
Abstract
The gypsy moth, Lymantria dispar L., is one of the most destructive forest pests in the world. While the subspecies established in North America is the European gypsy moth (L. dispar dispar), whose females are flightless, the two Asian subspecies, L. dispar asiatica and L. dispar japonica, have flight-capable females, enhancing their invasiveness and warranting precautionary measures to prevent their permanent establishment in North America. Various molecular tools have been developed to help distinguish European from Asian subspecies, several of which are based on the mitochondrial barcode region. In an effort to identify additional informative markers, we undertook the sequencing and analysis of the mitogenomes of 10 geographic variants of L. dispar, including two or more variants of each subspecies, plus the closely related L. umbrosa as outgroup. Several regions of the gypsy moth mitogenomes displayed nucleotide substitutions with potential usefulness for the identification of subspecies and/or geographic origins. Interestingly, the mitogenome of one geographic variant displayed significant divergence relative to the remaining variants, raising questions about its taxonomic status. Phylogenetic analyses placed this population from northern Iran as basal to the L. dispar clades. The present findings will help improve diagnostic tests aimed at limiting risks of AGM invasions.
Similar content being viewed by others


Introduction
The gypsy moth, Lymantria dispar (Linnaeus, 1758) (Insecta: Lepidoptera: Erebidae: Lymantriinae), is considered one of the most destructive forest defoliators over much of its range. In North America alone, losses and population suppression operations targeting this species are estimated at $3.2 billion each year1. Currently, L. dispar comprises three recognized sub-species2 based on morphological criteria, female flight capability and geographic origins: L. dispar dispar Linnaeus, found over most of western Europe (“EGM” for European Gypsy Moth) and North Africa, and accidentally introduced from France into North America in 18693, L. dispar asiatica Vnukovskij, distributed over much of continental Asia, including Russia, China and the Korean peninsula, and L. dispar japonica Motschulsky, which appears to be confined to Japan (Honshu, Shikoku, Kyushu and Hokkaido). For regulatory purposes, the latter two subspecies, along with three other closely related Japanese Lymantria species (i.e., L. umbrosa Butler, L. postalba Inoue and L. albescens Hori and Umeno), are generally considered “Asian gypsy moth” (AGM)2.
Unlike their European counterpart, the two Asian L. dispar subspecies have flight-capable females4,5 and a broader host range6, considerably increasing the risks of rapid propagation relative to EGM. With the current intensification of commercial trade with Asia, AGM introduction and establishment into North America are considered a very significant threat. AGM specimens are regularly intercepted at North American ports, and accidental introductions have occurred, resulting in costly eradication campaigns7.
The rigorous and rapid identification of intercepted gypsy moth samples suspected of being AGM and an assessment of their geographic origins are critical to the negotiations undertaken by North American regulatory agencies with their Asian trading partners in an effort to reduce risks of accidental introductions. However, morphological characters provide little useful information for species and subspecies delineation and for the identification of source countries, particularly when dealing with the immatures stages (e.g., eggs and larvae). As a consequence, many studies have examined the usefulness of molecular markers to help distinguish gypsy moth subspecies and to identify the geographic origins of intercepted specimens (see8). In this respect, a 658 bp fragment of the mitochondrial cytochrome c oxidase subunit I (COI) gene has received much attention, given its recognized effectiveness in delineating taxa9. However, although the COI 5’ barcode region could easily separate the European from the two Asian subspecies, it proved inadequate to distinguish L. d. asiatica from L. d. japonica 8,10,11. On the other hand, distinct regions of the mitochondrial genome, including the 3′ portion of the COI gene8 and five other genes12 (ND2, ND6, ATP6, ATP8 and CytB) provided resolution of the two Asian strains, pointing to the informativeness of alternative mitochondrial regions.
With respect to identifying the geographic origins of gypsy moth specimens, previous work has shown that the COI barcode alone could provide some clues as to the source of unknown samples10,11, and that other mitochondrial genes could also be useful for this purpose12. While analysis of nuclear markers such as microsatellites12 and genome-wide SNPs13 points to their remarkable value in identifying the geographic origins of gypsy moth specimens, mitochondrial genomes should not be underestimated as a potential source of markers to help identify geographic variants, especially in view of their high substitution rate relative to nuclear genomes14,15.
For the present study, we undertook the sequencing of the entire mitochondrial genome of 10 geographic variants of Lymantria dispar, with a good coverage of this species’ geographic range and inclusion of two or more variants of each subspecies; the closely related L. umbrosa was selected as outgroup. Only three L. dispar mitochondrial genomes had previously been sequenced and deposited in GenBank, each with limited information about the origin and subspecies designation of the insect from which they were obtained (accession numbers: FJ617240, GU994783, GU994784). Here, we present a detailed comparative analysis of gypsy moth mitochondrial genomes with the aim of assessing their usefulness for subspecies delimitation and identification of geographic origins. Interestingly, the mitochondrial genome of one of the geographic variants sampled in this study displayed important differences relative to the remaining variants. We thus conducted phylogenetic analyses to help shed light on the potential taxonomic position of this previously uncharacterized geographic variant.
Results
Lymantria dispar mitochondrial genome organization
A total of 11 mitochondrial genomes of Lymantria specimens (Table 1; Fig. 1) were completely sequenced, assembled and annotated (Table 2). While one of these specimens was L. umbrosa, the other 10 specimens were originally considered to be geographic variants of L. dispar. However, one sample from northern Iran (referred to here as L?_IR) displayed important differences relative to the other L. dispar mitogenomes, calling into question its taxonomic status (see details in the next section). To minimize the risk of misidentification, all specimens were assayed using a TaqMan qPCR method8 as well as cross-checked with their DNA barcodes in BOLD. At least two specimens identified here as L. dispar dispar, based on their mitochondrial genome sequences (Ldd_RB and Ldd_KZ; Table 1), are believed to be strongly admixted dispar-asiatica populations on the basis of their nuclear genotypes8,12,13.

Sampling locations for Lymantria specimens used in this study. Red symbols represent sampling locations for L. dispar subspecies (1–9), blue symbol is for L. umbrosa (10) and green symbol is for the specimen from Iran (11). For full location names, see Table 1. Background map is a cropped version of the one available at: https://commons.wikimedia.org/wiki/Maps_of_the_world#/media/File:BlankMap-World-v2.png (for information about license, follow the same link).
As expected, all 11 sequenced Lymantria mitochondrial genomes were of similar size (~15,600 bp; Table 2), falling within the size range of other lepidopteran mitogenomes sequenced earlier15. Most of the observed size variation was attributable to differences in the non-coding A + T-rich region, which is subject to a higher rate of mutation and/or rearrangement than other regions. Not surprisingly, all 11 mitogenomes yielded the same annotation, an example of which is provided in Fig. 2 for L. dispar dispar. As described for other insect mitogenomes15,16, the circular double-stranded DNAs contained a conserved set of 37 genes (Table 3), including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), a large (16 S) and a small (12 S) ribosomal RNA subunit gene (rrnL and rrnS rRNA), and a large non-coding A + T-rich region, located between tRNA-Met and rrns rRNA (Fig. 2). The A + T content (Table 2) and the arrangement and orientation of genes (Fig. 2) were similar to those reported for other lepidopteran mitochondrial genomes16,17. All PCGs were observed to use an ATN start codon, except for the COI gene, which used CGA as start codon, as reported for other Lepidoptera17. The genomes presented here also showed incomplete stop codons (T- or TA-) for the ND4, COI and COII genes, as observed earlier for other lepidopteran mitogenomes16,18. There were no structural differences among the nine L. dispar mitogenomes analyzed here, but there was a codon deletion in the Lu_JH ATP8 gene.

Circular map of mitochondrial genome of L. dispar dispar (Ldd_NJ). The tRNA genes are indicated using the single letter IUPAC-IUB abbreviation, corresponding to their amino acid. COI, II and III: cytochrome oxidase subunits; Cyt b: Cytochrome b; ND: NADH dehydrogenase; rrnL and rrnS rRNA correspond to ribosomal RNAs. Gypsy moth caterpillar photo credit: Catherine Béliveau©.
Comparative mitogenome analysis
Nucleotide sequences
To assess variability and divergence among the 11 gypsy moth mitogenomes examined here, we conducted three different analyses. First, we aligned full genomic sequences to identify positions of single nucleotide polymorphisms (SNPs), using the genome of Ldd_NJ as reference (Fig. 3), while more quantitative assessments were obtained through computation of pair-wise percent identities (Table S2) and haplotype network analysis (Fig. S1). A simple visual examination of the graphical representation shown in Fig. 3 (see also Fig. S1) immediately brings to light the high degree of similarity among the four Asian L. dispar sequences, which comprise the asiatica and japonica subspecies, relative to the five L. dispar dispar geographic variants. In total 53 SNPs were common to all four Asian L. dispar mitogenomes, enabling easy discrimination between these two subspecies and L. dispar dispar. Despite the high sequence identity observed between L. dispar asiatica and L. dispar japonica (Table S2 and Fig. S1), SNPs that could tell them apart were identified in ND1, ND2, ND4, ATP6 and COI (Fig. 3; note that the latter was reported earlier8). The two japonica mitogenomes displayed fewer substitutions (4; Fig. S1) than their two asiatica counterparts (22; Fig. S1), perhaps reflecting differences in geographical distance between the two populations sampled for each subspecies.

Graphical representation of a nucleotide alignment of the 11 mitochondrial genomes sequenced in the present study. Black vertical bars represent substitutions in the nucleotide sequence relative to the Ldd_NJ genome (top). Annotations: green boxes, PCGs; red boxes, rRNA subunit genes; grey box, A + T-rich region; pink arrows, tRNAs.
The overall level of nucleotide identity among the five L. dispar dispar geographic variants was slightly lower than that observed among the four Asian samples (Table S2; Fig. S1), a conclusion that is congruent with the more scattered distribution of SNPs seen among the L. dispar dispar variants (Fig. 3). Some regions of the L. dispar dispar mitochondrial genomes sampled here, notably COI and ND5, displayed SNPs that may be diagnostic of the populations these insects were drawn from (Fig. 3).
The mitogenome of L. umbrosa, which was considered a subspecies of L. dispar until the taxonomic revisions of Pogue and Schaeffer2, displayed a large number of substitutions (243; Fig. S1) relative to the mitogenomes of L. dispar (Fig. 3; Table S2), providing additional support for the elevation of its status as a distinct species. In comparison, the gypsy moth sample from northern Iran displayed a number of substitutions (123; Fig. S1) intermediate between L. umbrosa and those identified as L. dispar (Fig. 3; Table S2). Although only one specimen was used to generate the full mitochondrial genome for that population, we sequenced a region spanning the COI and ND2 genes from four additional samples, including another one from northern Iran and three from the Russian Caucasus (Fig. S2). The almost perfect sequence identity observed among these five samples suggests that they belong to the same, genomically distinct population, found in the vicinity of the Caucasus.
Amino acid sequences
Following conceptual translation of the above DNA sequences, some of the SNPs we identified proved to be non-synonymous, resulting in amino acid substitutions. As those could have an impact on enzymatic activity and energy metabolism, possibly affecting flight capacity, we identified all amino acid substitutions among the 11 mitogenomes that we sequenced and determined whether they were conservative or not (Fig. 4). Not surprisingly, more amino acid substitutions were detected in the Lu_JH (35 substitutions) and L?_IR (19 substitutions) mitogenomes than in the remaining L. dispar genomes (average of 8 substitutions), using the most common cross-taxa residue as a point of reference (i.e., those shaded in blue in Fig. 4). Four substitutions were unique to the four Asian L. dispar mitogenomes (in ND4, ND5 and ND6), while two additional ones were shared by the Asian L. dispar mitogenomes and that of L?_IR (ND1) or those of both L?_IR and Lu_JH (ND3); all but one (K→S in ND5) of these substitutions were deemed conservative (Fig. 4). Three substitutions were unique to the L. dispar japonica mitogenome (in ND1, ND4 and ATP6), including one considered semi-conservative (G→S in ATP6). Among the L. dispar dispar mitogenomes, there were several scattered substitutions including some non-conservative ones, but no clear pattern could be detected. Importantly, no amino acid substitutions were common to the four Asian L. dispar populations and those with an L. dispar dispar mitochondrial signature but whose females display flight capability (Ldd_LJ and Ldd_RB5), considerably decreasing the likelihood of a hypothetical substitution that would confer an energetic advantage to populations with flight-capable females.

Comparative analysis of amino acid substitutions among the 13 PCGs of the 11 mitochondrial genomes sequenced in the present study. Unsubstituted amino acids at a given position are shaded in blue; substitutions are shaded in different colors according to their conservativeness (see legend). To identify substitutions and assess their level of conservativeness, amino acid sequences were aligned in MAFFT, using the ClustalW output format.
Phylogenetic analyses
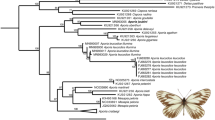
All analyses conducted here yielded the same topology for a given tree building approach (Maximum Likelihood [ML] and Bayesian inference [BI]), irrespective of the dataset (PCGs alone, PCGs + tRNA + rRNAs or PCGs + tRNA + rRNAs + A/T-rich region), with and without partitioning schemes, although there were differences in bootstrap (in ML) and posterior probability (in BI) support values among terminal taxa. None of the PCGs were found to be under positive selection and all but two (ND3 and ND4L) were considered to be under purifying selection (Table S3), as is expected for mitochondrial PCGs19. Figure 5 shows the BI and ML trees obtained using the full data set, with (BI) and without (ML) partitioning scheme.

Phylogenetic relationships among the nine L. dispar populations sampled here, plus a population from Iran (L?_IR) and L. umbrosa, based on all 37 mitochondrial genes, plus the A + T-rich region. (a) Bayesian inference (BI), MCMC consensus tree (10,000,000) with posterior probabilities shown on each branch. (b) Maximum Likelihood (non-partitioned data) with 1000 bootstrap replicates. L. umbrosa was used as outgroup.
In both BI and ML analyses, the representative from northern Iran formed a sister group to all L. dispar lineages, assuming a position intermediate between L. dispar and L. umbrosa. Indeed, the monophyly of L. dispar was not supported when the specimen from Iran was included in the analysis. Relationships among the three recognized subspecies were clear, although the two Asian subspecies formed a monophyletic group, separated by very short branches, but with strong branch support in both analyses (Fig. 5). The two analyses placed the European specimens within a strongly supported clade (bootstrap = 100; posterior probability = 1), but with unstable and poorly resolved internal relationships, where the Greek and Kazakhstan specimens formed a distinct, moderately supported clade (bootstrap = 71) in the ML tree, and the Greek specimen was basal to the rest of the group in the BI tree (Fig. 5).
Discussion
This study represents the first detailed analysis of multiple gypsy moth mitogenomes, featuring a taxon sample that comprises all three L. dispar subspecies and geographical variants thereof, plus the closely related L. umbrosa and a previously uncharacterized gypsy moth population displaying a mitochondrial haplotype similar to, but evidently distinct from, that of L. dispar. Previous molecular comparisons of gypsy moth variants relied primarily on the barcode region of the COI gene, which is here shown to be rich in nucleotide substitutions, some of which are useful for subspecies delineation. However, we found other mitochondrial genes to contain many additional informative SNPs with potential for segregating L. dispar dispar from the two Asian subspecies and for distinguishing L. dispar asiatica from L. dispar japonica (Fig. 3), which cannot be told apart using the barcode region alone10. Interestingly, ND2 and ATP6, two genes that we found to contain informative SNPs for this purpose, were among the five mitochondrial genes sampled by Wu et al.12 to generate a phylogeny of L. dispar variants in which the two Asian subspecies formed distinct clades.
Although the present analysis did not include multiple individuals from each of the populations sampled, thereby making it difficult to assess haplotype homogeneity within any given population, the number of SNPs observed among the five L. dispar dispar mitogenomes (Fig. 3) was large enough to justify the prediction that these genomes are likely to contain markers that will provide useful clues for identifying the geographic origins of unknown samples. The fact that more variability was observed among the five L. dispar dispar mitogenomes than among those of the four Asian samples, which comprise two subspecies, is not entirely surprising given the much greater geographic distance separating the L. dispar dispar populations than that separating the other four (Fig. 1). Within the L. dispar asiatica subspecies, COI-based evidence has recently been presented in support of population-specific mitogenome differences among samples obtained from different locations in China11, strongly suggesting that a broad geographic sampling of L. dispar asiatica would reveal inter-population differences similar to those observed here for L. dispar dispar mitogenomes. Thus, future population genomics studies aimed at developing markers for the purpose of identifying geographic origins should consider both nuclear and mitochondrial genomes.
Although mitochondrial markers have achieved great success for taxon delineation, their use for gypsy moth subspecies identification has proven somewhat tenuous when genotyping populations found primarily in central Asia, which display an L. dispar dispar COI haplotype8, but for which nuclear markers8,12,13 and phenotypes, including female flight capability5, suggest they have more in common with the asiatica than with the dispar subspecies. In fact, insects from some of these populations appear to have a dispar/asiatica mosaic nuclear genome13 (see also populations from Kazakhstan and Kyrgyzstan in12), suggestive of hybridization between the two subspecies, presumably after a period of reproductive isolation. Although we have not yet produced nuclear gene-based phylogenies equivalent to the ones presented here using mitogenomes (Fig. 5), the likelihood of discordance between such phylogenies for central Asian populations is high. This type of discordance is common20 and, in the case at hand, raises the question as to why central Asian hybrids would have a dispar instead of an asiatica mitogenome haplotype (note: it is not clear whether all populations in central Asia have a dispar-like mitogenome, but all those examined by our group do; see8).
Given that mitochondrial genomes are typically of maternal inheritance, the biogeographic discordance between nuclear and mitochondrial genomes observed in moths from central Asia could have resulted from (i) male-biased dispersal, resulting in a higher likelihood of crosses between L. dispar asiatica males and L. dispar dispar females than for the reciprocal cross21, and/or (ii) adaptive introgression of mitogenomes that strongly favors one mitogenome over the other20. If, as shown for other lepidopteran species21, gypsy moth males are the more mobile sex, irrespective of whether females are capable of flight or not, the situation observed here for central Asian populations suggests that L. dispar asiatica males could have colonized the range of L. dispar dispar, where asymmetric introgression took place from the local dispar population into the colonizing asiatica population, leading to the observed preponderance of the dispar mitogenome haplotype in such hybrids (see21). In addition, since mitochondrial OXPHOS proteins are subunits of larger proteins featuring both mitochondrial and nuclear components (N-mt genes), amino acid substitutions within these subunits can negatively affect protein assembly and lead to mitonuclear incompatibilities22. Alternatively, some mitochondria can accumulate a significant number of mildly deleterious mutations that are rescuable by replacement with a more fit haplotype20. Whether the latter scenario played a role in the mitochondrial haplotype pattern observed here is unknown. Although we found a significant number of amino acid substitutions among L. dispar mitogenomes, the available data do not permit predictions regarding levels of mitonuclear compatibility since the sequences of the N-mt genes are not currently available. Cointrogression of N-mt genes from L. dispar dispar into the central Asian genomes (featuring an otherwise asiatica genetic background) could also have favored the maintenance of the dispar mitogenomes in these populations20, a hypothesis that will be worth examining once the gypsy moth nuclear genome has been sequenced and annotated.
Interestingly, crosses between females of L. umbrosa (previously known as L. dispar praeteria) and males of L. dispar japonica yielded all-male broods, a result the authors attributed to some genetic incompatibility between the two taxa23. In view of our observation that the L. umbrosa mitogenome features 32 amino acid substitutions relative to that of L. dispar japonica, including 9 substitutions that are considered non-conservative (Fig. 4), the question arises as to whether the reported unviability of hybrid females23 could be explained, at least in part, by an incompatibility between a recessive paternal Z chromosome and maternal cytoplasm (e.g., mtDNA)24. Indeed, this type of female hybrid unviability fits Haldane’s rule, which predicts unviability of hybrids of the heterogametic sex (female in the case of Lepidoptera)24.
Irrespective of the method used, the phylogenetic analyses presented here (Fig. 5) generated trees for which the overall L. dispar branching pattern was similar to that obtained by Wu et al.12 using five mitochondrial genes (ND2, ND6, ATP6, ATP8 and Cytb), where the two Asian subspecies formed distinct, but more derived clades. In the latter study, the tree showed L. dispar dispar samples from the United States forming varying associations with those from France, Germany and Italy. However, that analysis did not include L. dispar dispar mitochondrial haplotypes from central Asia. Here, the North American haplotype clustered with those from Lithuania and Siberia, as opposed to that from Greece (i.e., closer to the presumed origin [France] of North American moths), which appeared basal to the L. dispar dispar group in the BI analysis and forming a basal cluster with the sample from Kazakhstan in the ML analysis (Fig. 5). A wider and finer sampling scheme across the entire range of L. dispar dispar mitochondrial haplotypes would provide a better assessment of connectivity among haplotypes.
As expected from the number of SNPs found in its mitogenome (i.e., compared with L. dispar and the population from northern Iran; Fig. 3), L. umbrosa occupied a basal position in the phylogenetic trees presented here, while the previously uncharacterized sample from Iran occupied an intermediate position between L. umbrosa and the L. dispar clade (Fig. 5). The latter observation raises important questions regarding the taxonomic status of this “Caucasian” gypsy moth population and its role in the evolutionary history of the L. dispar lineage. We currently have no information on biological attributes that may help distinguish this population from other gypsy moth populations, including morphometrics, female flight capability and host preference. A scan of the gypsy moth literature published in the USSR prior to 1991 (compiled in25) identified many publications devoted to studies of gypsy moth populations in the Caucasus (Russian Caucasus, Georgia, Armenia and Azerbaijan); however, because L. dispar and the “Caucasian” population may well be sympatric (and treated without distinction by earlier workers), no conclusion can be readily drawn from this literature. Thus, before establishing the taxonomic status of this population, i.e. whether it should be treated as a new subspecies of L. dispar or a distinct species altogether, additional information will need to be gathered on its unique biological traits, if any. In addition, a nuclear marker-based genomic comparison with gypsy moth samples identified as L. dispar will need to be conducted.
As to why the “Caucasian” population has such a distinct mitochondrial haplotype relative to L. dispar, important physical barriers such as the Greater and Lesser Caucasus Mountains, which form a deep valley in Azerbaijan, could have kept it in complete reproductive isolation for a long period, after which it may have expanded its range through paths along the Caspian and Black Seas. A similar type of reproductive isolation has been proposed as the mechanism that drove differentiation between the “Hokkaido gypsy moth” (L. umbrosa) and L. dispar asiatica. Although they are now reunited (creating zones of sympatry between the two species), the eastern portion of the Japanese island of Hokkaido was once separated by water from its western segment, thereby creating a reproductive barrier that appears to have led to the speciation of L. umbrosa 2.
Finally, although we cannot as yet establish whether the “Caucasian” population examined here is of significant biosecurity concern to North America, shipping vessels traveling from ports of the Black Sea could inadvertently carry these insects and, in doing so, favor their accidental introduction into Canada and the United States, therefore justifying the use of molecular tools to properly identify them. At the time we developed our TaqMan qPCR assay targeting Asian gypsy moths and related species posing a threat to North America8, we had no knowledge of the “Caucasian” population; as a consequence, it was not considered in the design of our assay. Here, when the five “Caucasian” samples were submitted to the TaqMan assay, they were all incorrectly identified as L. dispar dispar. Thus, we designed a new version of the “Simplex 3” sub-assay (corresponding to the “Is it Ldd?” node of Fig. 1 in8) to enable discrimination between L. dispar dispar and the “Caucasian” strain. To this end, we used SNPs within ND1 identified in the present work (manuscript in preparation).
Materials and Methods
Sampling strategy
Based on currently established criteria for the taxonomic placement of L. dispar and its subspecies2,26, we sampled one adult male from each of 10 populations distributed over the gypsy moth’s range, including five geographic variants of L. dispar dispar, two exemplars each of L. dispar asiatica and L. dispar japonica, and one specimen from northern Iran deemed to be of uncertain taxonomic affiliation (originally identified as L. dispar), together with the sister species L. umbrosa, used here as outgroup taxon (Table 1; Fig. 1). Sources of samples are listed in Table 1. To confirm identifications made based on morphological features, we used two molecular taxonomic tools: a multi-species TaqMan assay8 and DNA barcoding9. For the latter method, specimens were cross-checked with their DNA barcodes (COI 5′), as found in the BOLD (Barcode of Life Data System) database27, which contains a reference library for global gypsy moth populations, including the geographic variants used in this study. Two L. dispar specimens from central Asia (i.e., specimens 4 [Kazakhstan] and 5 [Russia; Siberia] as shown in Fig. 1) with uncertain subspecies affiliation are here treated as L. dispar dispar based on their mitochondrial haplotypes (Table 1).
DNA extraction
Total genomic DNA (nuclear and mitochondrial) was isolated from specimens that were either dry, preserved in 96% ethanol or frozen, using either the Qiagen Blood & Cell Culture DNA midi kit or the Qiagen DNeasy Blood & Tissue mini kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. For frozen material, the specimen’s wings and abdomen were first removed and the remaining body parts were ground in liquid nitrogen prior to DNA extraction as described8. For archival specimens, one or more moth legs were collected, ground in liquid nitrogen and submitted to DNA extraction with the following minor modifications: 56 °C incubation was conducted overnight and final DNA elution was done in 100–150 μL of sterile water. Eluted DNA was then quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific Inc.) and used either directly or diluted for mitochondrial genome PCR amplification.
Primer design, PCR amplification and sequencing
An initial assembly of the Ldd_NJ mitogenome was gleaned from a Newbler assembly generated during the sequencing of the L. dispar dispar nuclear genome, using Roche 454 sequencing technology. Similarly, initial assemblies of the Lda_TJ and Ldj_ID mitogenomes were obtained from nuclear genome assemblies generated using the Celera assembler on PacBio sequencing reads. Following error correction (see below), these three mitogenomes were used as a reference for the assembly of the mitochondrial genomes of the other specimens analyzed in this study.
To perform error correction on the three above mitogenomes and to obtain the sequences of the eight remaining genomes (Table 1), two specific primer pairs (P1533/P1365 and P1364/P1532, see Table S1) were designed (based on a multiple alignment of conserved regions of COI, Cytb and ND6) to amplify two long fragments (fragment 1: COI→Cytb and fragment 2: ND6→COI, 8,949 and 7,048 bp in size, respectively) overlapping in the ND6 region. Sequencing of different portions of these long fragments was achieved by either submitting these fragments directly to Sanger sequencing or by conducting prior amplification of shorter, overlapping portions using specific primer pairs (Table S1). All PCR amplifications were performed directly on total DNA extracts (0.5–5 ng) in a 25 µL final volume, using 0.5 U of Platinum SuperFi DNA polymerase (Invitrogen), 1x SuperFi buffer (Invitrogen), 0.33 mM of a dATP/dTTP mix, 0.10 mM of a dCTP/dGTP mix, 10 pmol of each primer and 9% DMSO. PCR conditions were: initial denaturation step at 95 °C for 2 min, followed by 40 cycles of denaturing (95 °C, 5 sec), coupled annealing and extension (60 °C, 10 min), and a final extension step at 60 °C for 10 min. Smaller fragments were also amplified to help resolve regions that were difficult to sequence (Table S1). One µL of PCR product was then analyzed by gel electrophoresis, and the remaining reaction volume used directly for Sanger sequencing. Primers used for the latter purpose were either those designed for PCR amplification or new ones especially designed for sequencing (see Table S1 for the complete list of primers used). Sequencing was performed by the Genome Sequencing and Genotyping platform of the CHUL, in Quebec City (Quebec, Canada).
Sequence assembly and genome annotation
Assembly and annotation were carried out using Geneious® software, version 9.1.2 (http://www.geneious.com)28, in combination with the NCBI Blast function to help predict PCGs. Transfer RNA genes were identified using tRNAscan-SE 1.2121,22,29,30, with the search mode and the cove cutoff score set as default, Mitochondria/Chloroplast as the search source and invertebrate mitochondrial genome as the genetic code for tRNA isotype prediction.
Assessment of the extent to which mitochondrial PCGs are under positive selection
To determine whether the mitochondrial PCGs considered in the present study were under positive selection, we used the codon based z-test, which compares relative abundances of synonymous and nonsynonymous mutations, as implemented in Mega 7.031.
Phylogenetic analyses
Matrices of mitogenomic data were analyzed using two model-based phylogenetic approaches, namely Maximum Likelihood (ML) and Bayesian Inference (BI). To explore phylogenetic signal in the data, the effects of varying gene combinations were first compared against analyses that included the full dataset, either concatenated or partitioned according to genes. On the basis of these preliminary explorations, we decided to include all genes and third codon positions in both ML and BI analyses; similarly, we decided to use non-partitioned data for ML analysis and partitioned data (1 partition for each gene; 38 partitions) for BI analysis. We found that inclusion of all genes/regions, including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), large (16S) and small (12S) ribosomal RNA subunit genes (rrnL and rrnS rRNA), and the large non-coding A + T-rich region, together with a partitioning scheme for BI, improved bootstrap (in ML) and posterior probability (in BI) support values among terminal taxa. BI and ML analyses were performed using the software MrBayes v3.132 and the recently developed IQ-TREE33, respectively. IQ-TREE searches were carried out using the default settings on the dedicated web-server IQ-TREE, available at http://iqtree.cibiv.univie.ac.at/ 34; the most appropriate models of sequence evolution were chosen using the Auto function on the IQ-TREE web server, following the authors’ recommendations. ML bootstrap analysis35 and clade robustness were then assessed using ultrafast bootstrap replicates with IQ-TREE. The Bayesian analyses were run independently twice for 10 million generations, with every 1000th tree sampled. The data matrix was split into 38 partitions: AT_rich, ATP6, ATP8, COI, COII, COIII, cyt-b, ND1, ND2, ND3, ND4, ND4L, ND5, ND6, rrnL, rrnS, trnA, trnC, trnD, trnE, trnF, trnG, trnH, trnI, trnK, trnL-1, trnL-2, trnM, trnN, trnP, trnQ, trnR, trnS-1, trnS-2, trnT, trnV, trnW, and trnY. We modelled the evolution of sequences according to the GTR + Γ model independently for the 38 partitions using the “unlink” command in MrBayes. Clade support was estimated by posterior probabilities in MrBayes. Convergence was determined when the average standard deviation of split frequencies (ASDSF = 0.013558) went below 0.05, the PSRF (Potential Scale Reduction Factor) approached 1, and both runs had properly converged to a stationary distribution after the burn-in stage (which was 1,000 sampled generations; burn-in = 7000).
Haplotype network analysis
Median joining networks were computed to examine intra- and interspecific divergence among the 11 mitogenomes examined here using default settings, as implemented in the software popart (Population Analysis with Reticulate Trees)36.
Data availability
All sequences reported in this paper have been deposited in GenBank. Accession numbers are provided in Table 2.
References
Bradshaw, C. J. A. et al. Massive yet grossly underestimated global costs of invasive insects. Nat. Commun. 7, 12986, https://doi.org/10.1038/ncomms12986 (2016).
Pogue, M. & Schaefer, P. W. A review of selected species of Lymantria Hübner (1819) (Lepidoptera: Noctuidae: Lymantriinae) from subtropical and temperate regions ofAsia, including the descriptions of three new species, some potentially invasive to North America (U.S. Dept. of Agriculture, Forest Health Technology Enterprise Team, Washington, D.C. 2007).
Forbush, E. H. & Fernald, C. H. The gypsy moth, Porthetria dispar (Linn.): a report of the work of destroying the insect in the Commonwealth of Massachusetts, together with an account of its history and habits both in Massachusetts and Europe. (Wright & Potter Printing Co., Boston, 1896).
Keena, M. A., Grinberg, P. S. & Wallner, W. E. Inheritance of female flight in Lymantria dispar (Lepidoptera: Lymantriidae). Environ. Entomol. 36, 484–494, https://doi.org/10.1603/0046-225X(2007)36[484:IOFFIL]2.0.CO;2 (2007).
Keena, M. A., Côté, M. J., Grinberg, P. S. & Wallner, W. E. World Distribution of Female Flight and Genetic Variation in Lymantria dispar (Lepidoptera: Lymantriidae). Environ. Entomol. 37, 636–649, https://doi.org/10.1603/0046-225X(2008)37[636:WDOFFA]2.0.CO;2 (2008).
Baranchikov, Y. N. Ecological basis of the evolution of host relationships in Eurasian gypsy moth populations. In: Wallner WE and McManus KA, tech. coords. Proceedings, Lymantriineae: A comparison of features of New and Old World tussock moth. (USDA Forest Service, Northeastern Forest Experiment Station, Broomall, PA. pp. 319–338 (1989).
USDA Pest alert – Asian gypsy moth. APHIS 81-35-027 leaflet. USDA Animal and Plant Health Inspection Service, Riverside Park, MD; 2016. Available online at: https://www.aphis.usda.gov/publications/plant_health/content/printable_version/fs_phasiangm.pdf.
Stewart, D. et al. A multi-species TaqMan PCR assay for the identification of Asian gypsy moths (Lymantria spp.) and other invasive lymantriines of biosecurity concern to North America. PLoS One 11, e0160878, https://doi.org/10.1371/journal.pone.0160878 (2016).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. P. Roy. Soc. Lond. B Biol. 270, 313–321, https://doi.org/10.1098/rspb.2002.2218 (2003).
deWaard, J. R. et al. Towards a global barcode library for Lymantria (Lepidoptera: Lymantriinae) tussock moths of biosecurity concern. PLoS One 5, e14280, https://doi.org/10.1371/journal.pone.0014280 (2010).
Chen, F. et al. DNA Barcoding of gypsy moths from China (Lepidoptera: Erebidae) reveals new haplotypes and divergence patterns within gypsy moth subspecies. J. Econ. Entomol. 109, 366–374, https://doi.org/10.1093/jee/tov258 (2016).
Wu, Y. et al. Genetic structure, admixture and invasion success in a Holarctic defoliator, the gypsy moth (Lymantria dispar, Lepidoptera: Erebidae). Mol. Ecol. 24, 1275–1291, https://doi.org/10.1111/mec.13103 (2015).
Picq, A. et al. Assessing the potential of genotyping-by-sequencing-derived single nucleotide polymorphisms to identify the geographic origins of intercepted gypsy moth (Lymantria dispar) specimens: a proof-of-concept study. Evol. Appl. in press, doi:10.1111/eva.12559.
Brown, W. M., George, M. & Wilson, A. C. Rapid evolution of animal mitochondrialDNA. P. Natl. Acad. Sci. USA 76, 1967–1971, https://doi.org/10.1073/pnas.76.4.1967 (1979).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117, https://doi.org/10.1146/annurev-ento-011613-162007 (2014).
Sun, Y.-X. et al. Characterization of the complete mitochondrial genome of Leucoma salicis (Lepidoptera: Lymantriidae) and comparison with other lepidopteran insects. Sci. Rep. 6, 39153, https://doi.org/10.1038/srep39153 (2016).
Cameron, S. L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 29, 400–411, https://doi.org/10.1111/syen.12071 (2014).
Dai, L.-S., Zhu, B.-J., Zhao, Y., Zhang, C.-F. & Liu, C.-L. Comparative mitochondrial genome analysis of Eligma narcissus and other lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. Sci. Rep. 6, 26387 (2016).
Castellana, S., Vicario, S. & Saccone, C. Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein-coding genes. Genome Biol. Evol. 3, 1067–1079, https://doi.org/10.1093/gbe/evr040 (2011).
Sloan, D. B., Havird, J. C. & Sharbrough, J. The on‐again, off‐again relationship between mitochondrial genomes and species boundaries. Mol. Ecol. 26, 2212–2236, https://doi.org/10.1111/mec.13959 (2017).
Petit, R. J. & Excoffier, L. Gene flow and species delimitation. Trends in Ecology & Evolution 24, 386–393, https://doi.org/10.1016/j.tree.2009.02.011 (2009).
Hill, G. E. Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecol. Evol. 6, 5831–5842, https://doi.org/10.1002/ece3.2338 (2016).
Higashiura, Y. et al. Male death resulting from hybridization between subspecies of the gypsy moth. Lymantria dispar. Heredity 106, 603–613, https://doi.org/10.1038/hdy.2010.92 (2011).
Presgraves, D. C. Patterns of postzygotic isolation in Lepidoptera. Evolution 56, 1168–1183, https://doi.org/10.1554/0014-3820(2002)056[1168:POPIIL]2.0.CO;2 (2002).
Baranchikov, Y. N., Nikitenko, G. N. & Montgomery, M. E. Russian and Ukrainian literature on the gypsy moth: an annotated bibliography. Gen. Tech. Rep. NE-253. Radnor, PA: U.S. Department of Agriculture, Forest Service, Northeastern Research Station. 166 p. https://www.fs.usda.gov/treesearch/pubs/3805 (1998).
Schintlmeister, A. The taxonomy of the genus Lymantria HÜBNER, [1819] (Lepidoptera: Lymantriidae). Quadrifina 7, 1–248 (2004).
Ratnasingham, S. & Hebert, P. D. N. BARCODING: bold: The Barcode of Life Data System (http://www.barcodinglife.org): BARCODING. Mol. Ecol. Notes 7, 355–364, https://doi.org/10.1111/j.1471-8286.2007.01678.x (2007).
Kearse, M. et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, https://doi.org/10.1093/bioinformatics/bts199 (2012).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964, https://doi.org/10.1093/nar/25.5.0955 (1997).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689, https://doi.org/10.1093/nar/gki366 (2005).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Ronquist, F. J. & Huelsenbeck, J. P. in Statistical methods in molecular evolution (ed R. Nielsen) (Springer, 2005).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235, https://doi.org/10.1093/nar/gkw256 (2017).
Felsenstein, J. Confidence-limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791, https://doi.org/10.1111/j.1558-5646.1985.tb00420.x (1985).
Leigh, J. W., Bryant, D. & Nakagawa, S. popart: full‐feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116, https://doi.org/10.1111/2041-210X.12410 (2015).
Acknowledgements
We thank M. Keena (USDA Forest Service), S.K. Korb (Russia), H. Nadel (USDA), C. Hideyuki (Bernice Pauahi Bishop Museum, Honolulu, USA) and H. Rajaei (State Museum of Natural History Stuttgart, Germany) for providing specimens used in this study. We also thank C. Béliveau (Cusson lab) for proving the larval picture used in Fig. 2. This work was funded by Genome Canada, Genome British Columbia, Genome Quebec, the Canadian Forest Service (Genomics Research and Development Initiative, GRDI) and the Canadian Food Inspection Agency, through a Genomics Applications Partnership Program (GAPP 6102; Genome Canada) grant.
Author information
Authors and Affiliations
Contributions
A.D., A.N., R.Z., and M.C. designed the work; A.D., A.N., R.Z., D.S., H.M. and L.F. performed the experiments and analyses; S.P. provided access to unpublished data and assisted in data interpretation; D.E.G.-R., M.E.S. and K.D. helped acquire initial mitogenome sequence data from independent NGS projects; R.C.L., R.C.H. and M.C. provided funding; all authors contributed to the drafting and/or editing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Djoumad, A., Nisole, A., Zahiri, R. et al. Comparative analysis of mitochondrial genomes of geographic variants of the gypsy moth, Lymantria dispar, reveals a previously undescribed genotypic entity. Sci Rep 7, 14245 (2017). https://doi.org/10.1038/s41598-017-14530-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14530-6
This article is cited by
-
Characterization and Molecular Phylogenetic Analysis of Subfamily Erebinae (Lepidoptera: Noctuoidea: Erebidae) Using Five Complete Mitochondrial Genomes
Biochemical Genetics (2023)
-
Characterization of complete mitochondrial genomes of five noctuoid moths (Lepidoptera: Noctuoidea) and their phylogenetic implications
Journal of Biosciences (2023)
-
Analyses of adult transcriptomes from four different populations of the spongy moth, Lymantria dispar L., from China and the USA
Scientific Reports (2022)
-
Characterization of four mitochondrial genomes from superfamilies Noctuoidea and Hyblaeoidea with their phylogenetic implications
Scientific Reports (2022)
-
Comparative mitochondrial genome analysis of Eudocima salaminia (Cramer, 1777) (Lepidoptera: Noctuoidea), novel gene rearrangement and phylogenetic relationship within the superfamily Noctuoidea
Molecular Biology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}