
Abstract
Cereal cyst nematode (Heterodera avenae) is attracted to and aggregated around wheat roots to initiate infection, but this interaction between wheat and the nematode is not fully understood. The transcriptional responses of both wheat and H. avenae were examined during their early contact stage by mRNA sequencing analysis; certain numbers of the differentially expressed genes (DEGs) were validated using quantitative real-time PCR. The immobile host wheat root only had 93 DEGs (27 up-regulated and 66 down-regulated), while the mobile plant parasitic nematode reacted much more actively with 879 DEGs (867 up-regulated and 12 down-regulated). Among them, a number of wheat DEGs (mostly down-regulated) were involved in biotic stress pathways, while several putative effector genes were up-regulated in the nematode DEGs. One putative chitinase-like effector gene of H. avenae was able to suppress BAX-triggered programmed cell death in Nicotiana benthamiana. Results of these experiments demonstrated that nematode responded more actively than wheat during the contact stage of parasitism. The parasite’s responses mainly involved up-regulation of genes including at least one anti-plant-defence effector gene, whereas the host responses mainly involved down-regulation of certain defence-related genes.
Similar content being viewed by others


Introduction
Plant-parasitic nematodes (PPNs) have caused extensive damage to many plant species1. Cyst nematodes, such as Heterodera spp. and Globodera spp. on cereal crops and soybean (Glycine max (L.) Merr.), and root-knot nematode (RKN, Meloidogyne spp.), are the most widely studied species because of their economic importance2. Heterodera avenae Wollenweber is globally one of the most important species of the cereal cyst nematodes (CCNs). It occurs in about 80% of the wheat (Triticum aestivum L.) growing areas in China3. Infestation of CCN has caused substantial yield losses of wheat ranging from 30 to 100%4,5. The discovery of the mechanisms underlying plant-nematode interactions will provide clues on the control of this destructive nematode.
Many parasitic nematodes produce larvae, which use sensory cues for locating their hosts. This complex behavior of the nematodes involves different sensory capabilities, for example olfaction and gustation, as well as temperature and humidity sensing. Although preventative measures can be applied during the attraction of the nematodes to plant roots, this initial step in the parasite-host interactions remains poorly understood6. PPNs can be attracted to plant roots7 and both volatile and non-volatile root extracts have been shown to attract potato cyst nematode (G. pallida Stone)8. Ethylene and auxin signaling pathways affect the attraction or repulsion of the roots to nematodes2,9,10,11. Increases in ethylene production reduced the attraction to the host by RKN10, while high auxin concentrations attracted Aphelenchoides besseyi Christie11 and RKN2. This might also be due to possible cross-talk between auxin and ethylene in plant12.
Meloidogyne species are attracted to low pH at the levels similar to the low pH environment produced by the growing roots13. Carbon dioxide (CO2) attracts a number of PPN species14,15,16,17,18. Plant-parasitic nematodes are also attracted to certain root volatiles, which are identical to those emitted by insect-damaged plants to attract entomopathogenic nematodes6,19. Nevertheless, little is known about the responses of either parasitic nematodes or their hosts during host-attraction/contact process. It was reported that RKN could activate subcellular reorganization and root-hair deformation in Lotus japonicus and tomato (Solanum lycorpersicum L.) roots via a signal that could be transmitted at a distance from the host20.
Studies on plant-nematode interactions have taken advantage of high-throughput techniques such as transcriptome sequencing, e.g., RKN21,22,23,24,25,26,27, cyst nematodes28,29,30,31,32,33,34 and other PPNs24,35. The first de novo transcriptomic analysis compared the gene expression of pre-parasitic infective juveniles (J2s) to adults in H. avenae, which resulted in the identification of some important genes that may be involved in either plant parasitism or nematode metabolism30. During the interaction between CCN and Aegilops variabilis Eig., the transcriptome of both CCN and Ae. variabilis roots were analyzed at 30 h, 3 d, and 9 d post inoculation28,32. Those studies identified 7,408 unigenes and three pathways in Ae. variabilis associated with plant stress resistance. They also detected 681 putative genes in the parasitic nematode, which included 56 putative effectors. Comparative transcriptome analysis of susceptible and resistant wheat cultivars was used to study the defence responses of wheat against H. avenae during the early infection stages (i.e., 24 h, 3 d and 8 d post infection), which resulted in the identification of 606 resistance genes and diverse defence-related pathways31.
To date, no transcriptome analysis has focused on the stage before infection of nematodes to their hosts, the initial contact stage of the host-nematode interaction. Previous studies have reported that H. filipjevi and H. avenae were attracted to and aggregated around wheat roots36,37. This study was conducted to identify transcriptomic responses of both wheat and its parasite H. avenae during the initial contact stage.
Results
Attraction of H. avenae to roots of wheat cultivar Wenmai 19
The CCN-susceptible wheat cultivar Wenmai 19 was used to attract infective J2s of H. avenae. The J2 nematodes, which were scattered in the Pluronic F-127 gels, migrated toward wheat roots and gathered in a large quantity around the root tips after 3 h (Fig. 1), except for a few that remained away from the wheat roots. The numbers of J2s gathering around wheat root tips peaked at 3 h, so samples of the wheat roots and the nematodes were collected at this time for transcriptome analysis. Simultaneously, it was confirmed that the H. avenae J2s did not penetrate the wheat roots, as no nematodes were observed inside the stained roots (data not shown).

Attraction of Heterodera avenae juveniles to the root tips of Wenmai 19 wheat. The figure shows the aggregation of H. avenae juveniles (indicated by arrows) around a wheat root tip when nematodes and wheat roots were incubated together in a Pluronic F-127 gel for 3 hours (scale bar = 1 mm).
Transcriptome data from the wheat roots
Six samples of wheat root tips, i.e., three replicates from wheat that had attracted nematodes (wheat treatment) and three from the wheat control, were separately subjected to RNA-sequencing analysis. A total of 52.23 Gb of clean data were obtained from the six root samples altogether, each of which contained ≥8.25 Gb with Quality Scores of Q30 ≥ 90.4% (Supplementary Table S1). A range of 64.0% to 67.3% clean reads of each sample were aligned onto the wheat reference genome and matched to either unique or multiple genomic locations (Table 1). In total, 109,496 unigenes including 9,152 new genes were mined in the wheat transcriptome. According to the databases of Non-redundant protein sequences (Nr), Swiss-Prot, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG), 6,780 new genes were annotated. The replicates of wheat roots repeated well with each other (r 2 = 0.98~1.00) (Supplementary Fig. S1a).
Wheat genes responding to CCN aggregation
Comparative analysis of gene expression was performed for the wheat roots that were exposed to the J2 nematodes and the negative control that did not contact with the J2s. Ninety-three unigenes, including 66 down-regulated and 27 up-regulated ones, were differentially expressed with false discovery rate (FDR) < 0.05 and fold change (FC) ≥ 1.5 (Fig. 2, Supplementary Table S2). Twelve differentially expressed genes (DEGs) were validated by quantitative real-time PCR (qPCR) and the expression patterns of eleven DEGs were consistent with those of the mRNA-Seq analysis (Table 2). These results demonstrated that the wheat roots responded to nematode aggregation even though they were not infected by the J2s. Results of the functional annotation indicated that all the DEGs had significant matches in the Nr database, and some of them also had annotation information in the Swiss-Prot, GO, KEGG and COG databases (Table 3).

Volcano plots of differentially expressed genes (DEGs) between the nematode-free wheat roots and those exposed to Heterodera avenae juveniles (a), and wheat root-free H. avenae juveniles and those exposed to wheat roots (b). Each dot represents one gene with the y-axis showing -lg (FDR) and the x-axis showing log2 (FC), respectively. The red, green and normal dots represent the up-regulated DEGs, down-regulated DEGs (FDR < 0.05 and FC ≥ 1.5 for wheat; FDR < 0.01 and FC ≥ 2 for nematode) and not significantly changed genes, respectively. FC: fold change; FDR: false discovery rate.
Based on the functional annotation obtained from GO enrichment analysis, the wheat DEGs were separated into 28 functional groups, which belong to three main categories: biological processes (60 DEGs), cellular components (54 DEGs), and molecular functions (68 DEGs) (Fig. 3a). In the biological process category, greater percentages of DEGs were involved in metabolic processes, single-organism processes and responses to stimuli compared to all unigenes of wheat roots. More proportions of DEGs in the cellular component category were localized to the extracellular region. The DEGs in the molecular function category were more enriched in GO class of nutrient reservoir activity, antioxidant activity, electron carrier activity, enzyme regulator activity and catalytic activity than all unigenes.

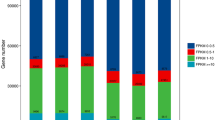
Gene Ontology (GO) categories of all unigenes and differentially expressed unigenes (DEG unigenes) in the wheat roots (a) and Heterodera avenae juveniles (b) in the study. The number and percentage of genes in each subcategory for the three main categories of biological process, cellular component, and molecular function are indicated for all the unigenes and DEG unigenes, respectively. On the right y-axis, blue and black numbers are DEG unigenes and all unigenes, respectively.
Analysis of KEGG pathways was performed to determine the biological functions of the DEGs. Thirty-one DEGs were allocated to 21 KEGG pathways (Fig. 4a, Supplementary Table S3). The phenylpropanoid biosynthesis pathway accounted for the highest number of DEGs, followed by glutathione metabolism, phenylalanine metabolism and starch and sucrose metabolism (Supplementary Table S3). Six DEGs, i.e., Wheat_newGene_1897, gene:Traes_1AS_F9013A945, gene:Traes_2AS_EE549925C, gene:Traes_2DS_2CCCA54C1, gene:Traes_7DL_0D9D56EC9, and gene:Traes_7DL_4C9B51BF6, were involved in the phenylpropanoid related pathways (phenylpropanoid biosynthesis or phenylalanine metabolism) (Supplementary Table S2). They were all down-regulated in the wheat roots that were exposed to the nematodes, and two of them, gene:Traes_1AS_F9013A945 and gene:Traes_2AS_EE549925C, were validated by qPCR with similar expression patterns (Table 2).

Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of differentially expressed genes (DEGs) of wheat roots (a) and Heterodera avenae juveniles (b) in the study. Cellular processes and metabolic pathways were enriched in wheat DEGs. Cellular processes, environmental information processing, genetic information processing, human diseases, metabolism and organismal system pathways were enriched in H. avenae DEGs. The x-axis shows the percentage of the annotated genes in each category and the number of genes is indicated at the top of the bar.
Using COG annotation, 29 wheat DEGs were grouped into six COG functional classes, which included energy production and conversion; secondary metabolite biosynthesis, transport and catabolism; general function prediction only; posttranslational modification, protein turnover, chaperones; amino acid transport and metabolism; and carbohydrate transport and metabolism (Fig. 5a). All but four of these DEGs were down-regulated (Supplementary Table S2).

Clusters of Orthologous Groups (COG) function classification of differentially expressed genes (DEGs) of wheat roots (a) and Heterodera avenae juveniles (b) in the study. The y-axis shows the number of genes in each function class (in different colours on the x-axis).
Among the DEGs of wheat roots, two protease inhibitor genes and one protease gene were annotated (Supplementary Table S2). Both Wheat_newGene_1218 and gene:Traes_1DL_A6553EC96 had GO annotation in the molecular function category of serine-type endopeptidase inhibitor activity (GO:0004867) and they were both localized in the cellular component of extracellular region (GO:0005576). Another DEG Wheat_newGene_2674 was annotated as xylem cysteine proteinase 1 by Nr_annotation. These three DEGs were all down-regulated.
Visualization of biotic stress pathways in the wheat DEGs
A number of wheat DEGs were mapped to the biotic stress pathways as revealed by MapMan analysis (Fig. 6, Supplementary Table S4). Specifically, 33 data points that showed putative involvement in biotic stress were mapped for 29 wheat DEGs (Fig. 6), involving peroxidases, glutathione S transferases, hormone signaling (auxins and jasmonic acid), pathogenesis-related proteins, and secondary metabolites. Most of these DEGs were down-regulated in the biotic pathways (Fig. 6). This was confirmed by qPCR analysis of eight of those DEGs, as their expression patterns were consistent with that of mRNA-Seq in all cases but one (Table 2).

Visualization of biotic stress pathways in wheat differentially expressed genes (DEGs) by MapMan. Thirty-three data points showing putative involvement in biotic stress were mapped for 29 DEGs of wheat (see detailed information on the genes in Supplementary Table S6). Red and blue dots represent the genes that were up- and down-regulated in wheat roots when they were exposed to nematodes compared to the control (wheat roots without contact with the juvenile nematodes), respectively. The magnitude of the change in gene expression is associated with deepness of the colour.
In the redox reaction, three DEGs (annotated as peroxidase 2, root peroxidase and class III peroxidase) and seven DEGs (annotated as putative glutathione-S-transferase, peroxidase and In2.1 protein) were mapped for peroxidases and glutathione-S-transferase, and they were all down-regulated, indicating weakening of the redox reaction. In the hormone signaling pathway, auxin and jasmonic acid (JA) signaling each contained two down-regulated DEGs, which were annotated as probable aldo-keto reductase 3, auxin-induced protein, lipoxygenase and 12-oxophytodienoate reductase, respectively. Seven data points mapped by the six down-regulated DEGs annotated as phenylalanine ammonialyase, agmatine coumaroyltransferase-2, tyrosine decarboxylase, leucoanthocyanidin dioxygenase, S-norcoclaurine synthase 1, flavonoid 3 & apos; 5 & apos;-hydroxylase 2 and S-norcoclaurine synthase 1 were almost involved in metabolism of phenylpropanoids and flavonoids. Defense genes coding for PR-proteins had three DEGs, which were all down-regulated with annotation as putative disease resistance RPP13-like protein and xylanase inhibitor. A down-regulated DEG (annotated as fasciclin-like protein FLA11) was mapped to cell wall protein and a predicted expansin-A13-like DEG up-regulated was related to cell wall modification pathway. The proteolysis pathway involved protease and ubiquitin were mapped by two down-regulated DEGs (annotated as xylem cysteine proteinase and ubiquitin) and two up-regulated DEGs (annotated as NEP1-interacting protein and E3 ubiquitin-protein ligase RING1-like). The respiratory burst involved one up-regulated DEG (annotated as respiratory burst oxidase homolog protein B). A transcription factor belongs to ethylene-responsive element binding protein family mapped by gene:Traes_5DL_41E3B1B23 was an up-regulated DEG (annotated as ethylene-responsive transcription factor ERF071).
Transcriptome data of CCN
Similar to the wheat roots, six samples of CCNs, from three replicates that were exposed to wheat roots and three for the controls without exposure to wheat roots, were separately subjected to RNA-sequencing analysis. The transcriptome analysis produced a total of 30.47 Gb clean data and ≥4.13 Gb per sample with the Quality Score of Q30 ≥ 89.1% (Supplementary Table S5). Altogether, 194,662 transcripts and 80,124 unigenes were obtained (Table 4). The total length, N50 length, and mean length of unigenes were 61,659,712 bp, 955 bp, and 769.55 bp, respectively (Table 4). A total of 15,197 unigenes were longer than 1 kb (Table 4). The proportions of mapped clean reads with the assembly data for each CCN sample ranged from 73.2% to 79.2%. A total of 43,741 unigenes were annotated according to the Nr, Swiss-Prot, GO, KEGG, COG, Clusters of Protein homology (KOG) and Pfam databases. Additionally, the replicates of CCNs showed good repeatability with each other (r 2 = 0.86~1.00) (Supplementary Fig. S1b).
CCN genes responding to wheat root attraction
The expression of genes of nematodes exposed to wheat roots was compared with the control (without exposure to wheat roots). A total of 879 unigenes were regarded as DEGs (FDR < 0.01 and FC ≥ 2). Most DEGs (867) were up-regulated and only 12 DEGs were down-regulated (Supplementary Fig. S2b, Supplementary Table S6). In addition, ten DEGs were analyzed by qPCR and their expression patterns were consistent with those of the mRNA-Seq results in all cases (Table 2). These results indicated that the CCNs were activated by the stimulation of wheat roots with up-regulation occurring for most of the genes detected. Results of functional annotation for the DEGs indicated that 742 of them (84.4%) were annotated in one or more of the Nr, Swiss-Prot, GO, KEGG, COG, KOG and Pfam databases (Table 3).
On the basis of their functional annotation, GO enrichment analysis classified 258 DEGs into 36 functional groups in three main categories: biological processes (159 DEGs), cellular components (107 DEGs), and molecular functions (223 DEGs) (Fig. 3b). The percentages of DEGs in GO class of metabolic processes and immune system processes were more than those of all unigenes of H. avenae juveniles. More proportions of DEGs in the cellular component category were localized to the macromolecular complex, organelle (part), and cell (part) compared to all the unigenes. The DEGs in the molecular function category were enriched more in the structural molecule activity, antioxidant activity and catalytic activity than all the unigenes.
Results of KEGG pathway analysis allocated 247 DEGs to 125 KEGG pathways (Supplementary Table S7), and 50 of the most significant pathways are shown in Fig. 4b. The pathways with the highest number of DEGs involved ribosomes (64 DEGs), and other pathways with a large numbers of DEGs included metabolism of xenobiotics by cytochrome P450, glutathione metabolism, and toll-like receptor signaling pathways (Supplementary Table S7). The ribosome pathway was more active in the responses of CCN exposed to and contacting with wheat roots than were the other pathways, which indicates that protein translation is much more active. Xenobiotics and drug metabolism were also considerably activated. The toll-like receptor signaling pathway (ko04620) involved 15 DEGs (Fig. 4b) and the DEGs related to IRAK1 (K04730; interleukin-1 receptor-associated kinase 1) and IRAK4 (K04733; interleukin-1 receptor-associated kinase 4) might lead to chemotactic effects (Supplementary Fig. S2).
A total of 386 DEGs were classified into 22 COG functional classes. The top four COG classes with the highest number of DEGs included those involved in general function prediction only (90 DEGs), translation, ribosomal structure and biogenesis (65 DEGs), energy production and conversion (45 DEGs), and amino acid transport and metabolism (44 DEGs) (Fig. 5b). The DEGs in those functional categories were the most strongly induced in the nematodes responding to exposure to the wheat roots.
Effector prediction
A total of 351 currently known effector gene sequences of PPNs were collected (Supplementary Table S8), and the DEGs of CCN were blasted against those sequences. Six DEGs were predicted to be homologous to the known effector genes 14-3-3, chitinase, beta-1,4-endoglucanase, pectate lyase, or cathepsin (Table 5). The description of their hit effector genes and the Nr annotations of the DEGs were consistent. The structural domains of those DEGs were predicted and they were also consistent with the gene descriptions (Fig. 7, Table 5). The DEGs that encode candidate effector proteins were all up-regulated when J2 nematodes were exposed to the wheat roots.

Structural domains of the six candidate effector genes of Heterodera avenae predicted through National Center for Biotechnology Information (NCBI). Both c66622.graph_co and c74386.graph_c0 contain a putative 14-3-3 domain; c68622.graph_c0 contains a GH18_chitinase-like domain or a glyco_18 domain; c72010.graph_c0 contains a putative glyco_hydro_1 domain; c72543.graph_c0 contains a putative pectate_lyase domain; and c78853.graph_c0 contains a peptidase_C1 domain and an inhibitor_I29 domain (cathepsin propeptide inhibitor domain (I29)).
Plant defence suppression by a predicted effector
Two putative effector genes c68622.graph_c0 (hit chitinase [H. glycines]) and c72543.graph_c0 (hit pectate lyase [H. glycines]) were selected for the BAX-triggered programmed cell death (BT-PCD) suppression assay in Nicotiana benthamiana Domin to verify their ability to suppress the plant defences. No obvious necrosis was observed on the infiltration spot of c68622.graph_c0 followed by BAX, while that of c72543.graph_c0 was obviously as necrotic as the infiltration buffer followed by BAX (Fig. 8). The two replicated results of the BT-PCD suppression assay in N. benthamiana were consistent. These findings suggest that the former gene suppresses BT-PCD while the latter gene does not. Therefore, c68622.graph_c0 is a candidate effector gene from the H. avenae DEGs that may play a role in suppressing the plant’s defences.

Assay for suppression of BAX-triggered cell death (BT-PCD) by the candidate Heterodera avenae effectors (a) c68622.graph_c0 and (b) c72543.graph_c0 in Nicotiana benthamiana. Leaves of N. benthamiana were infiltrated with the infiltration buffer or Agrobacterium tumefaciens cells containing a pGR107 vector carrying the candidate effector gene either alone or infiltration with A. tumefaciens cells carrying a mouse Bax gene 24 h later. Photos of the phenotypes of infiltrated N. benthamiana leaves were taken 6 days after infiltration. The spots with label 3 on the leaf show that the gene c68622.graph_c0 suppressed the necrosis induced by Bax, but the gene c72543.graph_c0 did not suppress necrosis.
Discussion
The analysis of the DEGs provided an outline of the transcriptome responses of both wheat and H. avenae during the first step in the host-nematode interaction process (i.e., the contact stage), which has been largely overlooked in previous studies of the host-nematode interactions. Wheat is one of the most important crops in the world and cereal cyst nematodes are causing more and more damage to wheat crops3. Control of the nematodes before infection stage would be of great importance. This study focused on the contact stage of nematodes with wheat roots in an effort to investigate their early interaction mechanism. Because wheat has a reference genome while H. avenae does not, the transcriptome data of wheat was analyzed based on the reference genome and that of H. avenae was assembled and analyzed without a reference genome. With the following characteristics: well repeated biological replicates, high Quality Scores of Q30, well aligned clean reads of wheat with the reference genome, good quality of the assembled data of nematode and so on, the sequenced transcriptome data in the present study were believed to be reliable for the DEGs analysis. The mobile plant parasitic nematode H. avenae was shown to react much more actively with 879 DEGs, while the immobile wheat root tips only activated 93 DEGs. The response of the wheat root tips might have been induced by contact with the CCNs during their aggregation and/or some signal transduction occurred between the two organisms.
The GO annotation indicated that the DEGs induced in wheat and the CCN were mostly responsible for metabolism processes (Fig. 3). While the molecular functions of DEGs were divergent for the host and parasite. In the wheat host, nutrient reservoir, antioxidant activity, electron carrier activity and enzyme regulator activity were the top four enriched function categories of DEGs. In the CCN parasite, DEGs were enriched in the function categories of structural molecule activity and antioxidant activity. However, both wheat host and CCN parasite responded to each other with the DEGs enriched in the molecular function category of antioxidant activity. KEGG analysis of the wheat DEGs revealed that phenylpropanoid pathways associated with phenylpropanoid biosynthesis and phenylalanine metabolism were involved in the plant responses to nematode aggregation. It is increasingly clear that phenylpropanoid pathway is important in plant defence38. Previous research has demonstrated that the up-regulated components of phenylpropanoid pathway were also closely related to plant defence against infestations of nematodes, such as G. rostochiensis, H. glycines, and H. avenae 31,39,40,41. While the suppressed expression of genes in the phenylpropanoid pathway were related to susceptible reactions of plants to nematode infestation, for example, tomato to G. rostochinensis 39 and soybean to H. glycines 42,43,44. In the present study, components of this pathway were also transcriptionally regulated in the host responses to the aggregation of CCNs, indicating that plant defence might has already been affected when nematodes massed around plant roots (Fig. 4a). The wheat DEGs involved in this pathway were all down-regulated during the contact stage (Supplementary Table S2), which was consistent with previous studies on susceptible interactions during infection39,40. In addition, MapMan analysis revealed that a number of wheat DEGs were mapped in biotic stress pathways (Fig. 6, Supplementary Table S4). Those DEGs were mostly down-regulated indicating weakening of defense pathways. The DEGs in those pathways including redox reaction (involved peroxidase and glutathione-S-transferase), metabolism of phenylpropanoids and flavonoids, auxin and JA hormone signaling, and defense PR-proteins were all down-regulated. Modulation (up- or down-regulation) of peroxidase genes including class III peroxidase genes were previously shown to be involved in the CCN infestation of wheat31,45, while results of the current study showed that the three peroxidase genes were down-regulated during the contact stage.
Three DEGs related to protease or protease inhibitor genes were detected. The protease DEG Wheat_newGene_2674 (annotated as xylem cysteine proteinase) was also involved in protein degradation of biotic stress pathway as revealed by MapMan analysis (Table S4). Cysteine protease was reported to be accumulated in maize vascular elements of leaf and root to defend against insect herbivores46. The xylem cysteine proteinase DEG in wheat roots was down-regulated and it might be related to weakening of wheat defense reaction to aggregation of nematodes. Protease inhibitors were involved in inducible defence in plants against herbivory including PPNs47. Serine protease inhibitors were also reported to confer resistance against nematode pests48,49. Two DEGs Wheat_newGene_1218 and gene:Traes_1DL_A6553EC96 annotated as serine protease inhibitors were down-regulated, indicating reduced defence of wheat at this stage.
The effector genes of PPNs have been reported to play important roles in their successful parasitism50,51,52, while it has not been reported whether the effector genes in CCN could be differentially expressed when nematodes approach the wheat roots. The results of transcriptome analysis identified six effector genes as DEGs from CCN during the contact stage and they were all up-regulated (Table 5), indicating that they were prepared in advance to promote subsequent infection by the nematodes. One candidate effector gene c68622.graph_c0 (hit chitinase [H. glycines]) from the DEGs was shown to suppress BT-PCD, which triggers a process that physiologically resembles a hypersensitive defence response (Fig. 8). Many studies have demonstrated that the effectors of nematodes (including H. avenae) could affect plant defence as they did in other pathogens53,54,55,56. The up-regulation of the candidate effector gene c68622.graph_c0 (hit chitinase [H. glycines]) during the contact stage as shown by both mRNA-Seq and qPCR analyses (Table 2) might be modulated by the nematodes in order to suppress plant immunity in the next infection stage. It appears that when they massed around wheat roots, the nematodes were armed already for fighting the plant defence to promote subsequent infection.
Other candidate effector genes in the H. avenae DEGs seemed to have different roles. For example, cell wall modification enzymes-like DEGs including c72010.graph_c0 (Nr annotation as beta-1,4-endoglucanase precursor of G. rostochiensis) and c72543.graph_c0 (Nr annotation as a pectate lyase of H. glycines) might degrade and soften the cell walls to allow the migration of PPNs inside the roots46. However, the DEGs for effectors perhaps were also effective during the host-parasite contact stage for signal induction or suppression in the host plant, as it has been reported that pre-parasitic J2s of RKN are stimulated to secrete quite a number of proteins, and could secrete low, but detectable levels of proteins even in the absence of stimulation57,58.
The phytohormones auxin and ethylene were reported to increase or decrease root attractiveness to nematodes, respectively2,9,10,11. During the contact stage of the nematodes with wheat roots after the attraction, some wheat genes related to phytohormones were also changed transcriptionally. For example, gene:Traes_5DL_41E3B1B23 (annotation as an ethylene-responsive transcription factor ERF071 of Arabidopsis thaliana) was up-regulated and two auxin related genes gene:Traes_2AL_1A870CE7B (annotation as probable aldo-keto reductase 3) and gene:Traes_6DS_768787FF4 (annotation as an auxin-induced protein) were down-regulated (Supplementary Table S4). Three DEGs related to flavonoid metabolism (gene:TRAES3BF009500080CFD_g annotated as leucoanthocyanidin dioxygenase, gene:TRAES3BF168600 030CFD_g annotated as S-norcoclaurine synthase 1, and gene: Traes_2BL_37005C9E0 annotated as flavonoid 3 & apos;, 5 & apos;-hydroxylase 2) were also found to be down-regulated (Supplementary Table S4), while flavonoids can affect auxin level59. All the six DEGs were related to the phytohormones auxin or ethylene production, which indicates that phytohormones not only affect the attraction of nematodes to the wheat roots, but also are influenced by the aggregation of nematodes. Besides, down-regulation of auxin related genes and up-regulation of an ethylene related gene might reduce the attractiveness of wheat roots to nematodes as a response of the wheat roots to nematode aggregation. In addition, these six genes might also participate in plant defence as they were included in the biotic stress pathways by MapMan analysis (Supplementary Table S4).
In the current study, 15 DEGs of CCN were involved in the toll-like receptor signaling pathway (ko04620) (Fig. 4b). The DEGs related to IRAK1 and IRAK4 might influence chemotactic effects (Supplementary Fig. S2). Tropism of nematodes to wheat roots might involve this pathway. However, the functions of those genes in nematodes remain unknown.
Methods
Sampling and the attraction assay
Cysts of H. avenae (Ha43 pathotype group60) were collected from a CCN-infested field in Xingyang, Henan Province. Infective J2s were obtained by hatching the cysts at 15 °C for about one week following incubation for at least 8 weeks at 4 °C. Seeds of wheat cultivar Wenmai 19, which is susceptible to the Ha43 pathotype group of H. avenae 61,62, were germinated on moist filter paper in Petri dishes for 2 days at room temperature. A Pluronic F-127 gel (Sigma, Saint Louis, MO) (23%, w/v) was used as the medium for observing attraction of nematodes to the host roots63. In each biological replicate, five to six germinated wheat seeds with roots 2–3 cm long were transferred into a Petri dish (9 cm in diameter) containing 25,000~30,000 J2s suspended in 15 ml Pluronic F-127 gel for the attraction assay, which was defined as the treatment. The treatments with wheat seedlings or nematodes alone in the gel were used as the negative controls. After 3 h at room temperature, wheat root tips and nematodes in each Petri dish were collected and separately frozen in liquid nitrogen for RNA extraction. Simultaneously, wheat roots of the treatment were stained in a sodium hypochlorite-acid fuchsin solution64 to visualize any penetration of J2 nematodes into roots with a microscope (Olympus CX31, Tokyo, Japan).
Wheat seedlings were removed from the gel, rinsed in sterile-double-distilled water three times and dried on absorbent paper. Wheat root tips (c. 1 cm long) were then cut and collected in 2-ml RNase-off centrifuge tubes. Sterile-double-distilled water was added into the Petri dishes containing nematodes to liquidize the gels. The mixtures were poured into 50-ml centrifuge tubes and centrifuged at 6000 rpm for 2 min. Then, the nematodes at the bottom of the tubes were transferred to 1.5-ml centrifuge tubes and rinsed in sterile-double-distilled water containing 2 drops of 0.05% Tween-20 (polyoxyethylene (20) sorbitan monolaurate, Sinopharm Chemical Reagent, Beijing, China) three times (2000 rpm, 1 min). Finally, the nematodes were collected in 2-ml RNase-off centrifuge tubes.
Bioinformatic analysis
Three replicates were included for each treatment or control. Total RNA was extracted from each sample of wheat roots or nematodes with the TRIzol reagent, which was used for library construction and mRNA-sequencing by HiSeq4000 (Biomarker Technologies Co. LTD, Beijing, China) independently. Data obtained from wheat roots and nematodes were analyzed by means of transcriptomic analysis with or without a reference genome (as H. avenae has no published reference genome).
For wheat, the raw reads of each sample were filtered to produce clean data, which were then aligned to the wheat genome using TopHat265 (http://ftp.ensemblgenomes.org/pub/plants/release-30/fasta/triticum_aestivum/dna/). All the multiple mapped reads were allocated to specific gene on the reference genome by Cufflinks software66 using Maximum Likelihood Model. The abundance of transcripts was calculated as fragments per kilobase of transcript per million fragments mapped (FPKM)67 using the Cuffquant and Cuffnorm components in Cufflinks software66, and Pearson’s correlation coefficient r was calculated in R packages by its formula using the counts of all the transcriptomic data to compare repeats68. The DEGs between the control and the treatment sample were screened using DESeq69 with an FDR threshold < 0.05 and FC ≥ 1.5. To annotate the functions of the transcripts, the unigenes were blasted against the databases of Nr, Swiss-Prot, KEGG, and COG using BLAST program70 with an E-value ≤ 1e-5. The Blast2GO program was used to annotate the major GO categories of genes with an E-value ≤ 1e-571. Furthermore, MapMan72 was used to visualize the biotic stress pathways of wheat DEGs after generating a mapping file of the wheat DEGs by Mercator73.
For CCN, the clean reads of each sample also were obtained after filtering, which were assembled together for all the nematode samples using Trinity software74. Clean data from each sample were aligned against the assembled transcripts or unigenes and the mapped reads were used for further analysis. The unigenes were annotated using BLAST program70 against Nr, Swiss-Prot, GO, KEGG, COG and KOG databases with an E-value ≤ 1e-5, and aligned with Pfam using HMMER75 with an E-value ≤ 10. To evaluate the expression levels (in the form of FPKM) of the unigenes, the reads of each sample were aligned with the assembled unigene database using Bowtie software76 and analyzed using RSEM program77. Repeat correlations were also tested by Pearson’s correlation coefficient68. The DEGs were identified by DESeq software69 with an FDR < 0.01 and FC ≥ 2.
The data described in this study have been deposited in the Gene Expression Omnibus78 of National Center for Biotechnology Information (NCBI) and are accessible through the GEO Series accession number GSE99228 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE99228).
Validation of mRNA-Seq by qPCR
Based on their potential functional importance, 12 and 10 DEGs were selected for validation by qPCR from wheat and CCN, respectively. The cDNA was prepared from the remaining total RNA after transcriptome sequencing according to the instructions of the SuperScript™ III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). A SYBR Green assay was used to quantify the expression of each gene using SYBR Premix Ex Taq (TaKaRa, Dalian, China) in a CFX Connect™ Real-Time PCR Detection System (Bio-Rad, Munich, Germany), with the primers for each DEG and the reference genes, actin for wheat45 and GAPDH-1 for CCN79 (Supplementary Table S9). Three nematode or wheat biological replicates were analyzed for each gene with three technical replicates. Data were processed using the 2−ΔΔCt method80 and analyzed statistically using the Student’s t-test in IBM SPSS Statistics 19 software (IBM Corp., Somers, NY, USA) to compare the difference between the treatments and the control samples at P < 0.05.
H. avenae effector prediction
The sequences of 351 known parasitism effectors of PPNs (Supplementary Table S4) were collected and aligned with the H. avenae DEGs. Putative effector genes were obtained using an E-value ≤ 1e-5. Structural domain prediction of the genes was conducted by blasting the gene sequence at NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Cell-death suppression assay in N. benthamiana
To detect plant defence suppression, two predicted effector genes from the H. avenae DEGs (i.e., c68622.graph_c0 and c72543.graph_c0) were constructed in a PVX vector pGR107 (with the CP promoter) containing a flag-tag fused at the N-terminal, following the instructions of the In-Fusion HD Cloning Kit (Clontech, Palo Alto, CA, USA), with the primer pairs c68622-107f-S1/AS1 and c72543-107f-S1/AS1 (Supplementary Table S9). Construct of pGR107::Bax was provided by Dr. B.Y. Xie of the Chinese Academy of Agricultural Sciences, China. The constructs were verified by sequencing prior to transformation into Agrobacterium tumefaciens strain GV3101.
Plants of N. benthamiana were grown in a greenhouse for 4 to 6 weeks at 25 ± 2 °C under a photoperiod of 16 h light/8 h dark. An assay of the suppression of BT-PCD was carried out as described by Chen et al.79, except that 10 mM 2-(N-Morpholino) ethanesulfonic acid, 4-morpholineethanesulfonic acid (MES, pH 5.6) and 200 μM acetosyringone were added to 10 mM MgCl2 as the infiltration buffer. This assay was repeated twice, using 5 plant replicates in which three leaves were inoculated per plant.
Data Availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Sasser, J. N. & Freckman, D. W. “A world perspective on nematology: the role of the society”, in Vistas on nematology: a commemoration of the twenty-fifth anniversary of the Society of Nematologists (eds Veech, J. & Dickson, D.). (Hyattsville, USA: Society of Nematologists), 7–14 (1986).
Kyndt, T. et al. Redirection of auxin flow in Arabidopsis thaliana roots after infection by root-knot nematodes. J. Exp. Bot. 67, 4559–4570 (2016).
Peng, D. et al. “Current knowledge of cereal cyst nematode (Heterodera avenae) on wheat in China,” In Cereal cyst nematodes: status, research and outlook (eds Riley, I. T., Nicol, J. M. & Dababat A. A.) (Antalya, Turkey: International Maize and Wheat Improvement Centre (CIMMYT)), 29–34 (2009).
Bonfil, D. J., Dolgin, B., Mufradi, I. & Asido, S. Bioassay to forecast cereal cyst nematode damage to wheat in fields. Precis. Agric. 5, 329–344 (2004).
Nicol, J. M., Elekçioğlu, İ. H., Bolat, N. & Rivoal, R. The global importance of the cereal cyst nematode (Heterodera spp.) on wheat and international approaches to its control. Commun. Agric. Appl. Biol. Sci. 72, 677–686 (2007).
Gang, S. S. & Hallem, E. A. Mechanisms of host seeking by parasitic nematodes. Mol. Biochem. Parasitol. 208, 23–32 (2016).
Rasmann, S., Ali, J. G., Helder, J. & van der Putten, W. H. Ecology and evolution of soil nematode chemotaxis. J. Chem. Ecol. 38, 615–628 (2012).
Farnier, K. et al. Novel bioassay demonstrates attraction of the white potato cyst nematode Globodera pallida (Stone) to non-volatile and volatile host plant cues. J. Chem. Ecol. 38, 795–801 (2012).
Curtis, R. H. C. et al. Do phytohormones influence nematode invasion and feeding site establishment? Nematology 9, 155–160 (2007).
Fudali, S. L., Wang, C. & Williamson, V. M. Ethylene signaling pathway modulates attractiveness of host roots to the root-knot nematode Meloidogyne hapla. Mol. Plant-Microbe Interact. 26, 75–86 (2013).
Feng, H. et al. The white-tip nematode, Aphelenchoides besseyi, exhibits an auxin-orientated behaviour affecting its migration and propagation. Nematology 16, 837–845 (2014).
Luschnig, C., Gaxiola, R. A., Grisafi, P. & Fink, G. R. EIR1, a root-specific protein involved in auxin transport, is required for gravitropism in Arabidopsis thaliana. Genes Dev. 12, 2175–2187 (1998).
Wang, C., Bruening, G. & Williamson, V. M. Determination of preferred pH for root-knot nematode aggregation using Pluronic F-127 gel. J. Chem. Ecol. 35, 1242–1251 (2009).
Klingler, J. et al. On the orientation of plant nematodes and of some other soil animals. Nematologica 11, 4–18 (1965).
Prot, J. et al. Migration of plant-parasitic nematodes towards plant roots. Revue De Nematologie 3, 305–318 (1980).
Dusenbery, D. B. Theoretical range over which bacteria and nematodes locate plant roots using carbon dioxide. J. Chem. Ecol. 13, 1617–1624 (1987).
Pline, M. & Dusenbery, D. B. Responses of plant-parasitic nematode Meloidogyne incognita to carbon dioxide determined by video camera-computer tracking. J. Chem. Ecol. 13, 873–888 (1987).
Robinson, A. F. et al. Optimal release rates for attracting Meloidogyne incognita, Rotylenchulus reniformis, and other nematodes to carbon-dioxide in sand. J. Nematol. 27, 42–50 (1995).
Ali, J. G., Alborn, H. T. & Stelinski, L. L. Constitutive and induced subterranean plant volatiles attract both entomopathogenic and plant parasitic nematodes. J. Ecol. 99, 26–35 (2011).
Weerasinghe, R. et al. Root-knot nematodes and bacterial nod factors elicit common signal transduction events in Lotus japonicus. Proc. Nat. Acad. Sci. USA 102, 3147–3152 (2005).
Jammes, F. et al. Genome-wide expression profiling of the host response to root-knot nematode infection in Arabidopsis. Plant J. 44, 447–458 (2005).
Barcala, M. et al. Early transcriptomic events in microdissected Arabidopsis nematode-induced giant cells. Plant J. 61, 698–712 (2010).
Damiani, I. et al. Plant genes involved in harbouring symbiotic rhizobia or pathogenic nematodes. New Phytol. 194, 511–522 (2012).
Kyndt, T. et al. Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytol. 196, 887–900 (2012).
Haegeman, A., Bauters, L., Kyndt, T., Rahman, M. M. & Gheysen, G. Identification of candidate effector genes in the transcriptome of the rice root knot nematode Meloidogyne graminicola. Mol. Plant Pathol. 14, 379–390 (2013).
Li, X. et al. De novo analysis of the transcriptome of Meloidogyne enterolobii to uncover potential target genes for biological control. Int. J. Mol. Sci. 17, 1442, https://doi.org/10.3390/ijms17091442 (2016).
Petitot, A. et al. Dual RNA-seq reveals Meloidogyne graminicola transcriptome and candidate effectors during the interaction with rice plants. Mol. Plant Pathol. 17, 860–874 (2016).
Xu, D. et al. De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode. BMC Genomics 13, 133, https://doi.org/10.1186/1471-2164-13-133 (2012).
Cotton, J. A. et al. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 15, R43, https://doi.org/10.1186/gb-2014-15-3-r43 (2014).
Kumar, M. et al. De novo transcriptome sequencing and analysis of the cereal cyst nematode, Heterodera avenae. PLoS ONE 9, e96311, https://doi.org/10.1371/journal.pone.0096311 (2014).
Kong, L. et al. Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genomics 16, 1–18 (2015).
Zheng, M. et al. RNA-seq based identification of candidate parasitism genes of cereal cyst nematode (Heterodera avenae) during incompatible infection to Aegilops variabilis. PLoS ONE 10, e141095, https://doi.org/10.1371/journal.pone.0141095 (2015).
Fosu-Nyarko, J. et al. Analysis of the transcriptome of the infective stage of the beet cyst nematode. H. schachtii. PLoS ONE 11, e147511, https://doi.org/10.1371/journal.pone.0147511 (2016).
Yang, D., Chen, C., Liu, Q. & Jian, H. Comparative analysis of pre- and post-parasitic transcriptomes and mining pioneer effectors of Heterodera avenae. Cell Biosci. 7, 11, https://doi.org/10.1186/s13578-017-0138-6 (2017).
Xiang, Y. et al. Transcriptome analysis of the chrysanthemum foliar nematode, Aphelenchoides ritzemabosi (Aphelenchida: Aphelenchoididae). PLoS ONE 11, e166877, https://doi.org/10.1371/journal.pone.0166877 (2016).
Cui, L. et al. Characterization of interaction between wheat roots with different resistance and Heterodera filipjevi. Acta Agron. Sin. 38, 1009–1017 (2012).
Hu, X. et al. Phenotype characterization of interaction between different wheat cultivars and Heterodera avenae. Plant Protect. 41, 78–83 (2015).
Dixon, R. A. et al. The phenylpropanoid pathway and plant defence-a genomics perspective. Mol. Plant Pathol. 3, 371–390 (2002).
Klink, V. P. et al. Syncytium gene expression in Glycine max[PI 88788] roots undergoing a resistant reaction to the parasitic nematode Heterodera glycines. Plant Physiol. Biochem. 48, 176–193 (2010).
Uehara, T. et al. Resistant and susceptible responses in tomato to cyst nematode are differentially regulated by salicylic acid. Plant Cell Physiol. 51, 1524–1536 (2010).
Mazarei, M. et al. Gene expression profiling of resistant and susceptible soybean lines infected with soybean cyst nematode. Theor. Appl. Genet. 123, 1193–1206 (2011).
Alkharouf, N. W. et al. Timecourse microarray analyses reveal global changes in gene expression of susceptible Glycine max (soybean) roots during infection by Heterodera glycines (soybean cyst nematode). Planta 224, 838–852 (2006).
Ithal, N. et al. Parallel genome-wide expression profiling of host and pathogen during soybean cyst nematode infection of soybean. Mol. Plant-Microbe Interact. 20, 293–305 (2007).
Puthoff, D. P., Ehrenfried, M. L., Vinyard, B. T. & Tucker, M. L. GeneChip profiling of transcriptional responses to soybean cyst nematode, Heterodera glycines, colonization of soybean roots. J. Exp. Bot. 58, 3407–3418 (2007).
Simonetti, E. et al. Analysis of class III peroxidase genes expressed in roots of resistant and susceptible wheat lines infected by Heterodera avenae. Mol. Plant-Microbe Interact. 22, 1081–1092 (2009).
Lopez, L. et al. Mir1-CP, a novel defense cysteine protease accumulates in maize vascular tissues in response to herbivory. Planta 226, 517–527 (2007).
Islam, A., Mercer, C. F., Leung, S., Dijkwel, P. P. & McManus, M. T. Transcription of biotic stress associated genes in white clover (Trifolium repens L.) differs in response to cyst and root-knot nematode infection. PLoS ONE 10, e0137981, https://doi.org/10.1371/journal.pone.0137981 (2015).
Urwin, P. E., McPherson, M. J. & Atkinson, H. J. Enhanced transgenic plant resistance to nematodes by dual proteinase inhibitor constructs. Planta 204, 472–479 (1998).
Vishnudasan, D., Tripathi, M. N., Rao, U. & Khuruna, P. Assessment of nematode resistance in wheat transgenic plants expressing potato proteinase inhibitor (PIN2) gene. Transgenic Res. 14, 665–675 (2005).
Haegeman, A., Mantelin, S., Jones, J. T. & Gheysen, G. Functional roles of effectors of plant-parasitic nematodes. Gene 492, 19–31 (2012).
Hewezi, T. & Baum, T. J. Manipulation of plant cells by cyst and root-knot nematode effectors. Mol. Plant-Microbe Interact. 26, 9–16 (2013).
Mitchum, M. G. et al. Nematode effector proteins: an emerging paradigm of parasitism. New Phytol. 199, 879–894 (2013).
Chronis, D. et al. A ubiquitin carboxyl extension protein secreted from a plant-parasitic nematode Globodera rostochiensis is cleaved in planta to promote plant parasitism. Plant J. 74, 185–196 (2013).
Lin, B. et al. A novel nematode effector suppresses plant immunity by activating host reactive oxygen species-scavenging system. New Phytol. 209, 1159–1173 (2016).
Liu, J. et al. Molecular characterization of a novel effector expansin-like protein from Heterodera avenae that induces cell death in Nicotiana benthamiana. Sci. Rep. 6, 35677, https://doi.org/10.1038/srep35677 (2016).
Niu, J. et al. Msp40 effector of root-knot nematode manipulates plant immunity to facilitate parasitism. Sci. Rep. 6, 19443, https://doi.org/10.1038/srep19443 (2016).
Masler, E. P. Responses of Heterodera glycines and Meloidogyne incognita to exogenously applied neuromodulators. J. Helminthol. 81, 421–427 (2007).
Bellafiore, S. et al. Direct identification of the Meloidogyne incognita secretome reveals proteins with host cell reprogramming potential. PLoS Pathog. 4, e1000192, https://doi.org/10.1371/journal.ppat.1000192 (2008).
Jacobs, M. & Rubery, P. H. Naturally occurring auxin transport regulators. Science 241, 346–349 (1988).
Yuan, H. et al. New pathotypes of Heterodera avenae (cereal cyst nematode) from winter wheat in Zhengzhou, Henan, China. Australas. Plant Pathol. 39, 107–111 (2010).
Xing, X. et al. Resistance to two species of cereal cyst nematode and evaluation methods in major wheat cultivars from Henan province, China. Acta Agron. Sin. 40, 805–815 (2014).
Wu, L. et al. Characterization of resistance to the cereal cyst nematode in the soft white winter wheat ‘Madsen’. Plant Dis. 100, 679–685 (2016).
Williamson, V., Wang, C. & Lower, S. Application of Pluronic gel to the study of root-knot nematode behaviour. Nematology 11, 453–464 (2009).
Bybd, D. W., Kirkpatrick, T. & Barker, K. R. An improved technique for clearing and staining plant tissues for detection of nematodes. J. Nematol. 15, 142–143 (1983).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Florea, L., Song, L. & Salzberg, S. L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2, 188, https://doi.org/10.12688/f1000research.2-188.v2 (2013).
Schulze, S. K. et al. SERE: single-parameter quality control and sample comparison for RNA-Seq. BMC Genomics 13, 524, https://doi.org/10.1186/1471-2164-13-524 (2012).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106, https://doi.org/10.1186/gb-2010-11-10-r106 (2010).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 25, 3389–3402 (1997).
Conesa, A. & Götz, S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 1–12 (2008).
Thimm, O. et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939 (2004).
Lohse, M. et al. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 37, 1250–1258 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Eddy, S. R. Profile hidden Markov models. Bioinformatics 14, 755–763 (1998).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25, https://doi.org/10.1186/gb-2009-10-3-r25 (2009).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323, https://doi.org/10.1186/1471-2105-12-323 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl. Acids Res. 30, 207–210 (2002).
Chen, C. et al. An ANNEXIN-Like protein from the cereal cyst nematode Heterodera avenae suppresses plant defense. PLoS ONE 10, e122256, https://doi.org/10.1371/journal.pone.0122256 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
The authors are grateful to Biomarker Technologies Co, LTD for providing the transcriptome sequencing technology services, and Prof. Xianchun Xia, Chinese Academy of Agricultural Sciences, for sharing the CFX Connect™ Real-Time PCR Detection System (Bio-Rad, USA). We also thank Dr. Robert Conner, Morden Research and Development Center, Agriculture and Agri-Food Canada and Dr. H. Jian of China Agricultural University for critical review of this manuscript. Financial support provided by the National Natural Science Foundation (31601621), CAAS Innovation Team Project and the National Engineering Laboratory of Crop Molecular Breeding are gratefully appreciated.
Author information
Authors and Affiliations
Contributions
H.J.L., C.L.C. and L.C. conceived and designed the research. C.L.C., Y.P.C., H.J.Z., P.L. and L.Y. performed the experiments. C.L.C., P.P.W., D.Q., J.W.Z. and D.Y. analyzed the data. C.L.C. and H.J.L. wrote the manuscript with contributions from Y.Z. and H.W.L.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, C., Cui, L., Chen, Y. et al. Transcriptional responses of wheat and the cereal cyst nematode Heterodera avenae during their early contact stage. Sci Rep 7, 14471 (2017). https://doi.org/10.1038/s41598-017-14047-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14047-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
