
Abstract
Hepatitis B virus (HBV) is a blood-borne pathogen responsible for chronic hepatitis, cirrhosis, and liver cancer. The mechanism of HBV entry into hepatocytes remains to be investigated. Recently, sodium taurocholate cotransporting polypeptide (NTCP) was discovered as a major HBV receptor based on an in vitro infection system using NTCP-reconstituted HepG2 cells. However, this infection system relies on the compound polyethylene glycol (4% PEG), which is not physiologically relevant to human infection. High concentration of heparin has been commonly used as an inhibitor control for in vitro infection in the field. Surprisingly, we found that heparin at physiological concentration can enhance HBV infection in a PreS1-peptide sensitive, NTCP-dependent manner in both HepaRG and HepG2-NTCP-AS cells. O-sulfation of heparin is more important for the infection enhancement than N-sulfation. This system based on the HepG2-NTCP-AS cells can support in vitro infection with HBV genotypes B and C, as well as using serum samples from HBeAg positive and negative chronic carriers. In summary, our study provides a PEG-free infection system closely resembling human natural infection. In addition, it points to a future research direction for heparin and heparin-binding host factor(s) in the blood, which are potentially involved in viral entry. To our knowledge, this is the first soluble and circulatory host factor which can enhance HBV in vitro infection.
Similar content being viewed by others


Introduction
HBV is an enveloped and partially double-stranded DNA virus which established chronic infection in around 240 million carriers worldwide. Current treatment for HBV cannot effectively eradicate the virus from chronic hepatitis B patients1. These patients have a high risk to develop liver cirrhosis and hepatocellular carcinoma2. Studies on the HBV life cycle have been hampered by the lack of an efficient and user-friendly in vitro infection system. Primary human hepatocytes (PHHs) and HepaRG cells had served as valuable tools for studying the early event of viral entry, albeit the viral receptor remained elusive3,4,5. One major concern of primary human hepatocytes is its expensive cost from the commercial source. In addition, it is not a robust system because the qualities of hepatocytes tend to vary from lot to lot. In contrast, the HepaRG cell system is relatively more reliable and consistent. However, the infection efficiency is not high, and it involves tedious work to grow and maintain well-differentiated HepaRG cells5. Recently, sodium taurocholate cotransporting polypeptide (NTCP) was identified as a functional receptor for HBV and HDV6. This NTCP-reconstituted HepG2 system has emerged as a powerful resource in the field of HBV research7,8,9.
One common way to achieve a higher efficiency in HBV infection is to include 4–5% of polyethylene glycol (PEG) during the incubation period of viral infection. For example, in the presence of PEG, the efficiency of HBV infection on PHH was elevated up to 20 times, mainly due to the enhanced adsorption between virus and hepatocytes4. Similarly, PEG was able to promote HBV infection in HepaRG and NTCP-reconstituted HepG2 hepatocytes5,6. On the other hand, the use of PEG is not without any reservations. For example, PEG is known to induce membrane fusion, such as its use in the generation of hybridoma10,11. Therefore, PEG might provoke non-specific fusion between the virions and the host cell membrane. Most importantly, PEG is a non-biological chemical not found in human body. It remains a concern whether the current PEG-containing infection protocol could faithfully mimic the viral entry event in vivo.
HBV infection is supposed to be initiated by HBV binding to the heparan sulfate proteoglycans (HSPGs) on the hepatocyte surface12. Soluble heparins, which share a similar structure with HSPGs, could therefore inhibit HBV entry by interrupting the interaction between HBV and HSPGs on target cells12. As a highly sulfated glucosaminoglycan, heparin is mainly secreted by basophils and mast cells13. The normal physiological concentration of heparin in human plasma ranges from 1 to 5 μg/ml14,15,16. The physiological roles of heparin and HSPGs are highly diverse, including anticoagulation, signaling, development, anti-inflammation and anti-metastatic17,18. To date, more than 400 heparin-binding proteins have been reported in literature19.
In this study, we revisited the effects of heparin and PEG on HBV infection. Consistent with a general impression about heparin in HBV infection, we also found that heparin at a higher concentration, can inhibit HBV infection12,20,21,22. However, to our surprise, at its physiological concentration (1 to 5 μg/ml), heparin could actually enhance HBV infection efficiency in both HepaRG and NTCP-reconstituted hepatocytes. This phenomenon was most pronounced when without PEG or with a reduced amount of PEG (1.2%). This enhancement effect of heparin could become obscured in the presence of 4% PEG, a concentration commonly used in HBV in vitro infection experiments. Our study established a PEG-free in vitro infection system which resembles more closely the physiological condition in the liver. One implication from our studies here is that heparin-binding host factors may deserve further attention in future studies of viral entry.
Results
Establishment of an HBV in vitro infection system using a HepG2-NTCP-AS cell line
The experimental procedure for an HBV in vitro infection is as outlined in Fig. 1a. To establish an NTCP-expressing HepG2 cell line for in vitro infection with HBV, HepG2 cells were stably transfected with a human NTCP-flag expression plasmid driven by a CMV promoter. A stable clone with the highest level of NTCP protein expression was named HepG2-NTCP-AS in this study. Glycosylated NTCP-flag protein is around 72 kD as detected by Western blot (Fig. 1b). Treatment with PNGase F removed the glycan from NTCP resulting in a 36 kD protein. We tested the infection efficiency of this HepG2-NTCP-AS cell line by using serum samples from HBV carriers (Supplementary Table S1) in the presence of 4% PEG. The serum sample B76 containing the highest HBV DNA titer was chosen for use in this study. In the time course experiments, increasing amounts of HBsAg (HBV surface antigen) and HBeAg (HBV e antigen) can be detected in the media by ELISA at various time points post-infection (Fig. 1c). No signals of HBsAg and HBeAg were detected in the negative control of parental HepG2 cells containing no NTCP. The trends of increasing levels of HBsAg and HBeAg in the post-infection time course strongly supports for the de novo synthesis of HBsAg and HBeAg in the infected HepG2-NTCP-AS cells. Consistent with the result from the ELISA assay, HBV replicative DNA intermediates can be detected on day 9 post-infection by Southern blot analysis (Fig. 1d). Moreover, expression of HBV core protein (HBc) can be visualized in 8–9% of infected cells by immunostaining and confocal microscopy using anti-HBc antibody (Fig. 1e). No HBc was detected in mock-infected cells. Taken together, our data provided evidence that we have successfully established an HBV in vitro infection system based on the NTCP reconstituted HepG2 cells. Furthermore, this HepG2-NTCP-AS cell system can support in vitro infection using serum samples from both HBeAg positive and negative patients, as well as using input viruses from HBV genotypes B and C (Supplementary Table S1; Fig. S1a). HBV DNA from the HBeAg-negative patient contains a hotspot stop codon mutation at precore codon 28, by PCR cloning and sequencing (Fig. S1b).

Establishment of an HBV in vitro infection system using a HepG2-NTCP-AS cell line. (a) An illustration of experimental design. (b) The expression of Flag-tagged NTCP in a HepG2-NTCP-AS stable cell line was detected by Western Blot using an anti-Flag antibody. De-glycosylation of NTCP by the PNGase F treatment shifted the MW from around 72 kD to 36 kD. The parental HepG2 cells served as a negative control for Flag-NTCP expression. GAPDH expression served as a loading control. (c) Similar trends of increasing expressions of both HBsAg and HBeAg were detected by ELISA only in HepG2-NTCP-AS cells infected with human HBV-containing serum. No detectable increase in HBsAg and HBeAg was noted in the negative controls using HepG2 cells or mock infection with no virus. (d) Total intracellular core particle-associated viral DNAs were analyzed by Southern blot at 9 dpi. Non-infected HepG2-NTCP-AS cells served as a negative control. RC: relaxed circle DNA; SS: single-strand DNA. (e) Confocal microscopy detected HBc protein (green) in HepG2-NTCP-AS cells infected with serum-derived HBV. Cell nuclei were counterstained with DAPI (blue).
Heparin at physiological concentration in human plasma can stimulate HBV in vitro infection
While NTCP has been identified as a receptor for HBV entry6, it is possible that other unknown host factors could also contribute to HBV viral entry. For example, hNTCP transgenic mice cannot support in vivo HBV infection or re-infection9. To look for any soluble host factors which might facilitate HBV infection of hepatocytes, we examined the plasma from a healthy individual for its potential effect on HBV in vitro infection. HepG2-NTCP-AS cells were infected with serum from an HBV chronic carrier (B76), in the presence or absence of 10% human plasma. However, dilution of human sodium citrated plasma in DMEM medium resulted in plasma clotting due to the diluted sodium citrate (anticoagulant) in human plasma. This clotting issue cannot be circumvented by replenishing sodium citrate to the infectious inoculum due to its calcium-chelating properties against cell attachment on the dish. To prevent plasma from clotting in cell culture, we mixed heparin into the infectious inoculum. Heparin has been known for its potent anticoagulant activity23. Unfortunately, it was also widely used as an HBV entry inhibitor12,20,21,22. To minimize the inhibitory effect of heparin on HBV in vitro infection, we used 4.5 μg/ml heparin as a minimum anticoagulant concentration in the infectious inoculum. Another unexpected problem in plasma clotting is about the use of 4% PEG. In the field, 4% PEG has been routinely included to enhance HBV in vitro infection efficiency4,12,20,21,22,24. Unfortunately, 4% PEG also promoted plasma clotting. Therefore, we reduced the PEG concentration down to 1.2% in the infectious inoculum in our experimental procedure of in vitro infection.
As outlined in Fig. 2a, we performed in vitro infection assay in search for potential enhancement factor(s) in human plasma. Interestingly, we detected reproducibly increased levels of HBsAg and HBeAg, when the infection experiment was supplemented with human plasma, compared to the serum only control (no plasma) (Fig. 2b). This phenomenon was observed whether or not human plasma was pretreated with heat. We fractionated human plasma from a healthy individual by Amicon Ultra centrifugal filters according to the molecular weights of 100 kD, 10 kD, and 3 kD. Plasma fractions do not clot due to the disrupted coagulation cascade. Different MW fractions of plasma, with or without heparin supplementation, were tested for HBV infection. To our surprise, none of these plasma fractions had any significant effect on HBV infection, if without heparin supplementation (Fig. S2a). In contrast, when heparin was supplemented during HBV infection, all fractions of human plasma exhibited an enhancing effect on HBV infection (Fig. S2b). These results led us to the hypothesis that heparin itself at lower concentrations might have an enhancing effect on HBV infection. We compared further the heparin effect without human plasma, and observed the infection enhancement in a plasma-independent manner (Fig. 2c). Our finding contradicts the current knowledge of heparin as a potent inhibitor of HBV infection12,20,21,22.

Heparin at physiological concentration in human plasma can stimulate HBV in vitro infection. (a) An illustration of experimental design. (b) Human plasma, with or without heat pre-treatment, enhanced HBV in vitro infection. HBsAg (left panel) and HBeAg (right panel) were measured at 9 dpi by ELISA. HepG2-NTCP-AS cells were infected with HBV serum at MOI 300–3000 in the presence of 1.2% PEG. Human plasma was supplemented with fresh heparin before adding to the cell culture to prevent coagulation. Data are shown as mean ± SEM of at least three independent experiments (**p < 0.01). Dashed lines represent the cutoff value of the background noise in the ELISA assays. (c) Heparin alone without human plasma can still enhance HBV infection. (d) Heparin at physiological concentrations (1 to 5 µg/ml)14,15,16 can enhance HBV infection in a dose-response experiment. HepG2-NTCP-AS cells were infected with HBV serum at MOI 300–3000 at 37 °C for 24 hours. Increasing concentrations of heparin (0, 1.5, 4.5, 13.5, 40, 120 μg/ml) were used in the presence of 1.2% PEG or 4% PEG. (*p < 0.05). (e) Treatment with PreS1 lipopolypeptide inhibited heparin-enhanced infection of HepG2-NTCP-AS cells. These HepG2-NTCP-AS cells were infected with HBV serum in the presence of 1.2% PEG with and without 4.5 μg/ml heparin. HepG2-NTCP-AS cells were pre-incubated with 500 nM PreS1 peptide for 30 minutes before infection. (*p < 0.05). (f) Heparin-enhanced HBV infection can be abrogated by the continuous presence of a nucleoside analog 3TC (10 uM), from 1 to 9 dpi. HepG2-NTCP-AS cells were infected with HBV serum in the presence of 1.2% PEG, with and without 4.5 μg/ml heparin.
To resolve this apparent discrepancy in the heparin effect on HBV infection, we titrated for the optimum concentration of heparin in the range from 0 to 120 µg/ml (Fig. 2d). We observed good enhancement for infection when heparin concentration was around 1.5–4.5 μg/ml, and at 40 μg/ml, heparin showed 50% inhibition of HBV infection in this HepG2-NTCP-AS cell system. Similar dose-response profiles were detected between HBsAg and HBeAg assays (Fig. 2d). The enhancement effect of heparin on HBV infection was most pronounced when the amount of PEG was reduced. At 4% PEG, heparin effect was then masked by the stronger effect from 4% PEG, suggesting that the mechanism of heparin enhancement could also be at the initial step of adsorption and attachment to the heparan sulfate proteoglycan (HSPG) on the cell surface of hepatocytes4,25. This heparin-mediated enhancement on HBV infection is most likely to be NTCP-dependent, since the PreS1 treatment (Myrcludex B) resulted in complete inhibition of HBV infection (Fig. 2e). Next, we examined whether 3TC (lamivudine, a nucleoside analog) treatment could inhibit heparin enhancement on HBV infection. Continuous presence of 3TC was included in the medium between HBV inoculation and day 9 post-infection (Fig. 2a). Significant differences in both HBsAg and HBeAg secretion were detected between treatments with vs. without 3TC (Fig. 2f). The abolishment of the heparin enhancement on HBV infection by 3TC treatment, provided experimental evidence for de novo synthesis of HBsAg and HBeAg in this HepG2-NTCP-AS in vitro infection system.
Altogether, our studies on human plasma uncovered unexpectedly an enhancement effect of heparin, a naturally occurring anticoagulant in the blood, at its physiological concentration.
Heparin facilitates PEG-free in vitro infection with HBV
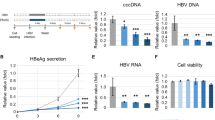
We observed the heparin enhancement in the presence of 1.2% PEG, but not at 4% PEG (Fig. 2). Here, we asked whether heparin can enhance HBV infection without PEG (Fig. 3a). Indeed, even in the absence of PEG, an increase of HBV viral antigen expression was observed, when infectious inoculum was supplemented with 4.5 μg/ml heparin (Fig. 3b). The results of Southern blot analysis were consistent with those from the ELISA analysis. HBV infection with neither heparin nor PEG showed no detectable HBV-specific replication signal (lane 3, Fig. 3c). However, heparin treatment strikingly increased HBV infection efficiency as measured by Southern blot analysis (compare lane 3 and lane 4, Fig. 3c). Infection efficiency was further increased in combination treatment with 1.2% PEG and 4.5 μg/ml heparin, relative to 1.2% PEG treatment alone (compare lane 5 and lane 6, Fig. 3c). Heparin treatment at high concentration (450 µg/ml) completely abolished the signal of HBV replicative intermediates (compare lane 6 and lane 7, Fig. 3c). This result is consistent with the known inhibitory effect of heparin on HBV infection in literature12,20,21,22. The results of heparin enhancement by ELISA and Southern blot analyses (Fig. 3b and c) were confirmed by immunofluorescence assay of HBV core protein (Fig. 3d). Combination of 4.5 μg/ml heparin and 1.2% PEG increased the percentage of HBc positive cells (Fig. 3d, left vs. right panels), compared to those with no heparin treatment in the absence or the presence of 1.2% PEG. We noted that heparin reduced the percentage of HBc positive cell number in the presence of 4% PEG (Fig. 3d, bottom).

Heparin facilitates PEG-free in vitro infection with HBV. (a) An illustration of experimental design. (b) Heparin alone can enhance HBV infection in a PEG-independent manner. The secretions of HBsAg and HBeAg were measured every other day by ELISA in a time course experiment (1–9 dpi). HepG2-NTCP-AS cells were infected with HBV serum pre-mixed with and without 4.5 μg/ml heparin. (c) A combinatory effect of heparin (4.5 µg/ml) and PEG (1.2%) can be detected by Southern blot analysis. Lane 1 and 2: Positive and negative controls were prepared from HBV DNA transfected HuH-7 cells and mock-infected HepG2-NTCP-AS cells, respectively. Heparin at a lower dose (4.5 μg/ml) enhanced HBV in vitro infection in the presence of 1.2% PEG (compare lanes 5 and 6), or in the absence of PEG (compare lanes 3 and 4). However, overdosed heparin (450 μg/ml) or treatment with PreS1 peptide (500 nM) was strongly inhibitory to HBV infection (lane 7 and 9). 4% PEG exhibited the most potent effect on HBV infection (lane 8). (d) In a PEG-free in vitro infection system, low dose heparin (4.5 µg/ml) significantly increased the percentage of HBc-positive HepG2-NTCP-AS cells by confocal microscopy. Cell nucleus was counterstained with DAPI. The estimated percentages of HBc positivity were based on the average from three different microscopic fields (Mean ± SD). Approximately 500 cells were scored in each field.
Heparin and heparan sulfate contain modifications like N-sulfation, N-acetylation or O-sulfation26. To determine whether heparin sulfation is required for heparin-enhancement on HBV infection, we compared the effects among sulfated heparin, O-desulfated (deO) heparin, and N-desulfated (deN) heparin using this HepG2-NTCP-AS-based in vitro infection assay (Fig. S3). While the N-desulfated heparin still maintained significant effect on HBV infection, O-desulfated heparin could no longer enhance HBV infection by the ELISA assay for HBsAg and HBeAg.
Encouraged by the results of heparin-mediated enhancement on HBV infection, we asked if the same heparin enhancement can be observed in hepatocytes other than the HepG2-NTCP-AS cells. As shown in Fig. 4, the same stimulatory effect of heparin on HBV infection can be observed in another human hepatoma cell line HepaRG5. Moreover, O-sulfation of heparin remains more important than N-sulfation using the HepaRG-based in vitro infection (Fig. S4).

Heparin enhancement on HBV infection using the HepG2-NTCP-AS cells were validated by using a HepaRG infection system. (a) A dose-dependent curve of heparin on HBV in vitro infection using human HBV-containing serum and adherent HepaRG cells. Increasing concentrations of heparin treatment (0, 1.5, 4.5, 13.5, 40, 120 μg/ml) were tested in the presence of 1.2% PEG. (b) Heparin can further enhance HBV infection using HepaRG cells in the presence of 1.2% or 4% PEG.
In summary, heparin at the physiological concentration in the blood stimulated HBV infection in vitro.
Discussion
The HepG2-NTCP system provides a new platform for the studies on the life cycle of HBV7,8,9. We established a similar HepG2-NTCP-AS system to search for potential host factors in the plasma involved in viral entry. To validate the in vitro infection system, we need to exclude the possibility of input virus contamination. In a time course experiment, we observed a reproducible trend of increasing signals of secreted HBsAg, HBeAg, and HBV DNA replicative intermediates. In addition, treatment with nucleoside analogs abolished these signals, suggesting that de novo synthesis post-viral entry is responsible for the generation of HBV specific proteins and DNA genome.
Heparan sulfate is known to serve as an attachment receptor for many bacteria, viruses, and parasites27. Many human viruses can initiate their infection by binding to cell surface heparan sulfate28. Heparin is structurally related to heparan sulfate, and can be produced by basophils and mast cells13. The best known function of heparin is to prevent blood from clotting. In most in vitro viral infection systems, soluble heparin was used as an inhibitor of infection at concentrations ranging from 1 μg/ml to 100 μg/ml, depending on the specific virus in each study28. On the other hand, heparin can also promote AAV-induced T cell activation and increase the uptake of AAV2 by dendritic cell29. In addition, heparin binding can induce conformational change at AAV230.
Our studies on human plasma led to the identification of heparin as an enhancement host factor for in vitro infection with HBV. In earlier reports, heparin has been commonly used as an control inhibitor for HBV infection (Table 1). The opposite effects of heparin on HBV infection between our current study and earlier reports can be related to a number of possibilities: 1) We detected heparin enhancement at physiological concentration (1–5 µg/ml), while earlier reports observed heparin inhibition at much higher concentrations (25 U/ml, 100 µg/ml, or 100 IU/ml)12,21,22. In our heparin dose-response experiment, we observed no enhancement at 13.5 µg/ml, and strong inhibition at 40 and 120 µg/ml (Fig. 2 d).2) Earlier reports used 4% PEG in their infection procedure12,21,22. It was shown that PEG can enhance the binding between the glycosaminoglycan (GAG) side chain of HSPG and the PreS1 domain of HBV large envelope protein (L-HBsAg)12. In our studies, we observed heparin enhancement only at no PEG or 1.2% PEG. At 4% PEG, we observed no heparin enhancement either (Fig. 3d). 3) One earlier report pre-treated primary Tupaia hepatocytes with heparin at 5 µg/ml for 1 hour before HBV infection, and observed 50% inhibition on HBV infection efficiency20. The difference in the heparin effects here could include differences between primary Tupaia hepatocytes and human HepG2-NTCP-AS cells, the sources of input viruses (purified vs. non-purified), as well as different protocols for the heparin treatment (1 hr vs. overnight), HBV binding and incubation time. 4) Another earlier report used 9.4 µg/ml heparin in their infection protocol, and obtained 50% inhibition of infection efficiency12. We noted that this report used 4% PEG and the source of HBV was purified from the medium of an HBV-producing cell line HepG2.2.15. In our low-PEG or PEG-free assay system, the source of HBV was directly from the unpurified human serum. Overall, the effect on the infection efficiency from our low-heparin and low/no-PEG protocol, is less strong than the effect from the 4% PEG protocol. However, our system is more closely related to human natural infection. It is our hope that such a system could provide a platform for the search of host factors important in viral entry.
What could then be the mechanisms for heparin enhancement and inhibition on the in vitro HBV infection? It is generally believed that HBV can bind to HSPGs as a low-affinity receptor before the initiation of HBV infection12,20,25,31 (Fig. 5, upper panel). Heparin can bind to the large envelope protein of HBV (L-HBsAg)12. Presumably, higher concentration of heparin can bind to the L-HBsAg on the virion surface and interfere with HBV attachment to HSPGs on the surface of hepatocytes. Recently, it was reported that, in addition to the PreS1 domain, the heparin binding site of HBV envelope is another determinant of HBV infectivity25. The heparin binding site is located close to an antigenic loop (AGL), which is separated from the PreS1 domain, and is structurally related to the group a-determinant of HBsAg. It is tempting to speculate that lower concentration of heparin can bind to the HBV virion surface and induce an active conformation, leading to the bypass of HSPG attachment, and thus allow more direct and effective interactions between the heparin-virion complex and the NTCP receptor (Fig. 5, middle panel). According to this conceptual framework, excessive amount of heparin binding could dampen the activation of the PreS1 domain, and prevent it from interaction with NTCP (Fig. 5, lower panel). Further studies would be required to elucidate the mechanism of heparin enhancement in molecular details.

Hypothetical mechanisms for the effects of heparin concentration on HBV entry via a two-step process. Dotted lines separate Fig. 5 into three panels. Upper panel: It is generally believed that HBV entry would undergo a 2-step process (see references in Table 1). In step 1, HBV virions would bind to heparin sulfate proteoglycan (HSPG) on the surface of hepatocytes12,20,25. This initial attachment step is then followed by step 2, where the PreS1 peptide of the L-HBsAg would undergo some kind of conformational change (more exposed), and become more “activated” for binding to a bile salt transporter NTCP on the cell surface, leading to the internalization of HBV25. Middle panel: Heparin and heparan sulfate share similar structures18. To explain the stimulatory effect of heparin at physiological concentration (1–5 μg /ml), we postulate here that once soluble heparin binds to the virion surface, it can also activate the PreS1 domain of L-HBsAg. In this scenario, step 1 is no longer necessary for HBV binding to NTCP, and can be bypassed. There are many heparin-binding protein factors in the serum19. It is possible that some unknown heparin binding factor(s) could play a positive role in this viral entry cascade. For example, complex formation of virions, heparin, and heparin-binding proteins, could facilitate either the conventional step 1-binding to HSPG, or the direct binding to NTCP. Lower panel: At non-physiologically high concentrations of heparin, the surface of HBV virions are fully decorated with excessive amounts of heparin. In this case, virions saturated with negatively-charged heparin cannot bind to negatively charged HSPG. We also speculate here that overly heparinated HBV virions cannot activate the PreS1 domain ligand for the NTCP receptor recognition.
In this regard, it is worth mentioning that we observed no significant enhancement on HBV infection by using non-physiological compounds, such as chondroitin sulfate and dextran sulfate (Fig. S5). In addition, in our preliminary studies, we found that heparin can significantly enhance HBV binding to HepG2-NTCP-AS hepatocytes in a 4-hr incubation period at 4 °C or 37 °C by 4% PEG, but not by low (1.2%) or no PEG (Fig. S6a). On the contrary, in a 24-hr incubation period at 37 °C condition, significant heparin-mediated enhancement on HBV binding to hepatocytes, can be more easily detected at low or no PEG than 4% PEG (Fig. S6b).
We noted that the O-sulfation of heparin is more important for the infection enhancement than N-sulfation (Figs S3 and S4). It is possible that O-sulfated heparin has a higher affinity for HBV virion than N-sulfated heparin. Alternatively, it is equally possible that O-sulfated heparin can recruit more or different host factors for infection. When completely de-N-sulfated heparin was studied for its inhibitory effect on HBV infection, no inhibition was observed12. Therefore, by inference, it is likely that O-sulfated heparin does not inhibit HBV infection, even when given in excess. Taken together, O-sulfated heparin may be considered as a positive factor for HBV infection.
In conclusion, we found that heparin at physiological concentration has a moderate, yet highly reproducible, stimulatory effect on HBV infection in the low or no PEG condition. Relative to the enhancement effect of 4% PEG, heparin enhancement is less potent. However, one merit from our study here resides in the establishment of a PEG-free HepG2-NTCP-AS platform, which is more closely mimicking HBV natural infection in vivo. It will be worth investigation in the future whether any of the hundreds of heparin-binding host factors could play a role in viral entry in the liver.
Methods
Ethics statement
Informed consent was obtained from patients. The study protocols were approved by the Institutional Review Board of the College of Public Health, National Taiwan University, Taipei, Taiwan and Kaohsiung Medical University Hospital, Kaohsiung, Taiwan (IRB number is KMUH-IRB950134). All methods in this study were performed in accordance with the relevant guidelines and regulations.
Reagents
CMV6-NTCP-flag tag expression vector was purchased from Origene.
Heparin, DeO heparin, DeN heparin and anti-flag M2 antibody were purchased from Sigma-Aldrich. PNGase F was purchased from Promega. PreS1 peptide was synthesized by Yao-Hong Biotechnology Inc. (Taiwan). PEG6000 was purchased from Millipore corporation. 30% stock solution was prepared in PBS buffer.
Establishment of HepG2-NTCP-AS cell line
Parental HepG2 cells were transfected with CMV6-NTCP-flag tag expression plasmid. Transfected cells were selected at 400 μg/ml Neomycin. Neo-resistant colonies were amplified and examined for NTCP expression by Western blot using anti-Flag M2 antibody. Colonies expressing a high level of NTCP were tested for HBV infection. HBV infection was evaluated by HBsAg, HBeAg ELISA, IFA of HBc, and Southern blot analysis.
Plasma fractionation
Normal human plasma was fractionated by Amicon Ultra Centrifugal Filters. A total of 2 ml normal human plasma was fractionated serially through 100 kD, 10 kD, and 3 kD filters. Concentrated fractions on each filter were diluted up to 10 ml with PBS, and the centrifugation process was repeated to further enrich plasma proteins in each MW fraction. Each concentrated fraction was adjusted up to 2 ml final volume by PBS, and stored at −80 °C. For HBV infection, plasma fractions were added into the infectious serum sample at a 10% final concentration (v/v).
HBV in vitro infection assay
HepaRG cells were purchased from Invitrogen and cultured according to the product manual. HepG2-NTCP-AS cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Before infection, HepG2-NTCP-AS cells were maintained in serum-free DMEM supplemented with 5 μg/ml transferrin, 3 μg/ml insulin, 5 ng/ml sodium selenite, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 2% DMSO for 1–2 days. Approximately 105 HepG2-NTCP-AS cells in one well of a 24-well plate were infected by inoculating a total of 0.5 ml/well of serum-free DMEM medium supplemented with HBV serum (300–3000 MOI), 1.2–4.0% PEG and 2% DMSO. PreS1 peptide treatment was conducted by incubating 500 nM PreS1 peptide 30 minutes before and during HBV infection. After HBV infection at 37 °C for 24 hrs, cells were washed with PBS twice at day 1 and day 3 post-infection. Cells were maintained in DMEM medium supplemented with 5% FBS and 2% DMSO after infection. Culture media were collected every other day starting from day 5 post-infection.
Western blot
NTCP-flag protein expression was detected by anti-flag M2 antibody from Sigma-Aldrich. Western blot assay procedures were performed as described previously32.
Immunofluorescence assay
HBc protein was stained by rabbit anti-HBc as reported previously32. Immunofluorescence assay procedures were performed as detailed elsewhere33.
Southern blot assay
HBV whole genomic DNA was purified from HBV-monomer plasmid DNA by EcoRI digestion. HBV DNA probe was radiolabeled by Red Prime DNA labeling system according to the manufacturer’s protocol (GE Healthcare). The Southern blot assay was performed according to the standard protocol as described previously34. The typical smearing signals on the Southern blot reflect characteristic HBV DNA replicative intermediates with different molecular weights, and are not due to overexposure on the Typhoon Imager or X-ray film.
HBsAg and HBeAg detection by ELISA
HBsAg and HBeAg ELISA kits were purchased from General Biological Corporation (Taipei, Taiwan). Determinations of HBsAg and HBeAg in the medium were performed according to the manufacturer’s protocol.
HBV binding assay
HepaRG cells were maintained following the product manual. HepG2-NTCP-AS cells were maintained in DMEM medium supplemented with 2% DMSO for 1 day before HBV inoculation. Human HBV serum (4%) was incubated with HepG2-NTCP-AS and HepaRG cells at 4 °C or 37 °C for 4 and 24 hours. After incubation, cells were washed extensively with ice cold PBS 5 times. Cell-bound HBV DNA was extracted together with cellular genomic DNA by Roche High Pure Viral Nucleic Acid Kit according to the manufacturer’s protocol. HBV binding to hepatocytes was counted by HBV copy number per well, using quantitative Real time PCR assay and HBV core specific primers “HBV-2279-F: TTCGCACTCCTCCAGCTTAT, HBV-2392-R: GAGGCGAGGGAGTTCTTCTT”. HBV copy number was calculated by comparing to the known amount of HBV monomer plasmid DNA. Quantitative real time PCR assay was performed by the 7500 real time PCR system using POWER SyBr green dye (Applied Biosystems).
References
Shih, C. et al. Control and Eradication Strategies of Hepatitis B Virus. Trends in Microbiology 24(9), 739–749, https://doi.org/10.1016/j.tim.2016.05.006 (2016).
Chen, C. J. et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 295(1), 65–73, https://doi.org/10.1001/jama.295.1.65 (2006).
Galle, P. R. et al. In vitro experimental infection of primary human hepatocytes with hepatitis B virus. Gastroenterology 106, 664–673 (1994).
Gripon, P., Diot, C. & Guguen-Guillouzo, C. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology 192, 534–540, https://doi.org/10.1006/viro.1993.1069 (1993).
Gripon, P. et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA 99, 15655–15660, https://doi.org/10.1073/pnas.232137699 (2002).
Yan, H. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and Dvirus. Elife 1, e00049, https://doi.org/10.7554/eLife.00049 (2012).
Watashi, K., Urban, S., Li, W. & Wakita, T. NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci. 15(2), 2892–905, https://doi.org/10.3390/ijms15022892 (2014).
Watashi, K. & Wakita, T. Hepatitis B Virus and Hepatitis D Virus Entry, Species Specificity, and Tissue Tropism. Cold Spring Harb Perspect Med. 5(8), a021378, https://doi.org/10.1101/cshperspect.a021378 (2015).
Li, W. & Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J Hepatol. 64(1 Suppl), S32–40, https://doi.org/10.1016/j.jhep.2016.02.011 (2016).
Karsten, U. et al. Direct comparison of electric field-mediated and PEG-mediated cell fusion for the generation of antibody producing hybridomas. Hybridoma 7, 627–633, https://doi.org/10.1089/hyb.1988.7.627 (1988).
Yang, J. & Shen, M. H. Polyethylene glycol-mediated cell fusion. Methods Mol Biol 325, 59–66, https://doi.org/10.1385/1-59745-005-7:59 (2006).
Schulze, A., Gripon, P. & Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46, 1759–1768, https://doi.org/10.1002/hep.21896 (2007).
Jaques, L. B. Heparin and the mast cell. Can J Biochem Physiol 39, 643–651 (1961).
Engelberg, H. Plasma heparin levels in normal man. Circulation 23, 578–581, https://doi.org/10.1161/01.CIR.23.4.578 (1961).
Cavari, S., Stramaccia, L. & Vannucchi, S. Endogenous heparinase-sensitive anticoagulant activity in human plasma. Thromb Res 67(2), 157–65, https://doi.org/10.1016/0049-3848(92)90135-W (1992).
Volpi, N., Cusmano, M. & Venturelli, T. Qualitative and quantitative studies of heparin and chondroitin sulfates in normal human plasma. Biochim Biophys Acta 1243(1), 49–58, https://doi.org/10.1016/0304-4165(94)00123-F (1995).
Francis, H. & Meininger, C. J. A review of mast cells and liver disease: What have we learned? Dig Liver Dis 42, 529–536, https://doi.org/10.1016/j.dld.2010.02.016 (2010).
Sasisekharan, R. & Venkataraman, G. Heparin and heparan sulfate: biosynthesis, structure and function. Curr Opin Chem Biol 4, 626–631, https://doi.org/10.1016/S1367-5931(00)00145-9 (2000).
Peysselon, F. & Ricard-Blum, S. Heparin-protein interactions: from affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis. Matrix Biol 35, 73–81, https://doi.org/10.1016/j.matbio.2013.11.001 (2014).
Leistner, C. M., Gruen-Bernhard, S. & Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol 10, 122–133, https://doi.org/10.1111/j.1462-5822.2007.01023.x (2008).
Watashi, K. et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59(5), 1726–37, https://doi.org/10.1002/hep.26982 (2014).
Iwamoto, M. et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochemical and biophysical research communications 443, 808–813, https://doi.org/10.1016/j.bbrc.2013.12.052 (2014).
Best, C. H. Preparation of heparin and its use in the first clinical cases. Circulation 19, 79–86, https://doi.org/10.1161/01.CIR.19.1.79 (1959).
Li, J. et al. Unusual features of sodium taurocholate cotransporting polypeptide as a hepatitis B virus receptor. J. Virol 90, 8302–8313, https://doi.org/10.1128/JVI.01153-16 (2016).
Sureau, C. & Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology 57, 985–994, https://doi.org/10.1002/hep.26125 (2013).
Nugent, M. A. Heparin sequencing brings structure to the function of complex oligosaccharides. Proc Natl Acad Sci USA 97, 10301–10303, https://doi.org/10.1073/pnas.97.19.10301 (2000).
Wadstrom, T. & Ljungh, A. Glycosaminoglycan-binding microbial proteins in tissue adhesion and invasion: key events in microbial pathogenicity. J Med Microbiol 48, 223–233, https://doi.org/10.1099/00222615-48-3-223 (1999).
Liu, J. & Thorp, S. C. Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev 22, 1–25, https://doi.org/10.1002/med.1026 (2002).
Vandenberghe, L. H. et al. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat Med 12, 967–971, https://doi.org/10.1038/nm1445 (2006).
Levy, H. C. et al. Heparin binding induces conformational changes in Adeno-associated virus serotype 2. J Struct Biol 165, 146–156, https://doi.org/10.1016/j.jsb.2008.12.002 (2009).
Zahn, A. & Allain, J. P. Hepatitis C virus and hepatitis B virus bind to heparin: purification of largely IgG-free virions from infected plasma by heparin chromatography. J Gen Virol 86, 677–685, https://doi.org/10.1099/vir.0.80614-0 (2005).
Yang, C. C., Huang, E. Y., Li, H. C., Su, P. Y. & Shih, C. Nuclear export of human hepatitis B virus core protein and pregenomic RNA depends on the cellular NXF1-p15 machinery. PLoS One 9, e106683, https://doi.org/10.1371/journal.pone.0106683 (2014).
Chou, S. F., Tsai, M. L., Huang, J. Y., Chang, Y. S. & Shih, C. The Dual Role of an ESCRT-0 Component HGS in HBV Transcription and Naked Capsid Secretion. PLoS Pathog 11, e1005123, https://doi.org/10.1371/journal.ppat.1005123 (2015).
Su, P. Y. et al. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Scientific Reports 6, 38959, https://doi.org/10.1038/srep38959 (2016).
Alexander, W. Heparin revisions: a call for heightened vigilance and monitoring. P T. 34(11), 634–635, 638 (2009).
Acknowledgements
This work was supported by Academia Sinica and Ministry of Science and Technology [MOST 104-0210-01-09-02、MOST 105-0210-01-13-01], Taiwan. We thank Dr. Wen Chang for dextran sulfate and chondroitin sulfate A, Dr. Chien-Jen Chen for the serum samples, An-Ting Liou and Ching-Chun Yang for discussions and careful reading of the manuscript.
Author information
Authors and Affiliations
Contributions
Experimental design: G.C., R.S.J., C.S. Conducted the experiments: G.C., R.S.J. Reagents: C.J.C., H.I.Y., M.L.Y., W.L.C., Y.Y.C. Data analysis: all authors. Paper writing: G.C., S.F.C. and C.S.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choijilsuren, G., Jhou, RS., Chou, SF. et al. Heparin at physiological concentration can enhance PEG-free in vitro infection with human hepatitis B virus. Sci Rep 7, 14461 (2017). https://doi.org/10.1038/s41598-017-14573-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14573-9
This article is cited by
-
Significance of hepatitis B virus capsid dephosphorylation via polymerase
Journal of Biomedical Science (2024)
-
Advances in HBV infection and replication systems in vitro
Virology Journal (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
