
Abstract
The introduction of different nutrient and energy sources during weaning leads to significant changes in the infant gut microbiota. We used an in vitro infant digestive and gut microbiota model system to investigate the effect of four commercially available cereal products based on either wheat, sorghum, rice or oats, on the gut microbiota of six infants. Our results indicated cereal additions induced numerous changes in the gut microbiota composition. The relative abundance of bacterial families associated with fibre degradation, Bacteroidaceae, Bifidobacteriaceae, Lactobacillaceae, Prevotellaceae, Ruminococcaceae and Veillonellaceae increased, whilst the abundance of Enterobacteriaceae decreased with cereal additions. Corresponding changes in the production of SCFAs showed higher concentrations of acetate following all cereal additions, whilst, propionate and butyrate varied between specific cereal additions. These cereal-specific variations in the concentrations of SCFAs showed a moderate correlation with the relative abundance of potential SCFA-producing bacterial families. Overall, our results demonstrated clear shifts in the abundance of bacterial groups associated with weaning and an increase in the production of SCFAs following cereal additions.
Similar content being viewed by others


Introduction
The human gut microbiota co-develops with the host in early life1. Initial microbial colonisation of the gut depends on various maternal and postnatal factors such as in utero environment, mode of delivery (vaginal or caesarean-section), gestational age, environment, antibiotic treatments, host genetics and diet (breast milk, formula milk or solid food)2,3,4,5,6. These factors shift the composition and functions of infant gut microbiota towards an established adult-like status within the first three years of life6. The adult gut microbiota is relatively more stable, higher in species diversity and lower in inter-individual compositional and functional variations compared to infants3,7. Accumulating data suggest a link between early life gut microbial colonisation and development of diseases, such as obesity, diabetes (type 1 and 2), food allergies and inflammatory bowel disease8,9,10,11,12,13,14,15. Therefore, establishment of the gut microbiota during infancy and maintenance thereafter likely plays a critical role for human health2.
The transition to solid food contributes significantly towards the infant gut microbiota development. This introduces infants to a larger range of plant and animal polysaccharides. Due to the lack of enzymes to digest most of these complex polysaccharides, infants largely depend on the gut microbiota to digest these otherwise non-digestible carbohydrates16. Therefore, weaning rapidly diversifies and alters the composition of the gut microbiota towards an adult-like composition, presumably to facilitate the metabolism of changing nutrients5. Exposure to new nutrients also leads to altered functions in the microbiota and production of different bacterial metabolites. For instance, introduction to solid food and more xenobiotics promote the growth of bacterial species associated with carbohydrate utilisation, vitamin biosynthesis and xenobiotic degradation3,7. Weaning is also linked with elevated levels of bacterial metabolic end products such as short chain fatty acids (SCFAs), possibly due to the high availability of non-digested dietary fibre7,17.
A number of previous studies have examined changes in the infant gut microbiota in relation to transition to solid food. Weaning in general is associated with decreased proportions of Bifidobacteria, Enterobacteria and some groups of Clostridium, whilst it promotes the growth of Bacteroidetes 3,7,18,19. The increase in Bacteroidetes could be due to their ability to digest a broad range of complex polysaccharides7. Very few in vivo studies have investigated the effect of specific dietary regimens on infant gut microbiota during the weaning phase. One such study reported the effects of feeding infants with commercially available pureed meat, iron- and zinc-fortified cereals or iron-only fortified cereals on the gut microbiota20. Infants fed pureed meat demonstrated enriched Clostridium group XIVa, whilst feeding iron-only fortified cereals resulted in decreasing the abundance of Lactobacilli and Bifidobacterium and promoting the abundance of Bacteroides 20.
Utilisation of in vitro models of the infant gut microbiota eliminates some of the issues associated with in vivo studies. In vitro studies reduce issues with ethical restrictions and volunteer compliance, while enabling more frequent sampling and providing a simplified system to study the gut microbiota without host interference21. In vitro infant gut microbiota model systems have been employed to investigate the effect of probiotics22, candidate probiotics23, milk lipid hydrolysis products24, iron25, milk oligosaccharides26,27,28, dietary polysaccharides and prebiotics29,30,31. Addition of short-chain fructo-oligosaccharides into an in vitro model of infant gut microbiota resulted in an increased abundance of the genus Lactobacillus while reducing the proportion of coliforms29. Shen et al. observed an increase in Bifidobacterium and Bacteroides upon addition of a prebiotic mixture of fructo-oligosaccharides and galacto-oligosaccharides into an in vitro model of infant gut microbiota30.
Lack of dietary fibre in modern Western diets has been associated with changing the gut microbiota composition, functions, diversity and spatial arrangement32,33,34,35. Bridging this gap in dietary fibre intake is of increasing interest as a therapeutic modulation of the gut microbiota in order to improve metabolic and inflammatory health36. Whole grain products generally contain a high amount of dietary fibre37. Although, whole grain cereals are among frequently introduced first food to infants15, the impact of cereals on infant gut microbiota is less well studied. In adults, consumption of whole grain maize based breakfast cereal promoted the growth of Bifidobacterium 38, whilst whole grain wheat cereal increased the abundance of Bifidobacterium and Lactobacillus/Enterococcus groups39. Consumption of whole grain barley and brown rice flakes increased the microbial diversity and reduced host markers associated with inflammation and postprandial glucose levels40.
Given the increasing popularity of whole grain cereals as an early weaning food and the impact on gut microbiota and disease development15, we chose to examine the effects of whole grain-based cereal products on the gut microbiota of infants. In this work, we investigated the effect of four commercially available cereal products, Weet-Bix™, Gluten free Weet-Bix™, Bellamy’s organic baby rice cereal and Real good food-Organic baby oat cereal on infant gut microbiota and SCFAs using an in vitro infant gut microbiota model system.
Results and Discussion
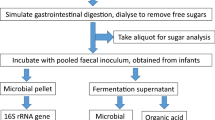
Samples of four commercially available cereal products (wheat, sorghum, rice and oats based) were treated using a series of pH controlled enzyme additions and a dialysis step to simulate infant digestion. Digested cereal products were introduced into an anaerobic basal medium to examine the effects of the cereal products on the infant gut microbiota. The basal growth medium without any cereal addition was run in parallel as a control, this is referred to as the no added cereal control. All cereal and control cultures were inoculated independently with fecal homogenate obtained from a healthy infant. A total of six biological samples (one each from six different infants) were analysed. Cultures were sampled at 0, 24 and 48 hours and V4 region amplicons of the 16 S rRNA gene were sequenced. A total of 21,231,850 reads were generated. After quality filtering and rarefaction 35,095 reads per each of the 270 samples were used for further analyses (270 = 6 biological samples × 3 time points × 3 technical replicates for 5 experimental groups including 4 cereal treatments and no added cereal control).
Each biological sample had a unique initial gut microbial composition
The bacterial phyla Firmicutes, Actinobacteria, Proteobacteria and Bacteroidetes dominated the gut microbiota of all infants at 0 hours. However, the relative abundance of these phyla differed between individuals. Similar variations in the composition were observed at a family level (Supplementary Fig. S1). The relative abundance of the family Veillonellaceae, which is associated with milk polysaccharide digestion was significantly higher (P < 0.05) in breast-fed infants compared to the formula-fed (Table 1). In agreement with our observation, Fan et al. 2014 have found a higher abundance of Veillonellaceae in breast-fed infants compared to that in formula or mixed-fed infants41.
The relative abundance of the family Lachnospiraceae was significantly higher (P < 0.001) in older infants (age > 6 months) compared to younger infants (age < 6 months). Samples obtained from older infants (age > 6 months) had a higher relative abundance of known plant polysaccharide digesting bacteria such as Lachnospiraceae, Ruminococcaceae and Bacteroidaceae. Differences in the abundance of these bacterial families in the infant gut microbiota due to age are largely in agreement with previous studies7,16,19,42,43.
Some bacterial families were highly variable between individuals. This is expected given that the composition of the infant gut microbiota varies depending on factors such as the mode of delivery (vaginal or caesarean section birth), usage of antibiotics, age, diet (breast milk or formula milk) and exposure to solid food1,2,5. The family Coriobacteriaceae was abundant in sample 2 (9.8%), 4 (4.1%) and 5 (14.4%) and not observed above 0.3% in other biological samples. The relative abundance of Porphyromonadaceae (12.2%) was high in sample 1, whilst sample 2 had a large proportion of Enterococcaceae (18.4%). In sample 4 Streptococcaceae was abundant (9%) and Ruminococcaceae was abundant in sample 6 (19.8%). The oldest biological sample (sample 6) showed the lowest relative abundance of Enterobacteriaceae (0.7%), whilst the lowest abundance of Bacteroidaceae (0.3%) was observed in biological sample (sample 3), obtained from an infant that had not been exposed to cereal grains.
All cereal additions altered the gut microbial composition
To determine the impact of different cereal additions on the gut microbiota at 0, 24 and 48 hours, non-metric multidimensional scaling (nMDS) plots were constructed based on the relative abundances of the Operational Taxonomic Units (OTUs) (Fig. 1). Samples at 0 hours in each biological sample clustered relatively close together irrespective of the treatments. All cereal additions resulted in different microbiota community structures at 24 and 48 hours compared to the samples at 0 hours and no added cereal control at 24 hours (global analysis of similarities (ANOSIM) R > 0.7, P < 0.0001) and 48 hours (global ANOSIM R > 0.8, P < 0.0001). The microbiota community structure of the no added cereal control also changed over time, however these remained distinct from the communities after cereal addition. The cereal additions showed similar shifts to each other in the nMDS plots (Fig. 1), and consistent with this there were no statistically significant differences in the microbial community structure between the cereal products.

Ordination of the gut microbiota in each biological sample (1–6) at 0, 24 and 48 hours. Data is shown as Bray-Curtis similarity of Log (X + 1) transformed relative abundance based nMDS plots. Treatments and time points are colour coded as shown in the legend. All cereal additions shifted the community structure at 24 and 48 hours (dotted line circle) compared to the samples at 0 hours (solid line circle) and no added cereal control at 24 and 48 hours (solid line square).
The bacterial diversity in each sample was determined using a Shannon diversity index. Biological sample 3 had a significantly (P < 0.0001) lower Shannon index (4.6 ± 0.05) at 0 hours compared to all other biological samples (Shannon diversity index of samples 1, 2, 4, 5 and 6 ranged from 4.8 ± 0.1 to 5.0 ± 0.04). The low Shannon index value in sample 3 was primarily due to the dominance of a single OTU of the common infant gut bacterium, Veillonella dispar (OTU 585419, relative abundance at 0 hours: 43.6% ± 7.3%). The diversity indices between the treatments were similar at 48 hours in all biological samples, except for sample 3. The diversity of this sample increased significantly at 48 hours with the addition of rice (P < 0.0001).
The relative bacterial abundance was examined at a family level and identifications across the samples were assigned into 33 bacterial families. Statistically significant differences in family abundance across the treatments in each biological sample were investigated using a two-way analysis of variance (ANOVA) test with Tukey’s multiple comparisons test. This identified 17 families with significantly (P < 0.05) different abundances in at least one treatment and time point combination (Fig. 2, Supplementary Fig. S2 and Supplementary Table S2). The impact of cereal additions on the microbiota composition was highly variable between the biological samples. However, for each biological sample the relative abundance of at least one potential Carbohydrate-active enzymes (CAZymes) producing bacteria (families: Bacteroidaceae, Bifidobacteriaceae, Lactobacillaceae, Prevotellaceae and Ruminococcaceae) increased with addition each of the tested cereal products.

Family level taxonomic compositions of the microbial communities for each biological replicate. The relative abundances of the families were determined using QIIME and GraphPad Prism (V7). Each bar is labelled first by treatment, followed by time point (0, 24 and 48 hours). No added cereal control is abbreviated as NAC. Major bacterial families are shown in different colours as indicated in the legend. Bacterial identifications that were not assigned to a family are categorised as “Unassigned”. Bacterial families that were not significantly differentially abundant comparing the treatment regimes in any of the six biological samples are categorised as “Other”. Significance (P < 0.05) was determined using a Tukey’s multiple comparisons test.
For three of the six biological samples the relative abundance of Bacteroidaceae increased upon addition of each of the four cereal products with the highest increase (P < 0.01) following addition of rice. In contrast, in biological sample 4 the relative abundance of this family significantly decreased (P < 0.001), while the abundance of Prevotellaceae significantly increased (P < 0.001) following all cereal additions. The families Bacteroidaceae and Prevotellaceae are members of the phylum Bacteroidetes, which are generally reported to degrade a wide range of dietary polysaccharides, due to their capacity to switch between energy sources depending on the availability44,45,46,47.
In all biological samples, the abundance of Veillonellaceae was significantly higher (P < 0.05) with addition of oats compared to all other treatments. For four out of the six samples, the abundance of this family also increased after the addition of all other cereals. Family Veillonellaceae is associated with utilising partial breakdown products of bacterial polysaccharide digestion and producing propionate and acetate, likely due to its limited ability to digest complex carbohydrates18,48,49,50,51. Previous studies have also reported an increase in the abundance of this family following in vitro fermentation of specific complex polysaccharides by the infant gut microbiota27,31.
The relative abundance of Enterobacteriaceae decreased following addition of each of the four cereal products, with the exception of biological sample 1, where this family significantly increased (P < 0.0001). According to previous studies, Enterobacteriaceae are usually more dominant in pre-weaned gut microbiota of younger infants and become less abundant due to weaning and age3,19,52,53,54. Therefore, the decrease in the abundance of Enterobacteriaceae with cereal supplementations may indicate the ability of the cereal products to aid the shift of the infant gut microbiota towards a mature status.
The relative abundance of Bifidobacteriaceae was significantly higher (P < 0.0001) with the addition of rice compared to other treatments. The family Lactobacillaceae was abundant in younger infants (age < 6 months) and the relative abundance significantly increased (P < 0.05) upon addition of rice. Previous observations of higher growth of Bifidobacteriaceae and Lactobacillaceae in the gut microbiota of adults and animal models upon addition of cereal grains38,39,40, particularly, brown rice55,56,57,58 are also in agreement with our results.
All four tested products have been obtained from cereal grains, which are naturally high in complex sugars such as starch, cellulose, arabinoxylans and glucofructans59, while oats are particularly rich in β-glucans59. The prevalence of Bacteroidaceae, Bifidobacteriaceae, Lachnospiraceae and Lactobacillaceae in all cereal additions is consistent with the ability of the members of these families to digest cellulose, starch and other polysaccharides45,60. The composition of the four tested cereal products varied in regards to dietary fibre, protein, iron, polyphenols and vitamins (Supplementary Table S1). Wheat and rice based cereal products are particularly rich in iron, which has been previously demonstrated to increase the abundance of enteropathogens in the family Enterobactericeae and modulate butyrate-producing bacteria25,61,62. While we did not observe significant changes in the abundance of the Enterobactericeae or butyrate-producing bacteria in samples with wheat supplementation, the abundance of potential butyrate-producing bacteria Bacteroidaceae and Bifidobacteriaceae was higher upon rice supplementation.
Biological sample 4, obtained from an infant who suffered from food allergies, showed a considerably higher initial relative abundance of Streptococcaceae than other biological samples. This sample displayed a notable expansion in the relative abundance of Steptococcaceae (P < 0.0001) upon all cereal additions, this family showed less than 1.0% relative abundance in other biological samples. As this is only a single individual, we cannot directly link the high abundance of the Streptococcaceae to the food allergies experienced by this individual. However, a high abundance of Streptococcus spp. in late infancy has been reported to be linked to allergic disease development63.
Microbial composition was also studied at OTU level and significant differences were determined using a two-way ANOVA with Tukey’s multiple comparisons test. Six OTUs that showed significantly different abundances (P < 0.01) between the treatments in at least three biological samples were identified (Supplementary Fig. S3 and Supplementary Table S3). All of these OTUs belonged to the families that are discussed above and showed similar trends in the relative abundances in each treatment.
Cereal products increased SCFA production
To investigate the effect of cereal addition on production of SCFAs, acetate, butyrate and propionate concentrations were measured from the samples collected at 0, 24 and 48 hours (Fig. 3, Supplementary Table S4). Addition of each of the four cereal products resulted in significantly higher (P < 0.01) concentrations of acetate across all biological samples at 24 and 48 hours compared to the no added cereal control.

Concentration (mmolL−1g−1) of acetate, butyrate and propionate in each treatment at 0 and 48 hours. Concentration measurements at 24 and 48 hours for all three SCFAs were similar, therefore only 48 hours are shown. Mean ± SD concentration for all treatments with each biological sample (sample 1–6) denoted by colour-coded bars. No added cereal control is abbreviated as NAC. The concentrations and results of ANOVA with Tukey’s multiple comparisons test for significance are provided in Supplementary Table S4.
Production of butyrate was higher following addition of wheat or sorghum for all biological samples except sample 3 at 48 hours. Butyrate was highly produced upon addition of rice and oats in at least four biological samples compared to the no added cereal control, however the increase in butyrate production upon supplementation with cereal products was statistically significant for only two biological replicates. Concentration of propionate was significantly higher (P < 0.01) in all cereal additions in biological samples 1, 2, 4 and 5 at 48 hours compared to the no added cereal control. The addition of oats significantly increased (P < 0.05) the concentration of propionate in biological samples 1, 3 and 6 compared to all other cereal additions.
The concentration of all three SCFAs positively correlated with the relative abundance of Bacteroidaceae (Spearman’s r = 0.21, P < 0.001), whilst the concentration of acetate positively correlated with Lactobacillaceae (Spearman’s r = 0.22, P < 0.0001) and concentration of propionate positively correlated with the relative abundance of Veillonellaceae (Spearman’s r = 0.20, P < 0.0001). Each of these families are known to produce SCFAs7,27,29,31,64. Higher production of SCFAs with cereal additions is in agreement with a number of previous studies that have also demonstrated an increase in the production of SCFAs upon gut microbial fermentation of cereal grains65,66,67. Furthermore, elevated production of SCFAs is also a characteristic weaning induced change in the infant gut microbiota during maturation to an adult-like composition7.
The pH of each of the culture vials with cereal additions at 48 hours showed significant reductions (P < 0.001) compared to the no added cereal control, which maintained the pH at the starting measurement of 7.0 ± 0.2 (Supplementary Fig. S4). Samples with rice demonstrated significantly lower (P < 0.001) pH levels compared to samples with oats, wheat and sorghum. pH has been previously been shown to impact gut microbiota composition, especially inhibiting the growth of pathogenic Escherichia coli 68. The metabolic activities of the major SCFA producing bacterial groups such as Bacteroidaceae, Bifidobacteriaceae and Lactobacillaceae have previously been reported to reduce the pH in the large intestine69,70. The higher abundance of at least one of these SCFA producing bacterial families and lower abundance of the family Enterobactericeae upon addition of all tested cereal products may be linked with the reduction in the pH.
Predicted functional changes in response to cereal products
In order to investigate the effect of cereal additions on the functions of the gut microbiota, the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology functional profiles in each treatment at 0, 24 and 48 hours were inferred from the 16 S rRNA gene abundances using phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt). This analysis predicted 12 functional pathways to be significantly differentially abundant in at least five biological samples following cereal additions (Fig. 4 and Supplementary Table S5). Based on the PICRUSt analyses, the functional category of fructose and mannose metabolism showed significantly decreased relative abundance (P < 0.05) in samples supplemented with oats. The Phosphotransferase system (PTS) functional category, responsible for membrane transport of simple carbohydrates, was significantly reduced (P < 0.01) in samples supplemented with oats, rice and wheat. There was a good positive correlation between the inferred relative abundance of the fructose and mannose metabolism functional group with the PTS functional group (Spearman’s r = 0.54, P < 0.0001), this is consistent with the primary uptake mechanism for fructose and mannose being via PTS transporters71. The reduction in the inferred relative abundance of these two functional pathways upon cereal addition could be linked to the addition of more complex sugars such as starch, hemicellulose, cellulose and other polysaccharides from the cereal products. Previous studies have also demonstrated a decrease in the PTS and fructose and mannose metabolism in adults and animal models upon consumption of dietary fibre72,73.

The predicted relative abundance of KEGG Orthology pathways for each sample with different cereal additions inferred using PICRUSt. The heat map shows the relative abundance of KEGG Orthology pathways (rows) with significant differences between treatments at 48 hours (columns) in at least five biological replicates. Significance was determined using an ANOVA with Tukey’s multiple comparisons test. Biological samples (Sample 1–6) were analysed independently. Blue and white represent the highest and lowest relative abundance respectively. Intensity of the colour denotes the level of the relative abundance (as shown in the legend). The inferred relative abundance of the predicted functional pathways and results of tests for significance are provided in Supplementary Table S5.
The inferred relative abundance of functional pathways for glycan degradation increased (P < 0.05) following addition of the cereal products, which could be linked to the presence of plant protein N-linked glycans, due to the availability of glycoproteins in all cereals74. The relative abundance of this pathway correlated with the abundance of Bacteroidaceae (Spearman’s r = 0.51, P < 0.0001). The correlation between glycan degradation and the family Bacteroidaceae is in line with the known ability of this family to digest a range of glycans44,75. Similar changes in these pathways have also been previously observed in animal models such as piglets, upon introduction to solid food76.
The inferred abundance of functional groups for lipopolysaccharide biosynthesis significantly increased (P < 0.05) upon addition of oats, wheat and sorghum, with the highest increase observed with the addition of oats. The inferred relative abundance of this pathway correlated with the relative abundance of the Gram-negative Veillonellaceae (Spearman’s r = 0.47, P < 0.0001), and negatively correlated with the Gram-positive Lachnospiraceae (Spearman’s r = −0.56, P < 0.0001), Rikenellaceae (Spearman’s r = −0.51, P < 0.0001) and Ruminococcaceae (Spearman’s r = −0.60, P < 0.0001). This is in agreement with the occurrence of lipopolysaccharides in Gram-negative bacterial cell wall77,78.
Conclusions
We observed clear shifts in the infant gut microbiota upon addition of each of the cereal products into a large intestine simulating basal medium inoculated with a fecal sample. The relative abundance of the families Bacteroidaceae, Veillonellaceae, Enterobacteriaceae, Bifidobacteriaceae, Lachnospiraceae and Lactobacillaceae significantly changed following cereal supplementation. There were corresponding changes in the concentrations of short chain fatty acids. The concentration of acetate increased with each cereal, whilst the concentrations of butyrate and propionate significantly changed only in specific biological samples with specific cereal additions.
Supplementation with all four cereal products was observed to promote the growth of plant polysaccharide digesting bacteria, reduce the abundance of dominant families in the pre-weaned gut and increase the production of SCFAs. Therefore, these cereal products may have the potential to aid the establishment of a mature gut microbial community. Utilisation of an in vitro gut mimicking model system in the present study facilitated frequent sampling without host interference. However, extension of this work in vivo would be useful as it would eliminate the inherent limitations of batch culture gut microbiota model systems such as accumulation of bacterial metabolites due to absence of host interactions. Therefore, similar in vivo studies using biological samples from a narrower age range would provide further insight into the impact of cereal products on the gut microbiota and host health parameters associated with different weaning diets.
Methods
In vitro digestion of cereal products
Cereal products used in this experiment are derived from whole grain wheat (Weet-bix™), whole grain sorghum (Gluten free Weet-Bix™), organic brown and white rice (Bellamy’s organic baby rice cereal) and organic oats (Real good food-Organic baby oat cereal) (full nutritional profile and ingredient list provided in Supplementary Table S1). Weet-Bix™, Gluten free Weet-Bix™, Bellamy’s organic baby rice cereal and Real good food-Organic baby oat cereal are referred as wheat, sorghum, rice and oats cereal products, respectively, here after.
All enzymes and reagents were purchased from Sigma Aldrich, Australia, unless otherwise stated. Wheat, sorghum, rice and oat based cereal products were purchased from a local Australian supermarket.
Wheat and sorghum based cereal products were ground with a mortar and pestle under sterile conditions prior to in vitro digestion. Each of the four cereal products and a sterile water (MilliQ, Millipore, Australia) sample as a no added cereal control was processed through simulated oral, gastric and small intestine digestion according to published protocols79 with slight modifications. Lower concentrations of salivary alpha amylase (150 U/mL)80, gastric pepsin (3125 U/mL)81, small intestine bile salt (2.5 × 10−3 M), pancreatic trypsin (10 U/mL), chymotrypsin (2.5 U/mL), lipase (2,000 U/mL), colipase (4,000 U/mL), amylase (20 U/mL)81,82 and a higher level of pH (3.0) in the gastric digestion step were maintained to accommodate the differences in infant digestive system81. Following the digestion, cereal products were dialysed at 5 °C in a 2000 MWCO dialysis membrane (Spectra/Por 6, Spectrum Labs) against a sterile NaCl (10 mM) dialysate for 12 hours, which was followed by an additional 2 hour incubation with fresh dialysate83. Dialysed cereal products and the no added cereal control were frozen at −80 °C and freeze dried prior to use.
Preparation of the basal medium
A basal medium was used which was designed to simulate large intestine conditions. The composition of the basal medium per litre was: Peptone 0.5 g, yeast extract 0.5 g, NaHCO3 6 g, Hemin solution (0.05% (w/v) Hemin and 0.2% (w/v) NaOH) 1 mL, L-cysteine HCl 0.5 g, Bile salts 0.5 g, Tween 80 2 mL, Resazurin solution (0.1% (w/v)) 1 mL, Vitamin stock84 1 mL, K2HPO4 0.228 g, KH2PO4 0.228 g, (NH4)2SO4 0.228 g, NaCl 0.456 g, MgSO4 0.0456 g, CaCl2.2H2O 0.0608 g and 1 mL trace mineral solution85 with additional NiSO4.6H2O (0.1 g/L), Na2SeO4 (0.19 g/L) and Na2WO2.2H2O (0.1 g/L). The pH of the medium was adjusted to 7.0 ± 0.2.
Preparation of the basal medium and subsequent culturing were performed under strict anaerobic conditions using a 25% carbon dioxide, 5% hydrogen and 70% nitrogen anaerobic chamber (Thermo Scientific model 1025 Forma). Anaerobic medium was aliquoted into airtight glass vials with rubber stoppers and aluminium lids prior to sterilisation.
Collection and preparation of fecal inocula
All experimental procedures and protocols were reviewed and approved by Macquarie University Human Research Ethics Committee (Reference number 5201400595) and all methods were performed in accordance with the relevant guidelines and regulations. One fecal sample each was collected from six healthy infants (4 female and 2 male) aged 5–11 months. None of the infants were given antibiotics in at least three months prior to sample submission. Infants were fed breast milk (n = 2), formula milk (n = 2) or both (n = 2). All infants were exposed to solid food prior to sample collection. Four infants were introduced to a wider range of food types compared with the other two infants (Table 1).
Fresh fecal samples were collected in a sterile container and immediately placed in an anaerobic jar (Anaero jar, Oxoid Limited, UK) with an Anaerogen sachet (Oxoid) and an anaerobic indicator (Oxoid). Samples were transported anaerobically and laboratory processing was commenced in less than two hours of collection. Fecal slurries were prepared from individual samples by homogenising in anaerobic sterile basal medium and filtering through a sterile nylon mesh cloth (985 µm) prior to using as an inoculum. Fecal slurry preparation was performed under strict anaerobic conditions as used for media preparation.
In vitro fermentation of the cereal products
In vitro digested and freeze dried samples of wheat, sorghum, rice and oats based cereals were added into separate sterile anaerobic vials with the basal medium. A control sample was run in parallel with no added cereal. The final concentration of the cereal additions was maintained at 1% (w/v). Each of these vials were then inoculated with filtered fecal homogenate to obtain a final concentration of at least 0.6% (w/v) in a final volume of 50 mL (0.3 g feces per vial). Experiments were performed in triplicate for each of the fecal samples obtained from six healthy infants. All culture vials were anaerobically incubated at 37 °C with agitation (100 rpm). Aliquots (2 mL) from these cultures were harvested at 0, 24 and 48 hours of incubation and were stored at −80 °C prior to further analyses. The pH of the cultures at 48 hours were measured using pH indicator strips universal pH 0–14 and pH 4.5–10 (Dosatest, VWR, Australia).
Analysis of the gut microbiota
Harvested cultures were used to collect microbial cells by centrifugation at 20,238 × g for 15 minutes. Total community DNA was extracted from cell pellets using a FastDNA spin kit (MP Biomedicals) according to the manufacturer’s instructions. The lysing matrix in the kit was replaced by Lysing matrix E (MP Biomedicals)86. The 16 S rRNA (V4 region) gene was amplified from extracted DNA using 515 (5′-GTGCCAGCMGCCGCGGTAA-3′) forward and 806 (5′-GGACTACHVGGGTWTCTAAT-3′) reverse primers with custom barcodes87,88. PCR amplification, amplicon quantification, purification and sequencing using an Illumina MiSeq V4 platform (2 × 250 bp paired-end sequencing) were conducted at the Ramaciotti Centre for Genomics, Australia.
Two independent Illumina Miseq sequencing runs were performed on all samples (n = 270) as technical replicates of sequencing. Quantitative Insights Into Microbial Ecology (QIIME) software (version 1.9.1)89 was used to process the raw sequence data. Full length and high quality (-q 19 and with other default parameters) reads were used to determine OTUs pre-clustered at 97% similarity using an open-reference protocol against the Greengenes database (version 13_8)90.
After confirming the reproducibility of the two Illumina MiSeq sequencing runs, raw data for each sample were combined and reanalysed using QIIME software according to the methods described above. This resulted in a total of 21,231,850 reads (mean 78,636 ± 16,684) prior to filtering out the OTUs with less than 0.005% reads. Reads per sample were rarefied at 35,095 reads prior to statistical analyses.
Functional prediction using PICRUSt
Functional genes in each treatment condition at 0, 24 and 48 hours were inferred from the 16 S rRNA gene sequences using PICRUSt, online galaxy version 1.1.091. All de-novo OTUs were removed from the open-reference picked OTUs (filtered and rarefied) and those with Greengenes database (version 13_8) identifications were retained for analysis in PICRUSt. These new OTUs were normalised by the 16 S rRNA copy number and functional genes were inferred using KEGG Orthology genes92. The inferred KEGG Orthology genes were grouped into functional pathways at the third BRITE hierarchy level using PICRUSt. A total of 5,516,828,518 (mean 20,432,698 ± 4,553,675) KEGG Orthology genes were predicted. Each of the 270 samples was rarefied at 15,198,942 KEGG Orthology genes. Functional pathways inferred to have >10% higher/ lower relative abundance in at least one cereal addition compared to the no added cereal control were identified. Biological samples were analysed individually and the inferred functional pathways that showed >10% change in at least five biological replicates were used for further statistical analysis.
Quantification of SCFAs
The supernatants (500 µl) of the samples collected at 0, 24 and 48 hours were spiked with an internal standard (4-methyl valeric acid). This was further diluted in a 70% (v/v) ethanol and 0.1% (v/v) trifluoroacetic acid (TFA) solution to obtain a final concentration of the internal standard in the mixture at 100 ppm. The solution was then vortexed and filtered through a 0.2 µm membrane (Millipore, Australia) prior to analysis using a gas chromatograph with a flame ionisation detector (GC-FID, Shimadzu GC-17A). Samples were separated on a 30 m × 0.25 × 0.5 µm i.d. HP-INNOWax fused silica column (Hewlett-Packard) as per the manufacturer’s instructions. GC-FID analysis for each of the 270 samples was performed with further instrument specific technical triplicates (n = 810). SCFA concentrations were normalised for the weight of the fecal inoculum in each biological sample.
Statistical analyses
Statistical analyses of the gut microbiota sequence data were performed on filtered and rarefied OTUs using PRIMER-7 software package93. Non-metric multidimensional scaling (nMDS) plots were constructed based on Bray-Curtis similarity matrices of Log (x + 1) transformed abundance of the OTUs. One-way ANOSIM was performed with 9999 permutations using the Bray-Curtis similarity matrix for each biological sample. An ANOSIM R-value closer to 1 indicates a higher separation of the microbiota structure between samples, whilst R closer to 0 indicates a lower separation. The Shannon diversity index for each sample was determined based on the OTU abundance using the PRIMER-7 software package.
Bacterial families and OTUs with more than 1% relative abundance in at least three biological samples were used for further statistical analyses. Significant differences in the relative abundance of 16 S rRNA gene identifications (family and OTU level), relative abundance of inferred KEGG Orthology pathways, concentration of SCFAs, Shannon diversity indices and pH measurements between treatments were identified using GraphPad Prism (version 7) software (GraphPad Software, USA). Two-way ANOVA with Tukey’s multiple comparisons tests were employed to compare each treatment. Biological samples were analysed individually.
The correlations between the relative abundance of bacterial families, SCFA concentrations, abundance of inferred KEGG Orthology pathways and pH measurements were determined using Spearman’s correlation analyses (two-tailed test) on GraphPad Prism (version 7) software. Correlation analyses were performed between all bacterial families, SCFA concentrations, abundance of inferred KEGG Orthology pathways and pH measurements, however, results of tests where the Spearman’s correlation (r) was −0.2 > r > 0.2 are presented.
References
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267 (2012).
Rodríguez, J. M. et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microbial Ecology in Health & Disease 26, https://doi.org/10.3402/mehd.v26.26050 (2015).
Backhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell host & microbe 17, 690–703, https://doi.org/10.1016/j.chom.2015.04.004 (2015).
Nuriel-Ohayon, M., Neuman, H. & Koren, O. Microbial changes during pregnancy, birth, and infancy. Frontiers in microbiology 7, 1031, https://doi.org/10.3389/fmicb.2016.01031 (2016).
Lim, E. S., Wang, D. & Holtz, L. R. The bacterial microbiome and virome milestones of infant development. Trends in microbiology 24, 801–810, https://doi.org/10.1016/j.tim.2016.06.001 (2016).
Tamburini, S., Shen, N., Wu, H. C. & Clemente, J. C. The microbiome in early life: implications for health outcomes. Nat Med 22, 713–722, https://doi.org/10.1038/nm.4142 (2016).
Koenig, J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. PNAS 108, 4578–4585, https://doi.org/10.1073/pnas.1000081107 (2011).
Eggesbø, M., Botten, G., Stigum, H., Nafstad, P. & Magnus, P. Is delivery by cesarean section a risk factor for food allergy? Journal of Allergy and Clinical Immunology 112, 420–426, https://doi.org/10.1067/mai.2003.1610 (2003).
Sevelsted, A., Stokholm, J., Bonnelykke, K. & Bisgaard, H. Cesarean section and chronic immune disorders. Pediatrics 135, e92–98, https://doi.org/10.1542/peds.2014-0596 (2015).
Huh, S. Y. et al. Delivery by caesarean section and risk of obesity in preschool age children: a prospective cohort study. Arch Dis Child 97, 610–616, https://doi.org/10.1136/archdischild-2011-301141 (2012).
Risnes, K. R., Belanger, K., Murk, W. & Bracken, M. B. Antibiotic exposure by 6 months and asthma and allergy at 6 years: Findings in a cohort of 1,401 US children. Am J Epidemiol 173, 310–318, https://doi.org/10.1093/aje/kwq400 (2011).
Saari, A., Virta, L. J., Sankilampi, U., Dunkel, L. & Saxen, H. Antibiotic exposure in infancy and risk of being overweight in the first 24 months of life. Pediatrics 135, 617–626, https://doi.org/10.1542/peds.2014-3407 (2015).
Kronman, M. P., Zaoutis, T. E., Haynes, K., Feng, R. & Coffin, S. E. Antibiotic exposure and IBD development among children: a population-based cohort study. Pediatrics 130, e794–803, https://doi.org/10.1542/peds.2011-3886 (2012).
Virtanen, S. M. et al. Microbial exposure in infancy and subsequent appearance of type 1 diabetes mellitus-associated autoantibodies: a cohort study. JAMA Pediatr 168, 755–763, https://doi.org/10.1001/jamapediatrics.2014.296 (2014).
Poole, J. A. et al. Timing of initial exposure to cereal grains and the risk of wheat allergy. Pediatrics 117, 2175–2182, https://doi.org/10.1542/peds.2005-1803 (2006).
Arrieta, M. C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M. & Finlay, B. The intestinal microbiome in early life: health and disease. Frontiers in immunology 5, 427, https://doi.org/10.3389/fimmu.2014.00427 (2014).
Tremaroli, V. & Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249, https://doi.org/10.1038/nature11552 (2012).
Valles, Y. et al. Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of Spanish infants. PLoS genetics 10, e1004406, https://doi.org/10.1371/journal.pgen.1004406 (2014).
Fallani, M. et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology 157, 1385–1392, https://doi.org/10.1099/mic.0.042143-0 (2011).
Krebs, N. F. et al. Effects of different complementary feeding regimens on iron status and enteric microbiota in breastfed infants. The Journal of pediatrics 163, 416–423, https://doi.org/10.1016/j.jpeds.2013.01.024 (2013).
McDonald, J. A. et al. Evaluation of microbial community reproducibility, stability and composition in a human distal gut chemostat model. Journal of microbiological methods 95, 167–174, https://doi.org/10.1016/j.mimet.2013.08.008 (2013).
Moroeanu, V. I. et al. Probiotic strains influence on infant microbiota in the in vitro colonic fermentation model GIS1. Indian journal of microbiology 55, 423–429, https://doi.org/10.1007/s12088-015-0542-8 (2015).
Arboleya, S. et al. Assessment of intestinal microbiota modulation ability of Bifidobacterium strains in in vitro fecal batch cultures from preterm neonates. Anaerobe 19, 9–16, https://doi.org/10.1016/j.anaerobe.2012.11.001 (2013).
Nejrup, R. G. et al. Lipid hydrolysis products affect the composition of infant gut microbial communities in vitro. The British journal of nutrition 114, 63–74, https://doi.org/10.1017/S0007114515000811 (2015).
Dostal, A. et al. Iron modulates butyrate production by a child gut microbiota in vitro. mBio 6, e01453–01415, https://doi.org/10.1128/mBio.01453-15 (2015).
Moon, J. S., Joo, W., Ling, L., Choi, H. S. & Han, N. S. In vitro digestion and fermentation of sialyllactoses by infant gut microflora. Journal of Functional Foods 21, 497–506, https://doi.org/10.1016/j.jff.2015.12.002 (2016).
Satoh, T. et al. In vitro comparative evaluation of the impact of lacto-N-biose I, a major building block of human milk oligosaccharides, on the fecal microbiota of infants. Anaerobe 19, 50–57, https://doi.org/10.1016/j.anaerobe.2012.12.007 (2013).
Vester Boler, B. M. et al. In vitro fermentation characteristics of select nondigestible oligosaccharides by infant fecal inocula. Journal of agricultural and food chemistry 61, 2109–2119, https://doi.org/10.1021/jf305056f (2013).
Cinquin, C., Le Blay, G., Fliss, I. & Lacroix, C. Comparative effects of exopolysaccharides from lactic acid bacteria and fructo-oligosaccharides on infant gut microbiota tested in an in vitro colonic model with immobilized cells. FEMS microbiology ecology 57, 226–238, https://doi.org/10.1111/j.1574-6941.2006.00118.x (2006).
Shen, Q., Tuohy, K. M., Gibson, G. R. & Ward, R. E. In vitro measurement of the impact of human milk oligosaccharides on the faecal microbiota of weaned formula-fed infants compared to a mixture of prebiotic fructooligosaccharides and galactooligosaccharides. Letters in applied microbiology 52, 337–343, https://doi.org/10.1111/j.1472-765X.2011.03005.x (2011).
Le Blay, G., Chassard, C., Baltzer, S. & Lacroix, C. Set up of a new in vitro model to study dietary fructans fermentation in formula-fed babies. The British journal of nutrition 103, 403–411, https://doi.org/10.1017/S0007114509991796 (2010).
Sonnenburg, E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215, https://doi.org/10.1038/nature16504 (2016).
Earle, K. A. et al. Quantitative imaging of gut microbiota spatial organization. Cell host & microbe 18, 478–488, https://doi.org/10.1016/j.chom.2015.09.002 (2015).
Desai, M. S. et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353 e1321, https://doi.org/10.1016/j.cell.2016.10.043 (2016).
O’Keefe, S. J. et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nature communications 6, 6342, https://doi.org/10.1038/ncomms7342 (2015).
Deehan, E. C. & Walter, J. The fiber gap and the disappearing gut microbiome: Implications for human nutrition. Trends in endocrinology and metabolism: TEM 27, 239–242, https://doi.org/10.1016/j.tem.2016.03.001 (2016).
Graf, D. et al. Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis 26, 26164, https://doi.org/10.3402/mehd.v26.26164 (2015).
Carvalho-Wells, A. L. et al. Determination of the in vivo prebiotic potential of a maize-based whole grain breakfast cereal: a human feeding study. The British journal of nutrition 104, 1353–1356, https://doi.org/10.1017/S0007114510002084 (2010).
Costabile, A. et al. Whole-grain wheat breakfast cereal has a prebiotic effect on the human gut microbiota: a double-blind, placebo-controlled, crossover study. The British journal of nutrition 99, 110–120, https://doi.org/10.1017/S0007114507793923 (2008).
Martinez, I. et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. The ISME journal 7, 269–280, https://doi.org/10.1038/ismej.2012.104 (2013).
Fan, W., Huo, G., Li, X., Yang, L. & Duan, C. Impact of diet in shaping gut microbiota revealed by a comparative study in infants during the six months of life. J Microbiol Biotechnol 24, 133–143 (2014).
Castanys-Munoz, E., Martin, M. J. & Vazquez, E. Building a Beneficial Microbiome from Birth. Adv Nutr 7, 323–330, https://doi.org/10.3945/an.115.010694 (2016).
Adlerberth, I. & Wold, A. E. Establishment of the gut microbiota in Western infants. Acta paediatrica 98, 229–238, https://doi.org/10.1111/j.1651-2227.2008.01060.x (2009).
Koropatkin, N. M., Cameron, E. A. & Martens, E. C. How glycan metabolism shapes the human gut microbiota. Nature reviews. Microbiology 10, 323–335, https://doi.org/10.1038/nrmicro2746 (2012).
Cockburn, D. W. & Koropatkin, N. M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. Journal of molecular biology 428, 3230–3252, https://doi.org/10.1016/j.jmb.2016.06.021 (2016).
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P. & Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut microbes 3, 289–306, https://doi.org/10.4161/gmic.19897 (2012).
Johnson, E. L., Heaver, S. L., Walters, W. A. & Ley, R. E. Microbiome and metabolic disease: revisiting the bacterial phylum Bacteroidetes. J Mol Med (Berl) 95, 1–8, https://doi.org/10.1007/s00109-016-1492-2 (2017).
Laursen M.F. et al. Infant gut microbiota development is driven by transition to family foods independent of maternal obesity. American society for microbiology 1, doi:https://doi.org/10.1128/mSphere.00069-15 (2016).
Rogosa, M. The genus Veillonella. Journal of Bacteriology 87, 162–170 (1964).
Wang, M. et al. Fecal microbiota composition of breast-fed infants is correlated with human milk oligosaccharides consumed. J Pediatr Gastroenterol Nutr 60, 825–833, https://doi.org/10.1097/MPG.0000000000000752 (2015).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environmental microbiology 19, 29–41, https://doi.org/10.1111/1462-2920.13589 (2017).
Wang, M., Ahrne, S., Antonsson, M. & Molin, G. T-RFLP combined with principal component analysis and 16S rRNA gene sequencing: an effective strategy for comparison of fecal microbiota in infants of different ages. Journal of microbiological methods 59, 53–69, https://doi.org/10.1016/j.mimet.2004.06.002 (2004).
Avershina, E. et al. Transition from infant- to adult-like gut microbiota. Environmental microbiology 18, 2226–2236, https://doi.org/10.1111/1462-2920.13248 (2016).
Azad, M. B. et al. Infant gut microbiota and food sensitization: associations in the first year of life. Clin Exp Allergy 45, 632–643, https://doi.org/10.1111/cea.12487 (2015).
Yoshimi Benno et al. Effect of rice fiber on human fecal microflora. Microbiol. Immunol 33, 435–440 (1989).
Sheflin, A. M. et al. Pilot dietary intervention with heat-stabilized rice bran modulates stool microbiota and metabolites in healthy adults. Nutrients 7, 1282–1300, https://doi.org/10.3390/nu7021282 (2015).
Kumar A. et al. Dietary rice bran promotes resistance to Salmonella enterica serovar Typhimurium colonization in mice. BMC Microbiol 12 (2012).
Henderson, A. J., Kumar, A., Barnett, B., Dow, S. W. & Ryan, E. P. Consumption of rice bran increases mucosal immunoglobulin A concentrations and numbers of intestinal Lactobacillus spp. J Med Food 15, 469–475, https://doi.org/10.1089/jmf.2011.0213 (2012).
Koehler, P. & Wieser, H. Chemistry of Cereal Grains. 11–45, https://doi.org/10.1007/978-1-4614-5425-0_2 (2013).
Sheridan, O. P. et al. Polysaccharide utilization loci and nutritional specialization in a dominant group of butyrate-producing human colonic Firmicutes. Microb Genom 2, e000043, https://doi.org/10.1099/mgen.0.000043 (2016).
Flint, H. J., Duncan, S. H. & Louis, P. The impact of nutrition on intestinal bacterial communities. Curr Opin Microbiol 38, 59–65, https://doi.org/10.1016/j.mib.2017.04.005 (2017).
Freestone, P. P., Walton, N. J., Haigh, R. D. & Lyte, M. Influence of dietary catechols on the growth of enteropathogenic bacteria. Int J Food Microbiol 119, 159–169, https://doi.org/10.1016/j.ijfoodmicro.2007.07.039 (2007).
Fujimura, K. E. & Lynch, S. V. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell host & microbe 17, 592–602, https://doi.org/10.1016/j.chom.2015.04.007 (2015).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54, 2325–2340, https://doi.org/10.1194/jlr.R036012 (2013).
Bach Knudsen, K. E. Microbial degradation of whole-grain complex carbohydrates and impact on short-chain fatty acids and health. Adv Nutr 6, 206–213, https://doi.org/10.3945/an.114.007450 (2015).
Metzler-Zebeli, B. U., Zijlstra, R. T., Mosenthin, R. & Ganzle, M. G. Dietary calcium phosphate content and oat beta-glucan influence gastrointestinal microbiota, butyrate-producing bacteria and butyrate fermentation in weaned pigs. FEMS microbiology ecology 75, 402–413, https://doi.org/10.1111/j.1574-6941.2010.01017.x (2011).
Zhong, Y., Marungruang, N., Fak, F. & Nyman, M. Effects of two whole-grain barley varieties on caecal SCFA, gut microbiota and plasma inflammatory markers in rats consuming low- and high-fat diets. The British journal of nutrition 113, 1558–1570, https://doi.org/10.1017/S0007114515000793 (2015).
Duncan, S. H., Louis, P., Thomson, J. M. & Flint, H. J. The role of pH in determining the species composition of the human colonic microbiota. Environmental microbiology 11, 2112–2122, https://doi.org/10.1111/j.1462-2920.2009.01931.x (2009).
Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Applied and environmental microbiology 74, 4985–4996, https://doi.org/10.1128/AEM.00753-08 (2008).
O’Callaghan, A. & van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Frontiers in microbiology 7, 925, https://doi.org/10.3389/fmicb.2016.00925 (2016).
Siebold, C., Flukiger, K., Beutler, R. & Erni, B. Carbohydrate trasnporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS). FEBS letters 504, 104–111 (2011).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563, https://doi.org/10.1038/nature12820 (2014).
Turnbaugh, P. J., Backhed, F., Fulton, L. & Gordon, J. I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell host & microbe 3, 213–223, https://doi.org/10.1016/j.chom.2008.02.015 (2008).
Strasser, R. Plant protein glycosylation. Glycobiology 26, 926–939, https://doi.org/10.1093/glycob/cww023 (2016).
Comstock, L. E. Importance of glycans to the host-bacteroides mutualism in the mammalian intestine. Cell host & microbe 5, doi:https://doi.org/10.1016/j.chom.2009.05.010 (2009).
Hu, J. et al. Gradual changes of gut microbiota in weaned miniature piglets. Frontiers in microbiology 7, 1727, https://doi.org/10.3389/fmicb.2016.01727 (2016).
Wang, X. & Quinn, P. J. Endotoxins: lipopolysaccharides of gram-negative bacteria. Subcell Biochem 53, 3–25, https://doi.org/10.1007/978-90-481-9078-2_1 (2010).
Vesth, T. et al. Veillonella, Firmicutes: Microbes disguised as Gram negatives. Stand Genomic Sci 9, 431–448, https://doi.org/10.4056/sigs.2981345 (2013).
Minekus, M. et al. A standardised static in vitro digestion method suitable for food - an international consensus. Food & function 5, 1113–1124, https://doi.org/10.1039/c3fo60702j (2014).
Nguyen, T. T. P., Bhandari, B., Cichero, J. & Prakash, S. A comprehensive review on in vitro digestion of infant formula. Food Research International 76, 373–386, https://doi.org/10.1016/j.foodres.2015.07.016 (2015).
Dupont, D. et al. Comparative resistance of food proteins to adult and infant in vitro digestion models. Mol Nutr Food Res 54, 767–780, https://doi.org/10.1002/mnfr.200900142 (2010).
Böttger, F. H., Etzel, M. R. & Lucey, J. A. In vitro infant digestion of whey protein–dextran glycates. Food. Digestion 4, 76–84, https://doi.org/10.1007/s13228-013-0032-6 (2013).
Connolly, M. L., Lovegrove, J. A. & Tuohy, K. M. In vitro evaluation of the microbiota modulation abilities of different sized whole oat grain flakes. Anaerobe 16, 483–488, https://doi.org/10.1016/j.anaerobe.2010.07.001 (2010).
Scheifinger, C. C., Linehan, B. & Wolin, M. J. H2 production by Selenomonas ruminantium in the absence and presence of methanogenic bacteria. Applied Microbiology 29, 480–483 (1974).
Balch, W. E., Fox, G. E., Magrum, L. J., Woese, C. R. & Wolfe, R. S. Methanogens: Reevaluation of a unique biological group. Microbial Reviews 43, 260–296 (1979).
Gillings, M. R. Rapid extraction of PCR-competent DNA from recalcitrant environmental samples. Methods in molecular biology 1096, 17–23, https://doi.org/10.1007/978-1-62703-712-9_2 (2014).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal 6, 1621–1624, https://doi.org/10.1038/ismej.2012.8 (2012).
Caporasoa, J. G. et al. R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America 108, 4516–4522 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336, https://doi.org/10.1038/nmeth0510-335 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72, 5069–5072, https://doi.org/10.1128/AEM.03006-05 (2006).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, https://doi.org/10.1038/nbt.2676 (2013).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114, https://doi.org/10.1093/nar/gkr988 (2012).
Clarke, K. & Gorley, R. PRIMERv7: User Manual/Tutorial. PRIMER-E, Plymouth (2015).
Acknowledgements
We thank Sanitarium Health and Wellbeing, Australia and Macquarie University, Australia for providing financial support through the Enterprise Partnership Scheme, pilot research grant (2016–17).
Author information
Authors and Affiliations
Contributions
H.K.A.H.G., S.G.T., I.T.P. and J.A. designed the study. H.K.A.H.G. prepared samples for the GC-FID and conducted all experiments including in vitro digestion, culturing, DNA extraction and bioinformatics analysis. R.W.W.C. performed the GC-FID quantification of SCFAs. All the statistical analyses were performed by H.K.A.H.G. H.K.A.H.G., S.G.T., I.T.P., N.P. and J.A. interpreted the results. H.K.A.H.G. drafted the manuscript with contributions of S.G.T. and I.T.P. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
J.A. is an employee of Sanitarium Health and Wellbeing, Australia, the producer of Weet-Bix™ and Gluten free Weet-Bix™. All other authors have no competing financial interests.
Additional information
Accession codes: The 16S rRNA gene sequence data generated during this study are available on the GenBank Sequence Read Archive database under accession number SRP107068.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gamage, H.K.A.H., Tetu, S.G., Chong, R.W.W. et al. Cereal products derived from wheat, sorghum, rice and oats alter the infant gut microbiota in vitro . Sci Rep 7, 14312 (2017). https://doi.org/10.1038/s41598-017-14707-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14707-z
This article is cited by
-
Gastrointestinal Health Benefits of Sorghum Phenolics
Plant Foods for Human Nutrition (2024)
-
A Comprehensive Review on Formulation of Baby Foods: Nutraceutical potential of Fruits, Vegetables, Cereals and Probiotics in Baby Food Preparation
Current Food Science and Technology Reports (2024)
-
Starch Digestion in Infants: An Update of Available In Vitro Methods—A Mini Review
Plant Foods for Human Nutrition (2022)
-
The sugar composition of the fibre in selected plant foods modulates weaning infants’ gut microbiome composition and fermentation metabolites in vitro
Scientific Reports (2021)
-
Clinical Aspects of Thickeners for Pediatric Gastroesophageal Reflux and Oropharyngeal Dysphagia
Current Gastroenterology Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
