
Abstract
Biotic interactions are often acknowledged as catalysers of genetic divergence and eventual explanation of processes driving species richness. We address the question, whether extreme ecological specialization is always associated with lineage sorting, by analysing polymorphisms in morphologically similar ecotypes of the myrmecophilous butterfly Maculinea alcon. The ecotypes occur in either hygric or xeric habitats, use different larval host plants and ant species, but no significant distinctive molecular traits have been revealed so far. We apply genome-wide RAD-sequencing to specimens originating from both habitats across Europe in order to get a view of the potential evolutionary processes at work. Our results confirm that genetic variation is mainly structured geographically but not ecologically — specimens from close localities are more related to each other than populations of each ecotype from distant localities. However, we found two loci for which the association with xeric versus hygric habitats is supported by segregating alleles, suggesting convergent evolution of habitat preference. Thus, ecological divergence between the forms probably does not represent an early stage of speciation, but may result from independent recurring adaptations involving few genes. We discuss the implications of these results for conservation and suggest preserving biotic interactions and main genetic clusters.
Similar content being viewed by others

Introduction
Biotic interactions represent essential components of ecosystems1. Ecological relationships such as host-parasite, resource-consumer, mutualism or competition, affect the realized ecological niche of species and, consequently, their reaction to habitat changes2,3. The impact of such changes should be higher in ecological specialists, rather than generalists, as specialists are usually more constrained by their specific biotic and abiotic requirements4. Strong interactions and more specific needs may thus lead to faster isolation of populations and development of reproductive barriers. The evolution of different ecotypes may be associated with genetic differentiation, catalyzing lineage divergence and eventually driving the speciation processes5,6,7,8.
Shifts in host-plant associations are found in many herbivorous insects, such as butterflies, in which it is often considered one of the main mechanisms producing their observed diversity9,10,11. However, in order to drive differential selection, the host-plant association character (most importantly adaptation to its defense secondary metabolites) should be inherited, variable, and conferring a local advantage. Whether or not host-plant association shifts occurred repeatedly within a species’ populations, or are associated with a deeper divergence of lineages, remains an open question in most herbivores showing multiple hosts12,13. So far, unraveling the evolutionary history of host-plant shifts has been rather limited due to the low resolution of classical genotyping and sequencing methods in a coalescent framework. However, with the advent of Next-Generation-Sequencing technologies, it is possible to analyse hundreds of loci from non-model species, and establish the extent to which host-plant association is linked with genetic differentiation, across the whole genome with a higher statistical power.
Here, we apply a genome-wide approach to address this longstanding matter for the highly specialized lycaenid butterfly Maculinea alcon (Denis & Schiffermüller, 1775), a «cuckoo» species of Myrmica ant colonies, feeding in the very early stages on a restricted number of plants and then imitating the ant brood and being fed by nurse ants. This strong ecological specialization potentially increases habitat patchiness and makes Maculinea a good model genus for the design of conservation strategies14, for understanding genetic variation structuring15, and for understanding the evolution of social parasitism16.
In Europe, M. alcon sensu lato is known to oviposit predominantly on three main larval host plants (Gentiana cruciata, G. pneumonanthe, and Gentianella rhaetica) and to parasitize at least five Myrmica ant species17,18,19 in contrasting habitats, hygric versus xeric, with few populations occupying a spatially restricted xeric habitat at high elevations. Some authors referred to the hygric ecotypes as “pneumonanthe” and to the xeric as “cruciata”, based on the host plant20; however, here, we assign the names based on their habitat15,21. Furthermore, to distinguish the low and high altitude xeric ecotypes, we refer to the populations at high xeric elevations as the “alpine“ ecotype. At the very few known syntopic localities (Răscruci, Romania and the Bükk Mountains, Hungary), the two low-elevation ecotypes also differ in their phenology, and their flight times only occasionally and partially overlap, limiting possibilities for gene flow20,22,23,24.
Two taxa were previously recognized within this system and often treated as different species: the hygrophilous M. alcon and the xerophylous M. rebeli (Hirschke, 1904), but see Kudrna and Frič25 vs. Tartally et al.26 for nomenclatural issues regarding the taxon rebeli.
It has been debated for nearly two decades whether these ecotypes are associated with genetic variation20,27. Moreover, adults lack unambiguous significantly segregating morphological traits20,28, so that individuals and populations are usually classified based on the larval host plant and locality type. Furthermore, the re-examination of M. alcon populations occurring at high altitude in the Alps revealed a series of differences compared to lowland populations—flight period (being likely caused by phenological differences associated with different elevations), larval host plants, ant hosts, wing pattern and genetic structure20,25,26,29— but these differences do not seem to justify the emergence of two distinct species20. Nevertheless, the different ecotypes and subspecies represent important evolutionary and conservation units20. We consider the existence of (at least) three ecotypes of M. alcon and provide the summary of their characteristics: 1) hygric (occurring in hygric habitats, feeding mainly on G. pneumonanthe, ovipositing mainly on the sepals of the flower buds and parasitising predominantly My. rubra, My. ruginodis and My. scabrinodis), 2) xeric (occurring in xeric localities up to 1,600 m, feeding mainly on G. cruciata, ovipositing predominantly on its leaves, and using as ant hosts mainly My. schencki and My. sabuleti), and 3) “alpine” (occurring at xeric localities above the coniferous tree level and, in the nominotypical population, feeding on Gentianella rhaetica and parasitising My. sulcinodis 26,29,30,31,32).
In addition to the differences mentioned above, the ecotypes can be distinguished by different flight periods (differing by one to a few weeks), which, however, vary slightly between the localities and years and are dependent on the flowering period of the host plant. Despite these ecological differences33, the morphological analyses revealed no or only very slight distinctive traits between the low-altitude ecotypes, with only the “alpine” ecotype differing by the wing pattern20,28,29,33. Similarly, classical molecular phylogenetic and population genetic studies from several localities of their area of occurrence, failed to reveal significant genetic differentiation between the low-altitude ecotypes20,27,34,35 despite their strong ecological differentiation. Minor ecotype-driven genetic differentiation in the few known syntopic/nearly syntopic populations was found, but it was not consistent with ecotype when other nearby populations were included20,30,36. The “alpine” population is usually referred to as «true rebeli». It shows a slight genetic differentiation and combined evidence based on a large number of phenotypic and genotypic markers suggested the existence of a subspecific differentiation20. The fact that no consistent genetic differences among the low-altitude ecotypes have been found so far does not necessarily mean they do not exist. Indeed, the current findings are based only on microsatellites, allozymes and classical sequencing of a reduced set of genes20. Theoretically, fine-grain differences could still be retrieved by performing genome-wide analyses. If differences among ecotypes are to be found, two hypotheses can be considered: (a) each of the forms is monophyletic and there is ongoing divergence between them, or (b) selection at a restricted number of loci occurred repeatedly with several independent evolutions of the ecotypes, indicating convergent evolution of habitat preference. Alternatively, if no genetic differences among ecotypes were found, even with a whole-genome sequencing approach, it would mean that the differences between the forms are likely caused by phenotypic plasticity or epigenetic variation only.
In order to test which scenario is at work in M. alcon, we investigated specimens of three ecotypes spanning the distribution of the species by applying Restriction site associated DNA sequencing (RAD-seq)37 and detecting single nucleotide polymorphisms (SNPs) at randomly distributed loci across the genome. We eventually discuss our results in light of conservation management.
Results
The filtered dataset for 26 individuals (14 xeric, 11 hygric and one “alpine” ecotype – see Methods for detailed explanation) included 1,393 RAD loci. The SNP matrix is deposited in Zenodo (http://doi.org/10.5281/zenodo.997960)38 . In the additional dataset, where Maculinea arion was used as outgroup in order to identify the earliest diverging lineage in M. alcon, 949 RAD loci were retained. This dataset contained many loci that were monomorphic for M. alcon and would have produced a phylogeny with branch lengths equal or close to zero for all M. alcon specimens (due to shallow variability between them). After removing those monomorphic loci, 156 loci remained for the phylogenetic analysis.
Population structure
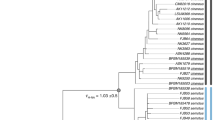
The Evanno method39 used to summarize the STRUCTURE outputs showed K = 4 as the most likely number of clusters. Three out of five runs for K = 4 showed the same pattern. We calculated mean cluster assignments from these three runs and used a threshold of 0.95 sample assignment to a given population (Table S1 and Fig. 1). A clear distinction among three xeric samples from central Italy (Monte Terminillo), three Spanish samples (two hygric and one xeric), three Serbian samples and 12 Romanian samples was observed. Five samples (all hygric, one from Spain, two from Romania and two from Italy) were characterized by large levels of admixture and could not be assigned to any group using the 0.95 assignment criterion.

Sampling localities and phylogenetic tree analysis of 26 samples that belong to three ecotypes of Maculinea alcon (hygric, xeric and “alpine”): (a) Map of sampling localities of the M. alcon samples created in QGIS v.2.12.1; http://qgis.org; (b) Phylogenetic tree based on 1,393 SNPs constructed in PhyML. The xeric individuals are highlighted in yellow and the single specimen of the “alpine” ecotype is underlined. Node numbers indicate bootstrap support and branch length represents the number of substitutions. Colours in each figure (pie-charts in a, vertical bars in b, correspond to the STRUCTURE clusters, i.e. red – Romania, orange – Serbia, blue – Italy, green – Spain. Specimens demonstrating high levels of admixture and not assigned to specific STRUCTURE clusters are shown with a dashed vertical bar.
Phylogenetic analyses
The phylogenetic tree reconstructed with PhyML revealed four main clades, corresponding to the clusters detected by STRUCTURE: Spanish, Italian, Serbian and Romanian (Fig. 1). Hygric and xeric forms were not monophyletic, and only the geographical origin of samples, not ecotypes, was a structuring factor of the phylogeny. Also, the “alpine” sample clustered with the other Italian samples. The phylogenetic tree using M. arion as an outgroup showed much lower node support and poorly resolved clades, most likely a consequence of the matrix containing only 156 loci (data not shown). However, it allowed us to determine that the earliest diverging lineage within M. alcon was the Italian cluster (bootstrap support = 1).
Correlation of genetic distances with geographical and ecological distances
The multiple regression on dissimilarity matrices explained nearly half of the overall variance (R2 = 0.492). Moreover, genetic distances were highly correlated with minimum path distance over land (regression coefficient = 0.700, p = 0.001) while they did not show any correlation with habitat type (regression coefficient = −0.020, p = 0.586).
Loci putatively under selection and Wolbachia infection
The BayeScan analysis revealed two significant SNP outliers suggesting the existence of ecotype-associated selection at those loci (false discovery rate, FDR = 0.07–0.10; loci no 29 and 629 in the dataset; Fig. 2, data shown for FDR = 0.10; Table S2). Exclusion of the “alpine” ecotype produced identical results. A detailed examination of sequence composition revealed that the match was stricter for SNP 29, with all hygric forms bearing a T (homozygous or heterozygous with G) and all xeric forms bearing a G (homozygous or heterozygous with T). For SNP 629, we found two mismatches (i.e., two xeric individuals bearing a homozygote G, instead of A, or A/G, as observed in the rest of xeric specimens; Table 1). When blasting these two contigs using the blastn tool against lepidopteran sequences40, hits against butterfly mRNAs were found: for sequence 29, blastn retrieved a mRNA sequence coding for RING finger and SPRY domain-containing protein 1-like from Papilio polytes (XM_013291785.1), as well as an mRNA coding for elongator complex protein 1 from Papilio machaon (XM_014513286.1); for sequence 629, blastn retrieved an anonymous mRNA from Bombyx mori (AK405939.1) as well as a putative uncharacterized protein from Papilio xuthus (XM_013315886.1).

BayeScan output plotted in R. Two outliers (SNP 29 and 629) show a clear association with M. alcon ecotypes and are potentially under selection (FDR = 0.10).
Wolbachia infection was found in all analyzed samples—32 among 2,116 contigs were of Wolbachia origin. For all 32 contigs, there was only one single allele in all analyzed specimens.
Discussion
In this study, we investigated the genetic coherence of three ecotypes of the emblematic butterfly Maculinea alcon, using Next-Generation-Sequencing methods. Contrary to previous studies, applying classical population genetics methods to a local-scale sampling encompassing two ecotypes, we used whole-genome RAD-seq and investigated the ecological genomics of three ecotypes at a large geographical scale. Furthermore, we applied BayeScan to search for loci under selection. Similarly to previous studies20,27,30,34,35,36, our genome-level SNP analyses did not reveal strong genetic differentiation between the three ecotypes but mainly geographical structuring (see Fig. 1). This suggests that ecological shifts within this species are not associated with lineage sorting and do not represent an early stage of speciation, in contrast to processes occurring in many butterflies and other herbivorous arthropods41,42,43. Our result is in agreement with previous analyses based on spatially more restricted datasets of M. alcon, showing no significant genetic structure associated with the respective ecotypes and higher local genetic relatedness of the two low-altitude ecotypes of M. alcon, including syntopically occurring forms, than of populations of each ecotype from geographically distant localities30,35,44,45. Similarly, two individuals from our study (one hygric - 10-B443 and one xeric - 10-C370), originating from the same locality as studied by Tartally et al.30 did not cluster together, also suggesting a mixed origin of this population. Such a pattern is paralleled by observations in other species of the genus Maculinea, as for instance in M. arion, in which the two phenological forms differ in morphology and ecology but not genetically46.
Initially, it was thought that Maculinea species or forms depend on a single host ant and it was suggested that social parasitism and associated specific life cycle may chiefly affect the evolution, speciation and genetic background of these butterflies32. However, it has been subsequently shown that the usage of ant hosts may differ among localities, and may be adapted to exploiting multiple species or be switched to other species when the preferred host is not present or occurs only in low densities17. Therefore, the hypothesis regarding the influence of the ant host was revisited and some authors suggested that adaptation to the host plant (and corresponding habitat) is more likely to drive the evolution of Maculinea than would do adaptation to the ant species30,46,47. The host plants indeed represent allochronic resources as they usually flower at different times of the year24, because of the microclimatic conditions at the respective xeric and hygric habitats.
Our genome-wide study suggests that hygric and xeric ecotypes are statistically associated with different alleles at at least two loci (29 and 629). In one of the loci (29), homology to two types of mRNAs of the family Papilionidae (related to Maculinea/Lycaenidae), was established: (1) the RING finger and SPRY domain-containing protein is associated with oogenesis in Lepidoptera48 and (2) the elongator complex protein 1 is likely involved in acetylation of histones49 — although this has not been confirmed in invertebrates — and is recognized as a fast-evolving gene in insects50. Ecological differences in occupied biotopes, as well as in host plant phenology may explain the association with discrete differences in the oogenesis process. Overall, our results may be viewed as the footprint of multiple independent evolution of the two ecotypes, involving a few genes only. Similarly to Bereczki et al.20, we rule out a possible effect of Wolbachia infections in the segregation of ecotypes, given that all the samples analyzed were infected by the same bacterial strain.
The evolutionary history of the third ecotype found in an alpine habitat cannot be retrieved from our data, although the fact that it clusters with geographically close populations from Italy may suggest a similar process of recurring evolution. Interestingly, a recent study20 found that specimens from the Styrian Alps (the area from where the “alpine” ecotype has been described) are slightly genetically differentiated from the other populations of M. alcon analyzed. Further investigation including more specimens from this ecotype is needed to identify putative genetic differentiation between this and other ecotypes of M. alcon.
Several studies suggested the need to focus conservation efforts separately for each ecotype (despite the fact that, at least the low altitude ones, do not fulfill the criterion of evolutionary significant units), and indicated a higher vulnerability of the alpine and xeric ecotypes26,30,36. Although we did not find evidence for lineage sorting or early-stage speciation, we found at least two statistically associated markers for the hygric and xeric ecotypes, at least at the scale of the current study. Ecotypic distinctiveness is therefore associated with population-level evolutionary processes rather than with ongoing divergence. This case raises the question how to treat the intra-specific variation from a conservation perspective. The massive use of molecular methods in the last decades enabled researchers to find even minor genetic variations within species or populations, but it also triggered questions concerning the minimal genetic variation and minimal taxonomic unit that should be considered to define conservation units51,52. Since the 1990s the idea that even intraspecific variation should be considered for conservation gained more and more support53. However, levels of within-species genetic differentiation should not be the only criterion used when planning conservation strategies54. Although the M. alcon ecotypes may only differ at few loci, they each have a different ecological strategy and very specific interactions with other species, and thus they may likely differ in the factors affecting their survival, the adequate management, and the impact of their potential extinction in the ecosystems.
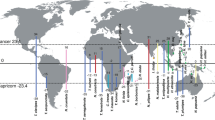
Maculinea species have become a model group not only for the study of parasitism and for insect conservation but also for preserving biotic interactions55, and we strongly support this view. Accordingly, we advocate a strategy aiming at conserving these ecotypes (each associated with specific biotic interactions) as well as the main genetic clusters (four detected in our study) despite the extremely low divergence within the species, this approach being the sole that guarantees that all diversity is protected (see Fig. 3). Moreover, we suggest further mapping of all the potential host ants and plants for these ecotypes, as gaining additional knowledge is crucial for appropriate conservation strategies. Additionally, experiments testing survival rates on non-preferred host plants (and ants) are key to test if natural selection could actually be generating divergence between ecotypes. Similarly, a wider sampling of ecotypes using all the potential variety of host ants (especially the so far undersampled populations using My. rubra and My. ruginodis) would be interesting to further confirm that there is actually no significant genetic divergence driven by the ant host specificity.

Schematic view illustrating optimal conservation focus in M. alcon, targeting ecotypes as well as genetic clusters. Colours indicate the four main lineages (as in Fig. 1), and an X indicates a population from xeric habitat. Only by protecting lineages and ecotypes (and thus associated biotic interaction), we can conserve both genetic and ecological processes. For simplicity we included only the hygric and xeric ecotypes. The map was created in QGIS v.2.12.1; http://qgis.org.
Methods
Field sampling
Twenty-six samples of M. alcon were collected from localities in Spain (3 individuals), Italy (5 individuals), Romania (14 individuals) and Serbia (4 individuals) between 2006 and 2014 (Table S3). The elevation of the localities ranged from 50 to ca. 1,800 m a.s.l. Fourteen specimens belonged to the xeric ecotype, at sites below 1,400 m a.s.l. where the host plant G. cruciata occurred (Fig. 4), 11 to the hygric ecotype at sites below 800 m a.s.l. where the host plant G. pneumonanthe occurred (Fig. 5), and one specimen was tentatively assigned to the “alpine” ecotype (collected at 1,817 m a.s.l. in Monte Chiadenis, Italian Carnic Alps, where the potential host plant Gentianella is present). However, without further study of more individuals from this latter population and comparison to the nominotypic alpine populations of the taxon rebeli, we cannot ascertain that our high-altitude sample belongs to this taxon and thus conservatively denominate it as “alpine” ecotype. One hygric and one xeric sample originated from a well-known locality near Răscruci, Romania, where both ecotypes are syntopic (Fig. S1).

Low-altitude xeric habitat. The inset images show the host plant Gentiana cruciata (left) and its detail with M. alcon eggs (right). Locality: Romania, Caraș-Severin county, near Dobraia, 29. 7. 2007. Photo: Vlad Dincă.

Hygric habitat. The inset images show the host plant Gentiana pneumonanthe (left) and its detail with M. alcon eggs (right). Romania, Brașov county, near Purcăreni, 14. 8. 2007. Photo: Vlad Dincă.
Adult individuals were captured using insect nets and stored in 98% ethanol at −20 °C. The field sampling was done under permits from the relevant authorities. All individuals are stored in the Butterfly Diversity and Evolution Lab at the Institute of Evolutionary Biology (CSIC-UPF) in Barcelona, Spain.
DNA extractions and RAD-sequencing
Extractions
For most of the samples, whole genomic DNA was extracted from the thorax or abdomen, using BioSprint 96 robot with BioSprint 96 DNA Blood Kit (Qiagen, Hilden, Germany). The samples for which only legs were available (because other parts of the body had already been used for other studies) were extracted using the DNeasy Blood & Tissue Kit (Qiagen), in order to maximise the DNA yield. Obtained DNA isolates were diluted or concentrated to reach a final range of 20–50 ng/ul, based on measurements obtained with the PicoGreen dsDNA Assay kit (Thermo Fischer Scientific, Waltham, MA, USA) and FLUOstar Galaxy fluorescence microplate reader (BMG Labtech, Offenburg, Germany).
RAD-library preparation
The RAD-seq library preparation was performed using the double-digestion RAD protocol37, with slight modifications. The whole genomic DNA was digested in 9 ul reaction with 1 U MseI (New England Biolabs-NEB, Ipswich, MA, USA) and 2 U SbfI-HF (NEB) restriction enzymes, 1x CutSmart buffer (NEB) and 6 ul of genomic DNA, at 37 °C for 3 hours. Single strand adapter oligonucleotides were annealed by heating to 95 °C and slow gradual cooling. Adapter ligation was performed at 16 °C for 3 hours in a 20 ul reaction comprising 9 ul of the digested DNA, 0.5 uM of both RAD-P1 and RAD-P2 (Table S4) adapters, 1x T4 ligase buffer and 400 U of T4 DNA ligase (NEB). The reaction products were purified using AMPure XP (Beckman Coulter, Brea, USA), with a reaction/AMPure ratio of 0.856. The purified template DNA was amplified by PCR (denaturation at 98 °C for 30 s, 13 cycles of 98 °C for 20 s, 60 °C for 30 s, 72 °C for 40 s and final extension at 72 °C for 10 minutes). The 10 ul reaction mix contained 1 ul of adaptor-ligated DNA, 0.2 U Q5 Hot-start polymerase, 1x Q5 buffer (both NEB), 0.2 mM of each dNTP, 0.6 uM of each Illumina primer (Table S4). Results of the reaction were verified using standard agarose gel electrophoresis. Samples were pooled and purified using the same protocol as before, with the AMPure ratio of 1.0. Fragments with a length ranging from 300 to 400 bp were selected using the Pippin Prep electrophoresis platform (Sage Science, Beverly, USA) and verified using Fragment Analyzer (Advanced Analytical). Libraries were sequenced on one lane of Illumina HiSeq using single-end protocol, at the Lausanne Genomic Technologies Facility (LGTF).
DNA assembly and SNP calling
Raw sequence reads were demultiplexed using the process_radtags program57 and assembled de novo following the dDocent.FB pipeline (v.2.2.16)58. Final SNP data sets were filtered according to the dDocent filtering tutorial59: i.e. all loci present in less than 50% of the individuals, with Phred quality lower than 30 and minimum depth for a genotype call lower than 3, as well as samples having more than 90% of missing data, were removed. Minor allele frequency and allele balance at heterozygous sites filtering was applied. Additionally, we kept only biallelic loci, and discarded loci with missing information in more than 3 samples and all samples with more than 3 missing loci.
Phylogenetic analyses and population structure
PGD Spider v. 2.1.0.360 was used to convert the resulting VCF file to other formats. Phylogenetic analyses were performed with PhyML v. 3.3.2.061 with the following changes to the default settings: GTR substitution model, estimated base frequencies, best of NNI and SPR tree topology search. Two runs, one without outgroup and one with four samples of M. arion as outgroup were performed. The M. arion samples, collected in the same way as the M. alcon samples, are deposited at the same institution and the same protocols were used for library preparation and calling (in addition to the previous filtering steps, the HWE filter was used).
The genetic clustering of individual genotypes was assessed using the Structure software version 2.3.462, with the following settings: K = 2–10, burn-in = 100,000; number of MCMC repetitions after burn-in = 2,000,000; admixture model, 5 runs for each K. The results were visualized in Pophelper v1.0.1063.
Correlation of genetic distances with geographical and ecological distances
Genetic distances among specimens were calculated in R v. 3.3.164 within RStudio v. 0.99.90365 using Provesti’s distance as implemented in the poppr package v. 2.2.166. A binary distance matrix was calculated for the ecological distance, attributing 0 to pairs of specimens belonging to the same ecotype, and 1 to those from different ecotypes. A geographical distance matrix encompassing the minimum path distance among specimens over land was produced—the minimum path distance was calculated by using the costDistance function as implemented in the gdistance R package (v. 1.2-1)67. The relative contribution of ecological and minimum path distances in explaining overall genetic distances was tested by multiple regression of dissimilarity matrix by using the MRM function implemented in the ecodist R package (v. 1.2.2)68. Since the variables were not normally distributed and the relationships not necessarily linear, a Spearman correlation was applied. P-values were calculated based on 1000 permutations.
Loci putatively under selection and detection of Wolbachia bacteria
Candidate loci diagnostic of ecotypes were investigated with BayeScan v. 2.141. The dataset was divided into three groups according to the three forms (xeric, “alpine”, and hygric) and two runs including and excluding the single “alpine” sample were performed. The results were plotted in R v. 3.3.164 using the script plot_R.r implemented in BayeScan. FDR 0.5–0.10 was used, as the 0.10 threshold for q-values in BayeScan is more stringent than the same threshold for p-values in classical statistics41.
Additionally, because Maculinea butterflies are very often infected by Wolbachia bacteria, which is known to manipulate reproduction and to affect genetic variation structuring69,70, contigs obtained by dDocent for all specimens were blasted against available Wolbachia sequence data deposited in GenBank with the blastn tool40.
Data Availability
The datasets supporting the conclusions are available in Zenodo (http://doi.org/10.5281/zenodo.997960).
References
Smith, S. A. & Donoghue, M. J. Rates of molecular evolution are linked to life history in flowering plants. Science 322, 86–89 (2008).
Pfenninger, M., Salinger, M., Haun, T. & Feldmeyer, B. Factors and processes shaping the population structure and distribution of genetic variation across the species range of the freshwater snail Radix balthica (Pulmonata, Basommatophora). BMC Evol. Biol. 11, 135 (2011).
Wimp, G. M. & Whitham, T. G. Biodiversity consequences of predation and host plant hybridization on an aphid–ant mutualism. Ecology 82, 440–452 (2001).
Charrette, N. A., Cleary, D. F. R. & Mooers, A. O. Range-restricted, specialist Bornean butterflies are less likely to recover from ENSO-induced disturbance. Ecology 87, 2330–2337 (2006).
Hawthorne, D. J. & Via, S. Genetic linkage of ecological specialization and reproductive isolation in pea aphids. Nature 412, 904–907 (2001).
Bernatchez, L., Chouinard, A. & Lu, G. Integrating molecular genetics and ecology in studies of adaptive radiation: whitefish, Coregonus sp., as a case study. Biol. J. Linn. Soc. 68, 173–194 (1999).
Le Moan, A., Gagnaire, P.-A. & Bonhomme, F. Parallel genetic divergence among coastal–marine ecotype pairs of European anchovy explained by differential introgression after secondary contact. Mol. Ecol. 25, 3187–3202 (2016).
Bergelson, J., Stahl, E., Dudek, S. & Kreitman, M. Genetic Variation Within and Among Populations of Arabidopsis thaliana. Genetics 148, 1311–1323 (1998).
Downey, M. H. & Nice, C. C. A role for both ecology and geography as mechanisms of genetic differentiation in specialized butterflies. Evol. Ecol. 27, 565–578 (2013).
Ferrer-Paris, J. R., Sánchez-Mercado, A., Viloria, Á. L. & Donaldson, J. Congruence and diversity of butterfly-host plant associations at higher taxonomic levels. PLOS ONE 8, e63570 (2013).
Suchan, T. & Alvarez, N. Fifty years after Ehrlich and Raven, is there support for plant–insect coevolution as a major driver of species diversification? Entomol. Exp. Appl. 157, 98–112 (2015).
Stireman, J. O. et al. Climatic unpredictability and parasitism of caterpillars: Implications of global warming. Proc. Natl. Acad. Sci. USA 102, 17384–17387 (2005).
Craft, K. J. et al. Population genetics of ecological communities with DNA barcodes: An example from New Guinea Lepidoptera. Proc. Natl. Acad. Sci. 107, 5041–5046 (2010).
Mouquet, N. et al. Conserving community modules: A case study of the endangered lycaenid butterfly Maculinea alcon. Ecology 86, 3160–3173 (2005).
Pellissier, L., Litsios, G., Guisan, A. & Alvarez, N. Molecular substitution rate increases in myrmecophilous lycaenid butterflies (Lepidoptera). Zool. Scr. 41, 651–658 (2012).
Elmes, G. W., Thomas, J. A. & Wardlaw, J. C. Larvae of Maculinea rebeli, a large-blue butterfly, and their Myrmica host ants: wild adoption and behaviour in ant-nests. J. Zool. 223, 447–460 (1991).
Als, T. D., Nash, D. R. & Boomsma, J. J. Geographical variation in host–ant specificity of the parasitic butterfly Maculinea alcon in Denmark. Ecol. Entomol. 27, 403–414 (2002).
Sielezniew, M. & Stankiewicz, A. Differences in the development of the closely related myrmecophilous butterflies Maculinea alcon and M. rebeli (Lepidoptera: Lycaenidae). EJE 104, 433–444 (2007).
Thomas, J. A. et al. Mimetic host shifts in an endangered social parasite of ants. Proc. Biol. Sci. 280, 20122336 (2013).
Bereczki, J., Pecsenye, K., Varga, Z., Tartally, A. & Tóth, J. P. Maculinea rebeli (Hirschke) – a phantom or reality? Novel contribution to a long-standing debate over the taxonomic status of an enigmatic Lycaenidae butterfly. Syst. Entomol. n/a-n/a, https://doi.org/10.1111/syen.12259 (2017).
Pierce, N. E. et al. The ecology and evolution of ant association in the Lycaenidae (Lepidoptera). Annu. Rev. Entomol. 47, 733–771 (2002).
Goia, M. & Dincă, V. The structure and distribution of the butterfly fauna (Hesperioidea & Papilionoidea) in the surroundings of Cluj-Napoca and current aspects of the anthropic influences on their environment. Buletin informare entomologică 139–197 (2006).
Rákosy, L. & Vodă, R. Distribution of Maculinea genus in Romania. Entomol rom 9–17 (2008).
Timuş, N., Craioveanu, C., Sitaru & Rákosy, L. Differences in adult phenology, demography, mobility and distribution in two syntopic ecotypes of Maculinea alcon (cruciata vs. pneumonanthe) (Lepidoptera: Lycaenidae) from Transilvania (Romania). Entomologica romanica 21–30 (2013).
Kudrna, O. & Frič, Z. On the identity and taxonomic status of Lycaena alcon rebeli Hirschke, 1905 - a long story of confusion and ignorance resulting in the fabrication of a ‘ghost species’ (Lepidoptera: Lycaenidae). Nachrichten Entomol. Ver. Apollo 34, 117–124 (2013).
Tartally, A., Koschuh, A. & Varga, Z. The re-discovered Maculinea rebeli (Hirschke, 1904): Host ant usage, parasitoid and initial food plant around the type locality with taxonomical aspects (Lepidoptera, Lycaenidae). ZooKeys 25–40, https://doi.org/10.3897/zookeys.406.7124 (2014).
Als, T. D. et al. The evolution of alternative parasitic life histories in large blue butterflies. Nature 432, 386–390 (2004).
Steiner, F. M. et al. Maculinea alcon and M. rebeli (Insecta: Lepidoptera: Lycaenidae) – one or two Alcon Blues? Larval cuticular compounds and egg morphology of East Austrian populations. Ann. Naturhistorischen Mus. Wien Ser. B Für Bot. Zool. 107, 165–180 (2005).
Habeler, H. Die subalpin-alpinen Lebensräume des Bläulings Maculinea rebeli (Hirschke, 1904) in den Ostalpen (Lepidoptera, Lycaenidae). Joannea Zool. 143–164 (2008).
Tartally, A., Kelager, A., Fürst, M. A. & Nash, D. R. Host plant use drives genetic differentiation in syntopic populations of Maculinea alcon. PeerJ 4, e1865 (2016).
Czekes, Z. et al. Differences in oviposition strategies between two ecotypes of the endangered myrmecophilous butterfly Maculinea alcon (Lepidoptera: Lycaenidae) under unique syntopic conditions. Insect Conserv. Divers. 7, 122–131 (2014).
Thomas, J. A., Elmes, G. W., Wardlaw, J. C. & Woyciechowski, M. Host specificity among Maculinea butterflies in Myrmica ant nests. Oecologia 79, 452–457 (1989).
Pech, P., Fric, Z., Konvička, M. & Zrzavý, J. Phylogeny of Maculinea blues (Lepidoptera: Lycaenidae) based on morphological and ecological characters: evolution of parasitic myrmecophily. Cladistics 20, 362–375 (2004).
Fric, Z., Wahlberg, N., Pech, P. & Zrzavý, J. Phylogeny and classification of the Phengaris–Maculinea clade (Lepidoptera: Lycaenidae): total evidence and phylogenetic species concepts. Syst. Entomol. 32, 558–567 (2007).
Bereczki, J., Pecsenye, K., Peregovits, L. & Varga, Z. Pattern of genetic differentiation in the Maculinea alcon species group (Lepidoptera, Lycaenidae) in Central Europe. J. Zool. Syst. Evol. Res. 43, 157–165 (2005).
Sielezniew, M. et al. Differences in genetic variability between two ecotypes of the endangered myrmecophilous butterfly Phengaris (=Maculinea) alcon – the setting of conservation priorities. Insect Conserv. Divers. 5, 223–236 (2012).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLOS ONE 7, e37135 (2012).
Koubínová, D. et al. Genomics of extreme ecological specialists: multiple convergent evolution but no genetic divergence between ecotypes of Maculinea alcon butterflies [Data set]. Scientific Reports. Zenodo. http://doi.org/10.5281/zenodo.997960 (2017).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Evans, L. M., Allan, G. J., Meneses, N., Max, T. L. & Whitham, T. G. Herbivore host-associated genetic differentiation depends on the scale of plant genetic variation examined. Evol. Ecol. 27, 65–81 (2012).
Hernández-Roldán, J. L. et al. Integrative analyses unveil speciation linked to host plant shift in Spialia butterflies. Mol. Ecol. 25, 4267–4284 (2016).
Bereczki, J., Pecsenye, K. & Varga, Z. Geographical versus food plant differentiation in populations of Maculinea alcon (Lepidoptera: Lycaenidae) in Northern Hungary. Eur. J. Entomol. 103, 725–732 (2006).
Pecsenye, K. et al. Genetic differentiation among the Maculinea species (Lepidoptera: Lycaenidae) in eastern Central Europe. Biol. J. Linn. Soc. 91, 11–21 (2007).
Bereczki, J., Tóth, J. P., Sramkó, G. & Varga, Z. Multilevel studies on the two phenological forms of Large Blue (Maculinea arion) (Lepidoptera: Lycaenidae). J. Zool. Syst. Evol. Res. 52, 32–43 (2014).
Steiner, F. M. et al. Host specificity revisited: New data on. Myrmica. J. Insect Conserv. 7, 1–6 (2003).
Carter, J.-M. et al. Unscrambling butterfly oogenesis. BMC Genomics 14, 283 (2013).
Hawkes, N. A. et al. Purification and characterization of the human elongator complex. J. Biol. Chem. 277, 3047–3052 (2002).
Senatore, G. L., Alexander, E. A., Adler, P. H. & Moulton, J. K. Molecular systematics of the Simulium jenningsi species group (Diptera: Simuliidae), with three new fast-evolving nuclear genes for phylogenetic inference. Mol. Phylogenet. Evol. 75, 138–148 (2014).
Hedenäs, L. Intraspecific diversity matters in bryophyte conservation – internal transcribed spacer and rpl16 G2 intron variation in some European mosses. J. Bryol. 38, 173–182 (2016).
Newton, A. C., Allnutt, T. R., Gillies, A. C. M., Lowe, A. J. & Ennos, R. A. Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol. Evol. 14, 140–145 (1999).
Taberlet, P., et al.Genetic diversity in widespread species is not congruent with species richness in alpine plant communities. Ecol Lett, 15, 1439–1448 (2012).
Vodă, R. et al. Historical and contemporary factors generate unique butterfly communities on islands. Sci. Rep. 6, srep28828 (2016).
Thomas, J. A., Simcox, D. J. & Clarke, R. T. Successful conservation of a threatened Maculinea butterfly. Science 325, 80–83 (2009).
Rohland, N. & Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. gr.128124.111, https://doi.org/10.1101/gr.128124.111 (2012).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Puritz, J. B., Hollenbeck, C. M. & Gold, J. R. dDocent : a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2, e431 (2014).
Puritz, J. B. dDocent Filtering Tutorial (2016), https://github.com/jpuritz/dDocent/blob/master/tutorials/Filtering%20Tutorial.md.
Lischer, H. E. L. & Excoffier, L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Guindon, S. et al. New algorithms and methods to estimate Maximum-Likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Francis, R. M. pophelper: an r package and web app to analyse and visualize population structure. Mol. Ecol. Resour doi:https://doi.org/10.1111/1755-0998.12509 (2016).
R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2016).
RStudio Team. RStudio: Integrated Development for R. (RStudio, Inc., Boston, MA, 2015).
Kamvar, Z. N., Brooks, J. C. & Grünwald, N. J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Plant Genet. Genomics 6, 208 (2015).
R Package gdistance: Distances and Routes on Geographical Grids | van Etten | Journal of Statistical Software. doi:https://doi.org/10.18637/jss.v076.i13.
Goslee, S. C. & Urban, D. L. The ecodist package for dissimilarity-based analysis of ecological data | Goslee | J Stat Soft (2007). Available at: https://www.jstatsoft.org/article/view/v022i07. (Accessed: 15th December 2016).
Werren, J. H., Baldo, L. & Clark, M. E. Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751 (2008).
Ritter, S. et al. Wolbachia infections mimic cryptic speciation in two parasitic butterfly species, Phengaris teleius and P. nausithous (Lepidoptera: Lycaenidae). PLOS ONE 8, e78107 (2013).
Acknowledgements
We thank Keith Harshman and his team from the Lausanne Genomic Technologies Facility (LGTF) for the sequencing job and their assistance during library preparation. We also thank Juan Carlos Vicente, Sergio Montagud and Antonio Engra for help in collecting samples, and Miloš Popović and Boyan Zlatkov for information regarding the larval food plant of some specimens included in this study. The work was supported by the Swiss National Science Foundation (grant PP00P3_144870), a Sciex PostDoc Fellowship (14.002), BNF project of the Bern University, Switzerland, the Spanish Ministerio de Economía y Competitividad CGL2016-76322-P (AEI/FEDER, UE), and the European Union’s Seventh Framework programme for research and innovation under the Marie Skłodowska-Curie grant agreement No 609402-2020 researchers: Train to Move (T2M) postdoctoral fellowship to R. Vodă. V. D. was supported by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme (project no. 625997). Access to computing and storage facilities owned by parties and projects contributing to the National Grid Infrastructure MetaCentrum provided under the programme “Projects of Projects of Large Research, Development, and Innovations Infrastructures” (CESNET LM2015042), is greatly appreciated.
Author information
Authors and Affiliations
Contributions
D.K. co-designed the study, performed the analyses and wrote the manuscript; V.D., L.D., R. Vodă collected the samples, collaborated on the preparation of the manuscript and assisted in data analyses (L.D. - distance analyses); T.S. assisted in laboratory work and data analyses; R. Vila and N.A.: designed the study, collected the samples (R. Vila), assisted in preparing the manuscript and data analyses. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koubínová, D., Dincă, V., Dapporto, L. et al. Genomics of extreme ecological specialists: multiple convergent evolution but no genetic divergence between ecotypes of Maculinea alcon butterflies. Sci Rep 7, 13752 (2017). https://doi.org/10.1038/s41598-017-12938-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12938-8
This article is cited by
-
Towards a genomic resolution of the Phengaris alcon species complex
Conservation Genetics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
