is a key element in the case of conservative domains of unknown function (DUFs) which represent 20% of all currently annotated protein domains in Pfam database. The data obtained from the first member usually defines the primary characteristics of the entire protein family allowing a better understanding of the biology/function of many organisms.
In the present study, we describe a modular endolysin (rare for Gram-negative infecting phages) originating from B. cenocepacia AP3 phage. The domains arrangement of AP3gp15 (CBD at the N-terminus and EAD at the C-terminus) is characteristic for all eight endolysins originating from Gram-negatives specific phages with confirmed modular structure10,12,13,14,15,34. The AP3gp15 CBD element revealed a high structural similarity to CBDs of Zn-dependent amidase from C. acetobutylicum and Pseudomonas phage phiKZ endolysin. The latter domain has a high-affinity to Gram-negatives PG and is able to increase an enzymatic activity of phiKZ endolysin23. It should be noted that genomes of both phages: phiKZ (giant myovirus, 280,334 bp) and AP3 (Peduovirinae; 36,499 bp) belong to entirely different taxonomic lineages. Moreover, CBD of AP3 endolysin shows significant similarity to almost two and a half thousand homologues (phages and bacteria) in genome databases including a recently described endolysin from Salmonella phage 1015. Therefore, AP3gp15-like CBDs seem to be universal in general, irrespectively to the origin. It stays in contrast to endolysins encoded by Gram-positives specific phages, where CBDs demonstrate a large sequence diversity, and genus or even species specificity4,35.
The structural alignment highlights the similarity of DUF3380 of AP3gp15 to proteins having an activity of lysozyme and lytic transglycosylase (phage lambda endolysin). The catalytic mechanism of a vast majority of lysozymes relies on two residues: Glu and Asp. Catalytic centers of canonical lysozymes derived from T4 and P21 phages are based on the Asp residue separated in a sequence by eight residues relative to catalytic Glu. Although, the AP3gp15 also contains Asp at analogous sequence position (Asp110), at the structural level this residue is not equivalent. In T4 and P21 lysozymes, the residue forms the active site whereas in AP3gp15 it is positioned away from the catalytic center. The crystal structure analysis suggests that only a single catalytic residue (Glu101) is present at the active site of AP3 endolysin. Moreover, the comparative in silico analysis of AP3gp15 EAD homologues revealed that Asp is not a conserved residue in this protein family (present only in 123 out of 233 analyzed DUF3380 domains) what again might suggest an insignificant role in the catalysis. Instead of given Asp, other replacing amino acids: asparagine, threonine, serine, histidine, proline, tyrosine or alanine, can be found in sequenced DUF3380 homologues. Of functionally characterized member, a DUF3380 domain of Salmonella phage 10 endolysin has threonine residue at a position equivalent to 110 in AP3gp15 (Fig. 1A) and its active site most probably consists only of Glu101 residue, although no structure is available. Experimental data of AP3gp15 mutant Glu101Ala with a decline in activity by 99.4%, confirmed of Glu101 involvement in the catalytic reaction. However, based on the putative catalytic residues identified on other lysozymes by sequence and structure similarity, we were not able to identify a second catalytic residue. To date, only one phage endolysin (lysozyme Lys68) with Glu as a putative single catalytic residue, has been described, but no sequence homology to AP3gp15 is seen10. A single glutamic acid is characteristics for the catalytic centre of GEWL group of lysozymes20 and lytic transglycosylases36. Although the structural comparison of AP3gp15 EAD suggests a closer similarity to phage lambda endolysin (lytic transglycosylase) rather than GEWL, the biochemical analysis showed lysozyme-like cleavage products of PG digestion by AP3gp15. Similar results showing a lysozyme cleavage specificity were obtained for a related modular endolysin derived from Salmonella phage 10 by Rodríguez-Rubio and co-workers15. This indicates that DUF3380 has a HEWL-like fold, but a novel type of catalytic center basing on one residue as in GEWL and lytic transglycosylase. The structural similarity of the catalytic domain AP3gp15 to other glycosidases allows speculating on the catalytic mechanism. The substrate binding and related distortion of the cleaved bond, the formation of a covalent intermediate and the electrophilic movement of C-1 carbon along the reaction coordinate are the canonical basis30. HEWL was the first enzyme for which the three-dimensional structure was determined by X-ray diffraction37 and currently it is the best structurally characterized glycosidase in terms of the catalytic mechanism and substrate binding. The cleavage is preceded by the binding of PG hexasaccharide unit with concomitant distortion within MurNAc. MurNAc unit in the -1 (Glu) subsite adopts a half-chair conformation, thereby forming a kink in the oligosaccharide between sites -1 and + 1 (Glu and Asp). Glu35 of HEWL transfers a proton to the O1 position while the negative charge on Asp52 stabilizes the positively charged oxonium ion intermediate30,38,39. Despite the catalytic domain of AP3 endolysin being much larger than HEWL and the core of AP3gp15 aligns well with HEWL, the core of AP3gp15 contains additional protrusions which function is unknown. Although, the significantly different binding mode to HEWL is unlikely, the substrate binding surface of AP3gp15 may be extended in comparison to HEWL. If so, EAD of AP3 endolysin (DUF3380) could be the largest catalytic subunit described to date among lysozymes. The broad distribution and conservative character of DUF3380 domains in phages and bacteria advocate its important function. Moreover, the lack of amino acid sequence homology between AP3 endolysin and other glycosidases may reflect evolutionary convergence. That would imply that DUF3380-like and other classes of muralytic glycosidases had aroused in nature independently in different ancestral phages/bacteria as a result of an evolutionary trend in the development of peptidoglycan-degrading proteins. Particularly interesting is the fact, that despite differences in the known structures of lysozymes, the enzymatic specificity remains the same.
The AP3gp15 exhibits almost twice higher muralytic activity comparing to commercially available HEWL lysozyme, both tested on previously permeabilized bacterial cells. Compared to other endolysins derived from Gram-negatives specific phages, the AP3gp15 was classified as a medium effective enzyme, having 65-fold lower than the strongest 10gp110, to 6-fold higher activity comparing to the weakest KP32gp1510,13,14,15,31,34. Similarly to others, the AP3 endolysin shows a broad spectrum of activity against Gram-negative strains, but no PG-degrading properties with respect to Gram-positives10,13,14,31,40,41. This is consistent with the differences in PG structure. The thick PG of Gram-positive bacteria varies significantly in the peptide composition, crosslinks, and modifications of the glycan chain, whereas the PG of Gram-negatives is conserved having 1-3 layers of A1γ chemotype42. Like the vast majority of endolysins derived from Gram-negative specific phages, the AP3gp15 is not able to lyse intact cells and for this purpose requires the combination with the outer membrane permeabilizers or the binding to specific cationic peptide elements9,10,11. It is worth to mention that despite amino acid homology between AP3 and Salmonella phage endolysins, they differ in terms of bactericidal potential and thermostability. The AP3gp15 has 65-fold lower PG-hydrolysing efficacy than Salmonella phage endolysin (958,720 U/mg). Moreover, in contrast to the latter one, the AP3gp15 is unstable above 60 °C. The stability of Salmonella phage endolysin is probably related to the presence of disulphide bridges (DiANNA 1.1 web server prediction)43 which is absent in AP3gp15. The accurate determination of protein thermostability is an important issue in protein characterization and allows for the rational optimization of bioprocesses and for prospective protein modification to increase their thermal resistance44,45,46. Possibly, the introduction of disulphide bridges guided by Salmonella phage endolysin into AP3gp15 would increase its stability and thus the utility for industrial application. No adverse effect on eukaryotic cell lines (lack of cytotoxicity) promotes the AP3gp15 potential as an antimicrobial agent, as well.
Summarizing, in silico comparative analysis of AP3gp15 CBD and EAD (DUF3380) domains demonstrated unexpectedly a widespread occurrence within phages and bacteria and to a lesser extent also among archaea. The conserved character and wide distribution of both domains suggest their important function and evolutionary ancient origin. Therefore, the first report of DUF3380 domain structure and experimental characterization of its enzymatic activity presented in this study provides a significant contribution to the understanding of this widespread protein family with muralytic properties.
Methods
In silico analysis of AP3gp15
In silico analysis of AP3 endolysin included comparative protein sequence homology analysis, conserved domains recognition (NCBI’s BLASTP, HMMER, Phyre2, HHpred) and predicted physicochemical parameters (ExPASy ProtParam) using methodology described elsewhere47,48,49,50.
Recombinant endolysin preparation
The AP3 endolysin (AP3gp15) sequence was amplified from AP3 genomic DNA by PCR using Pfu DNA polymerase (Thermo Fischer Scientific) and specific primers: forward: 5′-ATGTATAAAACCCTGCGCCTCGGCG-3′, 10 pM; reverse: 5′-CGCGGCCGCCCGGCTGTAGCGATCG-3′, 10 pM. The PCR product was cloned into commercially available pEXP5-CT/TOPO expression vector (Thermo Fischer Scientific) according to the manufacturer’s instruction. The construct was verified by sequencing and subsequently transformed into Escherichia coli BL21-AI (Thermo Fischer Scientific) expression strain. The culture was performed in LB medium at 37 °C with shaking (200 rpm) to achieve of mid-exponential-phase (OD600 ~ 0,6). Next, the expression was induced with L-arabinose (to a final concentration of 50.2%) and the incubation was continued at 20 °C for 18 hours. Cells were pelleted at 6000 x g, resuspended in lysis buffer (500 mM NaCl, 20 mM NaH2PO4, pH 7.7) and disrupted by the combination of freeze-thawing (−80 °C/RT; three times) and sonication (5 cycles of 15 s pulse and 30 s rest, Bandelin Sonopuls). Subsequently, the lysate was clarified at 15000 x g and recombinant protein was purified from filtered supernatant on NGC Medium Pressure Chromatography Systems (Bio-Rad) combined with Bio-Scale Mini Profinity IMAC Cartridges (Bio-Rad). The fractions containing recombinant protein were eluted (500 mM NaCl, 20 mM NaH2PO4, 500 mM imidazole, pH 7.7) and pooled. Protein was dialyzed against phosphate-buffered saline (PBS; pH 7.4) using Spectra/Por ® Float-A-Lyzer (Spectrum Laboratories, Inc. Rancho Dominguez, CA, USA). The concentration of purified recombinant enzyme was then determined fluorimetrically using Qubit® Protein Assay Kit, (Thermo Fisher Scientific). Seleno-methionine (SeMet) labelled endolysin was expressed in PASM-5052 medium containing 10 µg/ml methionine, 125 µg/ml SeMet, and 100 nM vitamin B12 according to Studier’s method51.
Determination of muralytic potential
The PG degrading activity and antibacterial spectrum of AP3gp15 were determined on five Gram-negative strains: E. coli (ATCC 25922), K. pneumoniae spp. pneumoniae (ATCC 700603), P. aeruginosa PAO1 (ATCC 15692), S. enterica serovar Typhimurium LT2 (food isolate, Centre of Food and Microbial Technology of the KU Leuven, Belgium), B. cenocepacia lineage IIIA (clinical isolate 7780, PCM 2854) and two Gram-positive strains: S. aureus (ATCC 6538), and S. epidermidis (ATCC 35983) (Table 2). In Gram-negative strains the outer membrane was removed by 45 min incubation in chloroform-saturated 50 mM Tris–HCl buffer (pH 7.7), according to Lavigne et al.52 methodology. The muralytic activity of AP3 phage endolysin was assayed as described by Briers et al.32. 270 µl of bacterial culture was combined with 30 µl of endolysin (final concentration range of 50 ng/ml – 200 ng/ml). As a control, the same concentrations of commercially available lysozyme from chicken egg white (Sigma Aldrich) was used. The kinetic drop in culture turbidity in the presence of endolysin or commercial lysozyme was measured spectrophotometrically using microplate reader (Asys UVM340). The muralytic activity was quantified using a standardized method developed by Briers et al.32, where 1 unit corresponds to the amount of endolysin resulting in an OD600 decrease in culture turbidity of 0.001/min.
Determination of AP3 phage endolysin stability
Storage stability in PBS buffer (pH 7.4) was determined by incubating the endolysin for 1 week at RT and 1 month at 4 °C and −20 °C. The thermostability of enzyme was determined in PBS (pH 7.4) by 10 min incubation at 30 °C, 40 °C, 50 °C, 60 °C and 70 °C. The pH stability was defined in the universal buffer (50 mM KCl, 10 mM KH2PO4, 10 mM Na-citrate and 10 mM H3BO4) adjusted to different pH (from 3 to 10 using) by concentrated NaOH or HCl. In each case, the residual activity was determined by monitoring the PG degrading activity against permeabilized P. aeruginosa PAO.
PG isolation and analysis of endolysin cleavage specificity
E. coli cells from 0.2 l overnight culture were pelleted at 4,500 x g and resuspended in 5 ml of PBS. An equal volume of 10% SDS was added and the sample was incubated in a boiling water bath with vigorous stirring for 4 h. Following the sample was further stirred overnight at RT. The insoluble fraction (PG) was pelleted at 400,000 x g, 15 min, 30 °C (TLA-100.3 rotor; OptimaTM Max ultracentrifuge, Beckman). SDS was washed out and the PG was treated with Pronase E 0.1 mg/ml at 60 °C for 1 h and further boiled in 1% SDS for 2 h to stop the reaction. The sacculus was resuspended in 200 μl of 50 mM sodium phosphate buffer pH 4.9 and digested overnight with 30 μg/ml muramidase (Cellosyl) or with 0.4 mg/ml of AP3gp15 in Tris-HCL 20 mM buffer pH 8.0, 1 mM MgCl2 and 1 mM ZnCl2. Samples were incubated at 37 °C. PG digestion was stopped by 5 min incubation in a boiling water bath. Coagulated protein was removed by centrifugation. The supernatants were mixed with 150 μl 0.5 M sodium borate pH 9.5, and subjected to reduction of muramic acid residues into muramitol by sodium borohydride treatment (10 mg/ml final concentration, 30 min at RT). Samples were adjusted to pH 3.5 with phosphoric acid. Chromatographic analyses of muropeptides were performed on ACQUITY Ultra Performance Liquid Chromatography (UPLC) BEH C18 column (130 Å, 1.7 μm, 2.1 mm by 150 mm; Waters), and peptides were detected at Abs. 204 nm using ACQUITY UPLC UV-Visible Detector. Muropeptides were separated using a linear gradient from buffer A (PBS 50 mM, pH 4.35) to buffer B (PBS 50 mM, pH 4.95 methanol 15% (v/v)) in 20 min, and flow 0.25 ml/min. The peaks from chromatogram that did not show similarity to any of previously described muropeptides were considered as background noise and removed from the chromatogram. Eluting peak samples were analyzed with Agilent 6550 iFunnel Q-TOF LC/MS System (Agilent Technologies) using an organic separation method53.
Crystallization, X-ray data collection, and structure determination
Native and SeMet-labelled AP3gp15 was concentrated to 15 mg/ml in a buffer containing 0.01 M Tris-HCl (pH 8.0), 0.05 M NaCl and 5 mM DTT. Crystallization screening was performed by sitting-drop vapour diffusion method using1:1 ratio of protein and reservoir solution at RT. A thin needle-like crystal of SeMet-AP3gp15 was obtained from a solution containing 0.1 M Tris-HCl (pH 8.0), 0.15 M NaCl and 8% w/v PEG 6000 (ProPlex, Molecular Dimension). Native AP3gp15 crystals were obtained from 0.1 M SPG (pH 7.0), 25% w/v PEG 1500 (PactPremier, Molecular Dimension). Crystals were cryoprotected in 25% glycerol in the mother liquor and flash cooled in liquid nitrogen.
The diffraction data were collected at beamline P11 PETRA III at DESY. A native data set extending to 1.72 Å resolution was collected using 0.9184 Å wavelength, while a data set for SeMet-labelled protein crystals were collected at the peak wavelength (0.9795 Å) and extended to 2.42 Å resolution. Data were integrated and scaled using XDS54 and SCALA55 from the CCP4 suite, respectively. The native crystals belonged to space group H32, and the Matthews coefficient suggested the presence of two monomers in the asymmetric unit. SeMet-labelled crystals belonged to space group P1211 with four monomers in the asymmetric unit.
The initial structure was determined by single-wavelength anomalous dispersion (SAD) using AutoSol program included in the PHENIX suite56. The model was constructed and partially refined in an automated mode using AutoBuild and Buccaneer57 from PHENIX and CCP4 suites, respectively. Initial, partially refined structure was used as a search model for molecular replacement using native data and performed with Phaser58. Refinement was performed with REFMAC559 to complement manual rebuilding using COOT60. Rfree was used to monitor the refinement strategy. Water molecules were added in 2Fobs – Fcalc map at densities above 2σ at reasonable sites upon visual inspection. Data processing and refinement statistics are summarized in Table 1. The geometry of the final model was analyzed with Molprobity61. Structural similarity searches were carried out using Dali server25 and Z scores of > 10 were considered as significant.
Site-directed mutagenesis
To establish the putative catalytic residue within the endolysin sequence, the active site mutation (Glu101Ala) was introduced. Two overlapping specific primers: forward 5′-GCCGTCAATGAGGTTGCATCGAAAGGTGCCGGG-3′ (10 pM) and reverse 5′-CCCGGCACCTTTCGATGCAACCTCATTGACGGC-3′ (10pM) with mutation basepairs underlined, were applied. The previously constructed expression vector (pEXP5-CT/TOPO plasmid containing the AP3gp15 DNA) was mutated using above primers and the GeneArt® Site-Directed Mutagenesis System from Thermo Fischer Scientific. The mutant was prepared, purified using the same protocol as for the wild type of AP3gp15. The PG muralytic activity of the purified AP3gp15 Glu101Ala mutant was quantified using permeabilized P. aeruginosa PAO1 cells, and protocol described below in “determination of muralytic potential” section.
Determination of endolysin cytotoxicity
Determination of endolysin cytotoxicity against human cells has been chosen as preliminary safety evaluation. Cytotoxicity of AP3gp15 was examined with respect to lung carcinoma epithelial cell line A549 (ATCC CCL-185) and acute monocytic leukaemia cell line THP-1 (ATCC TIB-202) using trypan blue assay. Endolysin suspended in PBS buffer pH 7.4 was added to exponentially growing cells (1 × 105 cells/ml) to a final concentration of 50 µg/ml. Cells were incubated for 24 h and 48 h in a suitable medium: DMEM (for A549) or RPMI 1640 (for THP-1), both supplemented with 2 mM glutamax, 1% antibiotic–antimycotic, and 10% heat-inactivated fetal bovine serum (all media and supplements from Gibco BRL, Thermo Fischer Scientific). Cell viability was determined by trypan blue staining (0.4% in PBS; Sigma Aldrich, Germany) and compared to a number of cells in the control (PBS treatment only).
Data availability
The protein sequence of AP3gp15 can be found in GenBank under accession number: AKA61137.1. Coordinates and structure factors for the AP3gp15 crystal structure have been deposited in the Protein Data Bank under accession code PDB 5NM7. Other data supporting this study are available from the corresponding authors upon reasonable request.
References
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y. & Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 101, 3103–3119 (2017).
Rodríguez-Rubio, L., Martínez, B., Donovan, D. M., Rodríguez, A. & García, P. Bacteriophage virion-associated peptidoglycan hydrolases: potential new enzybiotics. Crit. Rev. Microbiol. 39, 427–434 (2013).
Young, R. Bacteriophage lysis: mechanism and regulation. Microbiol. Rev. 56, 430–481 (1992).
Nelson, D. C. et al. Endolysins as antimicrobials. Adv. Virus Res. 83, 299–365 (2012).
Lim, J. A., Shin, H., Heu, S. & Ryu, S. Exogenous lytic activity of SPN9CC endolysin against gram-negative bacteria. J Microbiol Biotechnol. 24, 803–811 (2014).
Lood, R. et al. Novel phage lysin capable of killing the multidrug-resistant Gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob. Agents Chemother. 59, 1983–1991 (2015).
Oliveira, H. et al. Characterization and genome sequencing of a Citrobacter freundii phage CfP1 harboring a lysin active against multidrug-resistant isolates. Appl. Microbiol Biotechnol. 100, 10543–10553 (2016).
Briers, Y., Walmagh, M. & Lavigne, R. Use of bacteriophage endolysin EL188 and outer membrane permeabilizers against Pseudomonas aeruginosa. J. Appl. Microbiol. 110, 778–785 (2011).
Briers, Y. et al. Engineered endolysin-based ‘Artilysins’ to combat multidrug-resistant Gram-negative pathogens. MBio 5, e01379–14 (2014).
Oliveira, H. et al. A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against Gram-negative pathogens in presence of weak acids. PLoS One 9, e108376 (2014).
Yang, H., Wang, M., Yu, J. & Wei, H. Antibacterial activity of a novel peptide-modified lysin against Acinetobacter baumannii and Pseudomonas aeruginosa. Front. Microbiol. 6, 1471 (2015).
Briers, Y. et al. Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages phiKZ and EL. Mol. Microbiol. 65, 1334–1344 (2007).
Walmagh, M., Briers, Y., dos Santos, S. B., Azeredo, J. & Lavigne, R. Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201φ2-1 and PVP-SE1. PLoS One 7, e36991 (2012).
Park, Y., Lim, J. A., Kong, M., Ryu, S. & Rhee, S. Structure of bacteriophage SPN1S endolysin reveals an unusual two-module fold for the peptidoglycan lytic and binding activity. Mol. Microbiol. 92, 316–325 (2014).
Rodríguez-Rubio, L. et al. DUF3380 Domain from a Salmonella phage endolysin shows potent N-acetylmuramidase activity. Appl. Environ. Microbiol. 82, 4975–4981 (2016).
Höltje, J. V., Mirelman, D., Sharon, N. & Schwarz, U. Novel type of murein transglycosylase in. Escherichia coli. J. Bacteriol. 124, 1067–1076 (1975).
Vollmer, W., Joris, B., Charlier, P. & Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 32, 259–286 (2008).
Wohlkönig, A., Huet, J., Looze, Y. & Wintjens, R. Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS ONE 5, e15388 (2010).
Sun, Q. et al. Regulation of a muralytic enzyme by dynamic membrane topology. Nat. Struct. Mol. Biol. 16, 1192–1194 (2009).
Weaver, L. H., Grütter, M. G. & Matthews, B. W. The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the ‘goose-type’ lysozymes lack a catalytic aspartate residue. J. Mol. Biol. 245, 54–68 (1995).
Oliveira, H. et al. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 87, 4558–4570 (2013).
Roszniowski, B. et al. The temperate Burkholderia phage AP3 of the Peduovirinae shows efficient antimicrobial activity against B. cenocepacia of the IIIA lineage. Appl. Microbiol. Biotechnol. 101, 1203–1216 (2017).
Briers, Y. et al. The high-affinity peptidoglycan binding domain of Pseudomonas phage endolysin KZ144. Biochem. Biophys. Res. Commun. 383, 187–191 (2009).
Fokine, A., Miroshnikov, Ka, Shneider, M. M., Mesyanzhinov, V. V. & Rossmann, M. G. Structure of the bacteriophage phi KZ lytic transglycosylase gp144. J. Biol. Chem. 283, 7242–7250 (2008).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
Chang, C., Cuff, M., Joachimiak, G., Endres, M. & Joachimiak, A. Crystal structure of fused Zn-dependent amidase/peptidase/peptidoglycan-binding domain-containing protein from Clostridium acetobutylicum ATCC 824. data sets. http://www.rcsb.org/pdb/explore.do?structureId=4XXT (2015).
Ogata, M. et al. A novel transition-state analogue for lysozyme, 4-O-β-tri-N-acetylchitotriosyl moranoline, provided evidence supporting the covalent glycosyl-enzyme intermediate. J. Biol. Chem. 288, 6072–6082 (2013).
Rose, D. R. et al. Crystal structure of t4-lysozyme generated from synthetic coding DNA expressed in Escherichia coli. Protein Eng. Des. Sel. 2, 277–282 (1988).
Leung, A. K. W., Duewel, H. S., Honek, J. F. & Berghuis, A. M. Crystal structure of the lytic transglycosylase from bacteriophage lambda in complex with hexa-N-acetylchitohexaose. Biochemistry 40, 5665–5673 (2001).
Vocadlo, D. J., Davies, G. J., Laine, R. & Withers, S. G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 412, 835–838 (2001).
Maciejewska, B. et al. Klebsiella phages representing a novel clade of viruses with an unknown DNA modification and biotechnologically interesting enzymes. Appl. Microbiol. Biotechnol. 101, 673–684 (2017).
Briers, Y., Lavigne, R., Volckaert, G. & Hertveldt, K. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Methods 70, 531–533 (2007).
Bieńkowska-Szewczyk, K. & Taylor, A. Murein transglycosylase from phage lambda lysate: purification and properties. Biochim. Biophys. Acta 615, 489–496 (1980).
Walmagh, M. et al. Characterization of five novel endolysins from Gram-negative infecting bacteriophages. Appl. Microbiol. Biotechnol. 97, 4369–4375 (2013).
Drulis-Kawa, Z., Majkowska-Skrobek, G., Maciejewska, B., Delattre, A.-S. & Lavigne, R. Learning from bacteriophages - advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 13, 699–722 (2012).
Höltje, J. V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62, 181–203 (1998).
Blake, C. C. F. et al. Structure of hen egg-white lysozyme, a three dimensional fourier synthesis at 2 Angstroms resolution. Nature 206, 757–761 (1965).
Chipman, D. M., Pollock, J. J. & Sharon, N. Lysozyme-catalyzed hydrolysis and transglycosylation reactions of bacterial cell wall oligosaccharides. J. Biol. Chem. 243, 487–496 (1968).
Phillips, D. C. The hen egg-white lysozyme molecule. Proc. Natl. Acad. Sci. USA 57, 483–495 (1967).
Lai, M.-J. et al. Antibacterial activity of Acinetobacter baumannii phage phiAB2 endolysin (LysAB2) against both Gram-positive and Gram-negative bacteria. Appl. Microbiol. Biotechnol. 90, 529–539 (2011).
Oliveira, H. et al. Structural and enzymatic characterization of ABgp46, a novel phage endolysin with broad anti-Gram-negative bacterial activity. Front. Microbiol. 7, 208 (2016).
Schleifer, K. H. & Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36, 407–477 (1972).
Ferre, F. & Clote, P. DiANNA 1.1: an extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res. 34, W182–W185 (2006).
Huang, L. T. & Gromiha, M. M. Reliable prediction of protein thermostability change upon double mutation from amino acid sequence. Bioinformatics 25, 2181–2187 (2009).
Montanucci, L., Fariselli, P., Martelli, P. L. & Casadio, R. Predicting protein thermostability changes from sequence upon multiple mutations. Bioinformatics 24, i190–i195 (2008).
Pucci, F., Bourgeas, R. & Rooman, M. Predicting protein thermal stability changes upon point mutations using statistical potentials: introducing HoTMuSiC. Sci. Rep. 6, 23257 (2016).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Gasteiger, E. et al. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook (Humana Press Inc., 2005).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 (2008).
Soding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005).
Studier, F. W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
Lavigne, R., Briers, Y., Hertveldt, K., Robben, J. & Volckaert, G. Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell. Mol. Life Sci. 61, 2753–2759 (2004).
Alvarez, L., Hernandez, S. B., de Pedro, M. A. & Cava, F. Ultra-Sensitive, High-Resolution Liquid Chromatography Methods for the High-Throughput Quantitative Analysis of Bacterial Cell Wall Chemistry and Structure. Methods Mol. Biol. 1440, 11–27 (2016).
Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. Scaling and assessment of data quality. in Acta Crystallogr. Sect. D Biol. Crystallogr. 62, 72–82 (2006).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 62, 1002–1011 (2006).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 (1997).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010).
Acknowledgements
B.M. was supported by the Ministry of Science and Higher Education of the Republic of Poland with a grant for young scientists UWR-0420/1397/16 and UWR-0420/2298/17. G.D. was supported in part by grants UMO-2011/01/D/NZ1/01169 and UMO-2012/07/E/NZ1/01907 from the National Science Center. Parts of this research were carried out at the light source Petra III at the German Electron Synchrotron (DESY, Hamburg, Germany). Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a partner of the Leading National Research Center supported by the Ministry of Science and Higher Education of the Republic of Poland. Cava lab was supported by the Laboratory for Infection Medicine Sweden (MIMS), the Knut and Alice Wallenberg Foundation (KAW), the Swedish Research Council and the Kempe Foundation.
Author information
Authors and Affiliations
Contributions
B.M. and Z.D.-K. initiated and designed the study. B.M. prepared recombinant endolysin (including SeMet-labelled protein for crystallization and protein mutant) and determined stability, antibacterial and cytotoxicity assays. K.Z., K.P.R., M.W., and G.D. performed the structural studies. A.E. and F.C. defined the cleavage specificity. B.M., Z.D.-K. and G.D. wrote the manuscript. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maciejewska, B., Źrubek, K., Espaillat, A. et al. Modular endolysin of Burkholderia AP3 phage has the largest lysozyme-like catalytic subunit discovered to date and no catalytic aspartate residue. Sci Rep 7, 14501 (2017). https://doi.org/10.1038/s41598-017-14797-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14797-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
