
Abstract
The temperate bamboos (tribe Arundinarieae, Poaceae) are strongly supported as monophyly in recent molecular studies, but taxonomic delineation and phylogenetic relationships within the tribe lack resolution. Here, we sampled 39 species (36 temperate bamboos and 3 outgroups) for restriction-site associated DNA sequencing (RAD-seq) with an emphasis on Phyllostachys clade and related clades. Using the largest data matrix for the bamboos to date, we were able to infer phylogenetic relationships with unparalleled resolution. The Phyllostachys, Shibataea, and Arundinaria clades defined from plastid phylogeny, were not supported as monophyletic group. However, the RAD-seq phylogeny largely agreed with the morphology-based taxonomy, with two clades having leptomorph rhizomes strongly supported as monophyletic group. We also explored two approaches, BWA-GATK (a mapping system) and Stacks (a grouping system), for differences in SNP calling and phylogeny inference. For the same level of missing data, the BWA-GATK pipeline produced much more SNPs in comparison with Stacks. Phylogenetic analyses of the largest data matrices from both pipelines, using concatenation and coalescent methods provided similar tree topologies, despite the presence of missing data. Our study demonstrates the utility of RAD-seq data for elucidating phylogenetic relationships between genera and higher taxonomic levels in this important but phylogenetically challenging group.
Similar content being viewed by others
Introduction
The temperate bamboos (tribe Arundinarieae, Bambusoideae, Poaceae) are a clade of diverse taxa containing 32 genera and about 600 species1,2,3,4. Bamboos in this tribe have considerable ecological and economic value as most of species are major components of the subtropical and temperate forests in eastern and southeastern Asia. Many bamboo species are important sources of food, pulp manufacture, and materials for housing construction and artwork, such as Moso bamboo (Phyllostachys edulis)5. With highly diversified morphology and lack of flowering characters due to long vegetative periods, this tribe is notorious for the complicated taxonomy1, 6.
Although unequivocal sets of characters for classifying species and genera have not been identified, monophyly of temperate bamboos has been strongly supported in many molecular studies7,8,9,10,11,12. According to biogeographic analyses13, Arundinarieae diversified during the middle to late Miocene, and followed by a rapid radiation especially within the clades containing largest genera and species. Such recent origin might make the temperate bamboos undergo very little molecular variation14 and result in the intricate phylogenetic relationships within Arundinarieae. Based on broad sampling and eight non-coding plastid regions, Zeng et al.15 divided it into ten major lineages, and confirmed that most genera within this tribe were highly heterogeneous and incongruent with current taxonomic circumscriptions. Subsequently, two additional clades were recovered in Yang et al.16 and Zhang et al.13, thus twelve lineages in all were currently recognized in Arundinarieae based on plastid phylogeny. The evolutionary relationships among lineages were almost resolved but within lineages the resolution remained low12, 17, mainly due to the extremely slow molecular evolutionary rate of plastid DNA15. While in phylogeny based on nuclear DNA marker GBSSI 18, 13 lineages were resolved and incongruence was revealed between the plastid and nuclear trees, indicating different evolutionary trajectories. Moreover, the nuclear tree provided a poorly resolved phylogenetic relationship within Arundinarieae16, 19 because of insufficient informative characters, and more nuclear DNA markers were suggested to be needed to infer the evolutionary history of it.
Next-generation sequencing has recently been used to address evolutionary problems in the Bambusoideae17, 20, 21. Ma et al.17 and Attigala et al.21 used plastid genome sequencing to resolve the phylogenetic relationships in Arundinarieae and obtained robust relationships among the major clades. However, studies of Arundinarieae employing next-generation sequencing have mainly focused on the plastid genome, few involved the nuclear genome. By analyzing whole-genome datasets from the Poaceae, one of them identified 74 putative nuclear single copy orthologous genes for phylogenetic studies of temperate bamboos22, but this method is labor-intensive. With the development of high-throughput sequencing technologies, reduced-representation methods have revolutionized the fields of phylogeography, population genomics, and phylogenomics by providing high-resolution genomic data for non-model organisms at a reasonable cost23,24,25, such as restriction site-associated DNA sequencing (RAD-seq)26. By reducing genomic representation, RAD-seq can identify tens of thousands of single nucleotide polymorphism (SNP) markers, and address the issue of phylogenetic reconstruction with unprecedented power and precision, even with limited, or no reference genome27,28,29,30,31,32. Many empirical studies have employed this method on plants and animals to reconstruct their phylogenetic relationships and demonstrated its power on phylogenetic resolution in non-model organisms30, 32,33,34,35. Therefore, RAD-seq provides an opportunity to solve the contentious relationships of Arundinarieae from nuclear evolutionary trajectory.
Mapping and grouping are two SNP-calling systems for obtaining large numbers of SNPs from RAD sequencing data. In mapping, RAD sequencing reads are aligned to a reference genome and genotyped using standard tools, such as BWA36 and Stampy37, and the output alignments are supported by several generic SNP callers such as Genome Analysis Tool Kit38 (GATK) and SAMtools39. In grouping, RAD sequencing reads are used de novo, generating large marker sets where no reference genome is available. Several tools have been developed to produce RAD marker sets de novo, including Stacks40 and RADtools41. Pan et al.42 tested and compared SNP calling using the UNEAK, Stacks and bowtie2-GATK pipelines for genotyping-by-sequencing (GBS) data in nine individuals of the three pine species, and found that both Stacks and bowtie2-GATK were more efficient than UNEAK for SNP calling. However, to date, there has been no comparison of the performance of mapping and grouping in terms of the variants obtained and downstream phylogenetic analysis of RAD sequencing data.
In a pilot study, we elucidated the phylogenetic relationship between two closely related species in temperate bamboos using RAD sequencing43. However, the utility of RAD-seq in building Arundinarieae phylogeny when more taxa sampled remains elusive. The Phyllostachys clade (clade V) is the largest clade in Arundinarieae, with ca. 16 genera and more than 330 species which comprises about 50% of the genera and more than 70% of the species of the tribe15, 44. The clade is remarkable for combining high morphological diversity with low plastid DNA variability. Therefore we adopt broad taxon sampling with an emphasis on Phyllostachys clade and related clades to elucidate their phylogenetic relationships, which would act as a valuable starting point for reconstructing a comprehensive phylogenetic framework for the whole tribe. The primary goals of this investigation were (1) to test the utility of RAD data in providing a high-resolution estimate of the phylogenetic relationships among temperate bamboos, where a broad sample was examined; and (2) to evaluate and compare mapping and grouping systems for SNP calling and phylogeny inference based on RAD sequencing data.
Results
RAD sequencing
We obtained an average of 11.0 million paired-end reads of 82–86 bp per sample and approximately 615 million reads in all after barcode trimming, cleaning and quality checking. Details of the sequencing output are provided in Supplementary Table S1.
Data matrices from mapping system
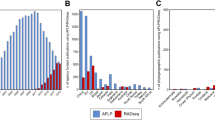
Using BWA, we were able to map between 6.58% (Guadua angustifolia) and 99.12% (Phyllostachys edulis) (mean = 57.55%) of the RAD tags to the genomic scaffold sequences. The reference-based GATK HaplotypeCaller identified 6,602,640 raw variants. Filtering for a coverage of 10 to 500 resulted in 5,934,688 variants being retained. Only 1390 variable sites were obtained when we set the strictest limit (0) for ‘number of no-called samples’ (matrix s56, requiring all the 56 samples to have data at each locus). In order to maximize the number of loci that could be analyzed we applied multiple threshold values for the ‘number of no-called samples’ (NCC) = 1, 5, 10 in further analyses. For example, matrix s55 (NCC = 1, requiring 55 of the 56 samples to have data at each locus) contains only 1.34% missing data, but only contains 5759 variable sites (Table 1). The characteristics of the data matrices produced in this method are outlined in Table 1. The four data matrices ranged from 2357 bp (1390 variable sites and 489 informative sites in s56) to 400,796 bp (272,254 variable sites and 97,104 informative sites in s45), and the proportion of missing data in the matrices ranged from 0 (s56) to 13.08% (s45) (Table 1).
Data matrices from grouping system
As Stacks only infers loci from forward reads, reverse reads were not included for genotyping. Setting the minimum coverage depth to 5 in ustacks produced between 39,291 (Indosasa sinica) and 496,805 (Dendrocalamus latiflorus) putative loci, with an average of 186,392 loci (Supplementary Table S2). Increasing the minimum stack depth to 10 produced between 6003 (I. sinica) and 308,783 (P. edulis) putative loci, with an average 57,507 (Supplementary Table S2). With the options -m 5 and 10, a total of 4,715,488 and 972,878 putative loci (cut sites), respectively, were produced among the 56 samples.
In this method, we generated a total of 14 SNP matrices (-m 5, 10 and -p 10, 15, 20, 25, 35, 45, 55) outlined in Table 2, which ranged in total sequence length from 0 bp to 362,823 bp (m = 5) and 9 bp to 42,788 bp (m = 10). Applying higher values of minimum stack depth tended to decrease the overall concatenated matrix length. The proportion of missing data in these matrices ranged from 0 to 72.64% at m = 5 and from 1.79% to 73.87% at m = 10 (Table 2). The coverage values are high, indicating that the sequencing is unlikely to be the main contributor to the high levels of missing data that we observed. One of the outgroups used in our study (G. angustifolia) consistently had the highest proportion of missing data. This may be caused by high levels of molecular divergence between G. angustifolia and the rest of the species analyzed.
Phylogenetic inference
We estimated phylogenetic trees (Fig. 1) for the temperate bamboos with nine representative data matrices selected from total eighteen matrices generated from both genotyping systems, which contained varying levels of missing data (Tables 1 and 2). Among the trees, eight monophyletic lineages can be discerned within the tribe with strong support. The eight lineages were designated: Drepanostachyum + Himalayacalamus, Gaoligongshania, Ferrocalamus + Indocalamus, Chimonobambusa, Sino-Japanese lineage, Chimonocalamus + Fargesia sect. Ampullares, alpine Bashania + Fargesia, and Yushania + Fargesia (detailed in Fig. 2).

Tree resolution increases as data are added to the phylogenetic analysis. The different colors represent eight major clades (see Fig. 2 for key). Values on branches are bootstrap support from 200 bootstrap replicates using RAxML’s rapid bootstrap algorithm. Topologies shown are the best tree from a full ML search. Bootstrap support values within major clades are not shown.

Phylogeny of the temperate bamboos based on an ML analysis (matrix s45: 400,796 base pairs, and 272,254 variable sites). Nodes with posterior probalilities ≥95 are unlabeled. Rhizome type is indicated by branch thickness: thick lines = pachymorph rhizomes and thin lines = leptomorph rhizomes.
For the reference-based genotyping data sets, phylogenetic analysis using the smallest data set (s55; 9416 bp; Table 1) produced topologies with low bootstrap support, and did not form eight major clades (Fig. 1 and Table 3). This pattern changed dramatically as more data were added to the analysis (Fig. 1). Analyses with data matrices of at least 147,524 bp resolved the eight principal lineages as monophyly with a bootstrap support of 100%, with one exception. The Gaoligongshania lineage had a bootstrap support of 79% and 87% in the s50 and s45 data matrix, respectively (Fig. 1 and Table 3).
Similar patterns were observed in the de novo assembly genotyping data sets. As the data matrix increased in size, the resolution of these clades also increased, along with the bootstrap support of the internal branches of the trees (Figs 1 and 3). Decreasing the minimum stack depth dramatically increased the size of the data matrix. Phylogenetic analysis using the smallest data set (m = 10, p = 20; 6356 SNPs; Table 2) produced topologies with very low bootstrap support, and only five of the eight lineages described above formed clades, with bootstrap supports of 87–100% (Fig. 1). The topology changed as more data were added. In the analysis of data matrix p15 (m = 5), the eight principal lineages were monophyletic, with a bootstrap support of 100%, with the exception of the Gaoligongshania lineage, which had a bootstrap support of 80%. The relatively low bootstrap support for the monophyly of this clade was not found in other analyses. For data matrices p15 (m = 5) and p10 (m = 5), we found only minimal differences in the supporting values at a few of the more derived branches. We noted there was a difference in topology in the derived Gaoligongshania lineage and Drepanostachyum + Himalayacalamus lineage between the largest data matrices p10 (m = 5, Stacks pipeline) and s45 (BWA + GATK pipeline) (Fig. 1).

The resolution of phylogenetic trees increases as a function of data matrix size. The colors indicate the proportion of branches with a bootstrap support above a given threshold.
We compared the support values for all shared bipartitions between data matrices with different proportions of missing data (Fig. 3). The total number of well-supported branches on the tree generally increased as a negative exponential function of the number of base pairs in the data matrix, with two exceptions (Fig. 3). In analyses of p15 (m = 5), the number of branches with 75% or greater support decreased slightly, while in p10 (m = 5) the number of branches with 50% or greater support slightly decreased. Notably, the tree length estimates obtained using the de novo assembly genotyping data matrices were substantially higher in comparison to those from the reference-based genotyping data matrices (Table 4).
Phylogenomics of Temperate bamboos
Summaries of the phylogenetic trees from analyses of the nine data matrices using RAxML are provided in the Table 4. The ML phylogenetic analysis of the largest data matrix (s45) is shown in Fig. 2. Neither the Shibataea (IV) nor Arundinaria (VI) clades formed monophyletic groups, nor did Phyllostachys (V). The current data provided a test of the monophyly of six genera (i.e., those for which at least two species were sampled). Of these, only three were strongly supported as monophyly (Chimonocalamus, Chimonobambusa, and Alpine Bashania), while three appear to be paraphyletic or polyphyletic (Fargesia, Yushania, and Phyllostachys), which corroborates earlier studies16, 18, 44. At the species level, Chimonobambusa ningnanica, Bashania fangiana, and all of the species within Yushania were resolved as monophyletic. Intrageneric relationships in Fargesia were unresolved, and none of the sampled species appeared to be monophyletic.
The species trees estimated using SVDquartets were largely similar to each other with four different data matrices, with a few notable exceptions (Fig. 4). First, the phylogenetic placement of the Sino-Japanese and Chimonocalamus + Fargesia sect. Ampullares lineages were not consistent. Second, the genera Ferrocalamus and Indocalamus did not form a clade when the smallest data matrix was used, but the two genera formed a clade in all other data matrices. Finally, the relationships within the Yushania + Fargesia and Sino-Japanese lineages varied across different data matrices. We noted that the phylogenetic placement of Drepanostachyum + Himalayacalamus and Gaoligongshania lineages were consistent across all data matrices. The species tree estimated using the largest matrix was largely similar to the expected ML phylogeny, except for some short internal branches in the Yushania + Fargesia lineage.

Species trees from temperate bamboos estimated using SVDquartets for data matrices s55, s45, p20 (m = 5), p10 (m = 5). The different colors represent eight major clades (see Fig. 2 for key). Bootstrap values (from 100 replicates) are shown on nodes.
Discussion
The temperate bamboos have long been considered a complex and taxonomically difficult group. Previous phylogenetic studies mainly based on plastid DNA and divided Arundinarieae into twelve clades, but few was known about the nuclear evolutionary history due to unusable of most single or low copy genes in bamboos and the low molecular variation14, 19, 45. In this study, we sampled 39 species and the results of our phylogenetic analyses with RAD-seq data provided new insights into the phylogenetic relationships of Arundinarieae, mainly involving the Phyllostachys clade and its closely related clades. Eight major lineages were identified in Arundinarieae with high support, two of which (Drepanostachyum + Himalayacalamus, and Chimonobambusa) are consistent with previous results based on nuclear genes16, 18, while others were recovered here for the first time. Such clear relationships among these clades of Arundinarieae demonstrated that RAD-seq is a powerful tool for solving this intricate group by providing tens of thousands of SNPs scatter in the whole genome.
Compared with plastid phylogeny, many inconsistencies were revealed in our study. The Phyllostachys, Shibataea, and Arundinaria clades defined from plastid phylogeny were not supported as monophyly anymore. Instead, they were separated into several different parts in the nuclear tree. Incomplete lineage sorting and/or hybridization (introgression) could be the cause for gene tree conflicts in temperate bamboos, as summarized by Zhang et al.18. In the plastid phylogeny, division of lineages was incompatible with traditional classification based on morphology, and the genera delimited based on these characters were not monophyletic, which has confused taxonomists for a long time. However, our nuclear phylogeny agrees with the morphology-based taxonomy to some extent, which has also been observed in other organisms46, 47. Two clades with leptomorph rhizomes were strongly supported as monophyletic (Fig. 2). One included the Sino-Japanese, Ferrocalamus + Indocalamus and Chimonobambusa, among them Chimonobambusa has leptomorph rhizomes and highly reduced culm-sheath blades. This genus was resolved as monophyletic group which is congruent with morphologic delimitation. The other was the alpine Bashania, three species from high mountains formed a strongly supported group. This result supported the combination of new genus Sarocalamus described by Stapleton et al.48 with a distribution from the eastern Himalayas to the Sichuan Province of China.
Fargesia and Yushania are two genera of alpine bamboos14, mainly distributed in the Hengduan Mt. They all produced pachymorph rhizomes and semelauctant synflorescences, and the length of culm-necks and the type of synflorescence are important characters for genera delimitation. However, such delimitation was confused because of the variability of the length of culm necks and the open degree of spathe subtending observed in the field. Whether to recognize Fargesia and Yushania as genera was still in debate. In our phylogeny, Yushania violascens was nested with Fargesia species with high bootstrap support. These taxa have intermediate morphology and hybridization may be a likely explanation for these taxonomic uncertainties in this group. Thus, Yushania might be a good genus for traditional taxonomists. With respect to Fargesia, it was separated into three parts. The Fargesia section Ampullares, as defined by Yi49 was resolved as monophyletic here and supported by morphological characters. All individuals in this section possess semi-orbicular (broadly ovoid) culm buds and numerous and congested branches. While Fargesia fractiflexa was transferred into Drepanostachyum as Drepanostachyum fractiflexum because of many branches with underdeveloped secondary branches in Flora of China6. According to the nuclear phylogeny, F. fractiflexa had a close relationship with Fargesia, and it was more reasonable to transfer D. fractiflexum back into Fargesia. Fargesia caduca was found in the South Yunnan of China and placed in section Fargesia because of bud characters49. As we observed in the field, F. caduca indeed exhibited the complete range of variation from ovoid to lanceolate buds, and might be an intermediate species.
Additionally, our work highlights the fact that different conclusions can be reached in phylogenetic inference using data sets generated from two different pipelines. Using data generated from 56 samples representing 36 species of temperate bamboo, we observed significant inconsistencies between the Stacks and BWA-GATK pipelines. These inconsistencies in the SNP datasets led to strong disagreement in downstream phylogenetic inferences. Both Stacks and BWA-GATK were efficient at SNP calling, identifying 2,255,239 and 5,934,688 SNPs in the samples, respectively. For the same level of missing data, the BWA-GATK pipeline produced much more SNPs in comparison with Stacks. One possible reason is that BWA-GATK used both forward and reverse reads, while Stacks only used forward ones. Moreover, compared to the BWA-GATK pipeline, Stacks suffered from a higher rate of missing data, with up to 100% of the SNPs recovered by Stacks having missing genotypes among the samples. The high proportion of missing genotypes of Stacks was linked to several characteristics of the pipeline, including non-gapped alignment, the requirement for a certain coverage depth per locus per sample, and an arbitrary and low sequence divergence threshold between alleles of a locus40. However, BWA-GATK pipeline allows for gaps and has a higher sequence divergence threshold in alignments50, which significantly reduces the proportion of SNPs with missing genotypes. A strategy incorporating both Stacks and BWA-GATK can take advantage of the unique properties of the two methods to perform more efficiently than either tool alone, especially in SNP calling for samples with high levels of genetic divergence.
RAD-seq was suggested as a powerful tool for interspecific phylogeny reconstruction by several simulated and empirical studies30, 33, 51. But calling SNPs without a reference genome is difficult and requires careful analysis to develop an optimal strategy. As seen here, the threshold of coverage depth required to create a stack had strong influence on the size of the final data matrix and inferred phylogenetic relationships (Table 2 and Fig. 1). We found conflicting topologies and variable levels of bootstrap support when changing the minimum stack depth and the minimum number of samples needed to retain a locus in the final alignment. Phylogenetic analyses using data matrices p20 and p15 (m = 10) produced different topologies with very low bootstrap support. When the minimum stack depth was reduced to five (m = 5), the phylogenetic relationships and bootstrap values were more stable across the different proportions of missing data (Fig. 1). This suggests that setting -m too high could miss too many information to cause incorrect inferences of sample differentiation52, 53. The possible cause for these missing information is that many reads with errors exist in duplicate and are labeled as stacks in the initial hashing stage of the algorithm when the minimum stack depth is five, but they would be forced into other loci when increasing the minimum stack depth to ten54. In this situation, stacks that truly have a depth of only five get lost, but including a larger sample of individuals with greater accuracy and precision for population parameters which would compensate for low coverage at individual loci in phylogenetic analyses27, 52. The results presented here demonstrate that setting the minimum stack depth to a higher value (e.g., ten) would not be beneficial for increasing precision and accuracy in phylogenetic analysis, but a lower one did.
Accompanying with the change of parameter setting, the number of loci containing missing data in the final sequence matrices altered and would influence the resolution and supports of the temperate bamboo phylogenies greatly. RAD loci usually contain large amounts of missing data, and this problem is more pervasive for distantly related species due to allelic dropout55. Assembling reads from next-generation sequencing data, either de novo or by mapping to a reference genome, relies on sequence similarity40. Hence, alleles with greater divergence among individuals (or relative to the reference genome) may be excluded, especially for more distantly related taxa. Meanwhile, the amount of missing data in the final data matrix is controlled by parameters which set by users, and applying a lower tolerance for missing data comes at the cost of retaining far fewer loci32. Thus, the number of SNPs acquired is positively correlated with missing data, matrices containing minimal missing data and relatively few SNPs produced topologies with extremely low bootstrap support (Fig. 1). In addition, as the tolerance for missing data becomes more stringent, the mutational spectrum represented in the sampled loci was truncated, leading to the disproportionately exclude of the loci with highest mutation rates56. However, concatenating more RAD loci generally obtained higher bipartition supports though with large amount of missing data, a phenomenon observed in both empirical24, 32, 33, 57 and simulated35, 51, 56 phylogenetic studies. Data processing directly impacts the size of the data matrix and therefore the phylogenetic reconstruction and generally larger data sets even if with high proportion of missing data can lead to more accurate inferences.
In conclusion, our study is one of the first to explore the utility of RAD sequencing technology in estimating phylogenetic relationships among Arundinarieae genera. With an emphasis on the Phyllostachys clade, we produce phylogenetic trees with unprecedented resolution for the temperate bamboos and demonstrate that RAD sequencing appears a promising tool for resolving difficult phylogenetic relationships for intractable plant groups. In future, with more broad taxa sampling the RAD sequencing could be used to reconstruct a comprehensive phylogenetic framework for the temperate bamboos. Our work also highlights the sensitivity of phylogenetic inferences to the parameter settings used during SNP genotyping. Careful attention to the analysis pipeline is vital for SNP calling and downstream phylogenetic analyses.
Materials and Methods
Taxon Sampling
Temperate bamboos are a diverse clade containing 19–31 genera and approximately 546 species. A total of 56 populations representing 39 species in 19 genera were sampled for this study (Supplementary Table S1). Thirty species were from clade V, two from clade IV, three from clade VI, and one from clade IX. Given the monophyly of the temperate bamboos, three species of tropical woody bamboos (Bambuseae), Bonia amplexicaulis, D. latiflorus, and G. angustifolia, were chosen as outgroup taxa. The tropical and temperate bamboos are hexaploids (2n = 72), tetraploids (2n=46) and tetraploids (2n = 48), respectively. All species were collected from natural populations, with two exceptions: Himalayacalamus asper, which was collected from Tradewinds Bamboo Nursery (http://www.bamboodirect.com/); and G. angustifolia, which was collected from the Xishuangbanna Tropical Botanical Garden of the Chinese Academy of Sciences. Vouchers of all collections were deposited in the herbarium of the Kunming Institute of Botany. Plant material was dried in silica gel to minimize DNA degradation. Four to six individuals per populations were sampled, with the exception of H. asper, B. amplexicaulis, D. latiflorus, G. angustifolia, and P. edulis, where only one individual was available.
RAD tag library construction and sequencing
Total genomic DNA was extracted from silica gel-dried leaf material using a modified CTAB procedure58. Genomic DNA was pooled from one to six individuals in each population to form population samples as described in Emerson et al.24 and Hohenlohe et al.59. In brief, sequencing adaptors and populations barcodes were ligated to EcoRI-digested total genomic DNA, and the resulting fragments were sequenced from the restriction sites. RAD libraries were prepared and sequenced according to Miller et al.60 and Baird et al.26. All libraries were sequenced on an Illumina HiSeq. 2000 platform, with paired-end sequencing and a read length of 91 bp. RAD sequences were pre-processed through two quality filtering steps to exclude reads which contained contaminated adapter sequences or had more than 50% of base calls with a low quality score (Q ≤ 5). Reads were then de-multiplexed by sorting them into the 56 samples according to their barcodes. Construction of RAD libraries, Illumina HiSeq. 2000 sequencing, raw data cleaning and quality control were performed at BGI Shenzhen, China.
Mapping system: reference-based genotyping approach
The genome of one species of temperate bamboo, P. edulis (Phyllostachys heterocycla), has recently been published61. We used the available P. edulis whole genome (http://www.ncgr.ac.cn/bamboo) as a reference for mapping and to identify SNPs. Sequencing reads (both paired-end 82–86 bp fragments) were aligned to the reference using BWA36 (v0.7.5). Default parameters were used, allowing up to four mismatches and one gap when aligning reads to the genome. Alignments were converted from sequence alignment map (SAM) format to sorted, indexed binary alignment map (BAM) files62 (SAMtools v0.1.18). The Picard tool (v1.103; http://broadinstitute.github.io/picard) was used to remove duplicate reads. Genotypes were called for all samples individually with HaplotypeCaller from the Genome Analysis Tool Kit (GATK)38 (v3.3.0) using the default settings. The variant calls were filtered using a minimum PHRED quality threshold of 20 and a minimum and a maximum variant coverage of 10 and 500 reads, respectively. To examine the balance between obtaining a large number of SNPs and minimizing missing data, we exported four data sets that contained varying levels of missing data by adjusting the “number of no-called samples” parameter (NCC = 0, 1, 5, 10).
Grouping system: de novo assembly genotyping approach
Reads with uncalled nucleotides were discarded using the process_radtags script from the Stacks pipeline40 (v1.08). Within each population, de novo assembly was conducted using ustacks. We set the maximum number of mismatches allowed between stacks (-M) to 3. The cstacks program was used to build a catalog from all populations with three mismatches among loci were allowed (n = 3), and then sstacks was used to search against the catalog produced by cstacks. Genotypes were called for all populations using the Stacks populations program. To assess the influence of coverage depth and the amount of missing data on the outcome of the data matrices and phylogenetic inferences, we used different combinations of parameters, including (1) two different minimum depths of coverage required to form a stack (-m 5, 10); and (2) seven different minimum numbers of populations required to process a locus (-p 10, 15, 20, 25, 35, 45, 55).
Maximum likelihood phylogenetic analyses
We conducted phylogenetic analyses with concatenated SNP datasets using the maximum likelihood (ML) method. The species B. amplexicaulis, D. latiflorus, and G. angustifolia were used as outgroups. Maximum likelihood analyses were implemented using RAxML63 (v7.2.8). Non-parametric bootstrapping was implemented using the fast bootstrap algorithm of RAxML with 200 replicates. The data matrices were analyzed using the GTR + Γ model of nucleotide evolution (GTRGAMMA), as recommended by the authors of the program. Trees were visualized and edited in FigTree (version 1.3.1; http://tree.bio.ed.ac.uk/software/figtree/). We extracted measures of total tree length from the results files (RAxML info files), as well as branch lengths and bootstrap values from the RAxML bipartition tree files.
Species tree estimation
We used the program SVDquartets64 (v1.0) to estimate the coalescent-based species tree using the RAD loci. An advantage of this approach for analyses of RAD data is that it seems to be able to handle large amounts of missing data35. We applied SVDquartets to six data matrices, p10 (m = 5), p15 (m = 5), p20 (m = 5), s45, s50, and s55. For each data matrix, we randomly sampled 100,000 quartets from the 56 populations. The quartet program Quartet MaxCut65 (v2.1.0) was used to infer the species tree from the sampled quartets. We used nonparametric bootstrapping with 100 replicates to measure uncertainty in bipartitions. The bootstrap values were mapped to the species tree estimated from the original data matrix using SumTrees66 (v3.3.1).
Data accessibility
The raw data of four samples (AEM, AWL, YEM, YWL) are available on Dryad (doi:10.2061/dryad.lmj31), and the rest data files are available from the National Center for Biotechnology Information (NCBI) Sequence-Read Archive (SRA) database with accession numbers SRR6001006-SRR6001057.
References
Bamboo Phylogeny Group. An updated tribal and subtribal classification of the bamoos (Poaceae: Bambusoideae). Bamboo Sci. Cult. 24, 1–10 (2012).
Stapleton, C. M. Bergbambos and Oldeania, new genera of African bamboos (Poaceae, Bambusoideae). Phyto Keys, 87 (2013).
Attigala, L., Kathriarachchi, H. S. & Clark, L. G. Taxonomic revision of the temperate woody bamboo genus Kuruna (Poaceae: Bambusoideae: Arundinarieae). Syst. Bot. 41, 174–196 (2016).
Attigala, L., Triplett, J. K., Kathriarachchi, H. S. & Clark, L. G. A new genus and a major temperate bamboo lineage of the Arundinarieae (Poaceae: Bambusoideae) from Sri Lanka based on a multi-locus plastid phylogeny. Phytotaxa 174, 187 (2014).
Jiang, Z. Bamboo and rattan in the world (China Forestry Publishing House, Beijing (2007).
Li, D. Z. et al. Bambuseae (Poaceae) in Flora of China (eds Wu, Z.Y., Raven, P.H., Hong, D.Y.) 7-9 (Science Press and Missouri Botanical Garden Press (2006).
Kelchner, S. A. & Clark, L. G. Molecular evolution and phylogenetic utility of the chloroplast rpl16 intron in Chusquea and the Bambusoideae (Poaceae). Mol. Phylogenet. Evol. 8, 385–397 (1997).
Zhang, W. Phylogeny of the grass family (Poaceae) from rpl16 intron sequence data. Mol. Phylogenet. Evol. 15, 135–146 (2000).
Bouchenak-Khelladi, Y. et al. Large multi-gene phylogenetic trees of the grasses (Poaceae): progress towards complete tribal and generic level sampling. Mol. Phylogenet. Evol. 47, 488–505 (2008).
Peng, S., Yang, H. Q. & Li, D. Z. Highly heterogeneous generic delimitation within the temperate bamboo clade (Poaceae: Bambusoideae): evidence from GBSSI and ITS sequences. Taxon 57, 799–810 (2008).
Sungkaew, S., Stapleton, C. M., Salamin, N. & Hodkinson, T. R. Non-monophyly of the woody bamboos (Bambuseae; Poaceae): a multi-gene region phylogenetic analysis of Bambusoideae ss. J. Plant Res. 122, 95–108 (2009).
Attigala, L., Wysocki, W. P., Duvall, M. R. & Clark, L. G. Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol. Phylogenet. Evol. 101, 111–121 (2016).
Zhang, X. Z. et al. Multi-locus plastid phylogenetic biogeography supports the Asian hypothesis of the temperate woody bamboos (Poaceae: Bambusoideae). Mol. Phylogenet. Evol. 96, 118–129 (2016).
Guo, Z. H., Chen, Y. Y., Li, D. Z. & Yang, J. B. Genetic variation and evolution of the alpine bamboos (Poaceae: Bambusoideae) using DNA sequence data. J. Plant Res. 114, 315–322 (2001).
Zeng, C. X., Zhang, Y. X., Triplett, J. K., Yang, J. B. & Li, D. Z. Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol. Phylogenet. Evol. 56, 821–839 (2010).
Yang, H. M., Zhang, Y. X., Yang, J. B. & Li, D. Z. The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol. Phylogenet. Evol. 68, 340–356 (2013).
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H. & Li, D. Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (poaceae). Syst. Biol. 63, 933–950 (2014).
Zhang, Y. X., Zeng, C. X. & Li, D. Z. Complex evolution in Arundinarieae (Poaceae: Bambusoideae): Incongruence between plastid and nuclear GBSSI gene phylogenies. Mol. Phylogenet. Evol. 63, 777–797 (2012).
Guo, Z. H. & Li, D. Z. Phylogenetics of the Thamnocalamus group and its allies (Gramineae: Bambusoideae): inference from the sequences of GBSSI gene and ITS spacer. Mol. Phylogenet. Evol. 30, 1–12 (2004).
Burke, S. V., Clark, L. G., Triplett, J. K., Grennan, C. P. & Duvall, M. R. Biogeography and phylogenomics of New World Bambusoideae (Poaceae), revisited. Am. J. Bot. 101, 886–891 (2014).
Wysocki, W. P., Clark, L. G., Attigala, L., Ruiz-Sanchez, E. & Duvall, M. R. Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evol. Biol. 15, 50–61 (2015).
Zhang, L. N. et al. Identification of putative orthologous genes for the phylogenetic reconstruction of temperate woody bamboos (Poaceae: Bambusoideae). Mol. Ecol. Res. 14, 988–999 (2014).
Xu, P. et al. Population genomic analyses from low-coverage RAD-Seq data: a case study on the non-model cucurbit bottle gourd. Plant. J. 77, 430–442 (2014).
Emerson, K. J. et al. Resolving postglacial phylogeography using high-throughput sequencing. P Proc. Natl. Acad. Sci. USA. 107, 16196–16200 (2010).
Keller, I. et al. Population genomic signatures of divergent adaptation, gene flow and hybrid speciation in the rapid radiation of Lake Victoria cichlid fishes. Mol. Ecol. 22, 2848–2863 (2013).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS one 3, e3376 (2008).
Cariou, M., Duret, L. & Charlat, S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol. Evol. 3, 846–852 (2013).
Darwell, C. T., Rivers, D. M. & Althoff, D. M. RAD-seq phylogenomics recovers a well-resolved phylogeny of a rapid radiation of mutualistic and antagonistic yucca moths. Syst. Entomol. 41, 672–682 (2016).
Herrera, S. & Shank, T. M. RAD sequencing enables unprecedented phylogenetic resolution and objective species delimitation in recalcitrant divergent taxa. Mol. Phylogenet. Evol. 100, 70–79 (2016).
Hipp, A. L. et al. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS One 9, e93975 (2014).
Hou, Y. et al. Thousands of RAD-seq loci fully resolve the phylogeny of the highly disjunct arctic-alpine genus Diapensia (Diapensiaceae). PLoS One 10, e0140175 (2015).
Wagner, C. E. et al. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 22, 787–798 (2013).
Eaton, D. A. & Ree, R. H. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62, 689–706 (2013).
Leache, A. D. et al. Phylogenomics of phrynosomatid lizards: conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genome Biol. Evol. 7, 706–719 (2015).
Leache, A. D., Banbury, B. L., Felsenstein, J., de Oca, A. N. & Stamatakis, A. Short Tree, long tree, right tree, wrong tree: newa cquisition bias corrections for inferring SNP phylogenies. Syst. Biol. 64, 1032–1047 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Lunter, G. & Goodson, M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939 (2011).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3 (Bethesda) 1, 171–182 (2011).
Baxter, S. W. et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS One 6, e19315 (2011).
Pan, J. et al. Optimization of the genotyping-by-sequencing strategy for population genomic analysis in conifers. Mol. Ecol. Resour. 15, 711–722 (2015).
Wang, X. Q., Zhao, L., Eaton, D. A. R., Li, D. Z. & Guo, Z. H. Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol. Ecol. Resour. 13, 938–945 (2013).
Triplett, J. K. & Clark, L. G. Phylogeny of the temperate bamboos (Poaceae: Bambusoideae: Bambuseae) with an emphasis on Arundinaria and allies. Syst. Bot. 35, 102–120 (2010).
Guo, Z. H., Chen, Y. Y. & Li, D. Z. Phylogenetic studies on the Thamnocalamus group and its allies (Gramineae: Bambusoideae) based on ITS sequence data. Mol. Phylogenet. Evol. 22, 20–30 (2002).
Xiang, Y. et al. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 262–281 (2017).
Massatti, R., Reznicek, A. A. & Knowles, L. L. A case study in Carex sect. Racemosae. Am. J. Bot. 103, 337–347 (2016). Utilizing RADseq data for phylogenetic analysis of challenging taxonomic groups.
Stapleton, C. M., Chonghaile, G. N. & Hodkinson, T. R. Sarocalamus, a new sino-himalayan bamboo genus (Poaceae: Bambusoideae). Novon 14, 345–349 (2004).
Yi, T. P. A study of the genus Fargesia from China. Journal of bamboo research 7, 6–15 (1988).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Rubin, B. E., Ree, R. H. & Moreau, C. S. Inferring phylogenies from RAD sequence data. PLoS One 7, e33394 (2012).
Alex Buerkle, C. & Gompert, Z. Population genomics based on low coverage sequencing: how low should we go? Mol. Ecol. 22, 3028–3035 (2013).
Mastretta-Yanes, A. et al. Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Resour. 15, 28–41 (2015).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Arnold, B., Corbett-Detig, R. B., Hartl, D. & Bomblies, K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Mol. Ecol. 22, 3179–3190 (2013).
Huang, H. & Knowles, L. L. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst. Biol. 65, 357–365 (2016).
Takahashi, T., Nagata, N. & Sota, T. Application of RAD-based phylogenetics to complex relationships among variously related taxa in a species flock. Mol. Phylogenet. Evol. 80, 137–144 (2014).
Doyle, J. J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Hohenlohe, P. A. et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS genet. 6, e1000862 (2010).
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. & Johnson, E. A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248 (2007).
Peng, Z. et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 45, 456–461 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Chifman, J. & Kubatko, L. Quartet inference from SNP data under the coalescent model. Bioinformatics 30, 3317–3324 (2014).
Snir, S. & Rao, S. Quartet MaxCut: a fast algorithm for amalgamating quartet trees. Mol. Phylogenet. Evol. 62, 1–8 (2012).
Sukumaran, J. & Holder, M. T. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Acknowledgements
We thank Xiaoyan Wang, Xuyao Zhao, Xianzhi Zhang, Lina Zhang, Xia Li, and Yichi Zhang for joining in the field work to collect samples. We also thank Dr. Chunxia Zeng, Yuxiao Zhang, Pengfei Ma, Xuemei Zhang, and Nian Wang for their assistance and support. Special thanks go to Dr. Linda E Neaves, at the Royal Botanic Garden Edinbrugh, for her providing helpful comments that improved this manuscript. This project was supported by the National Natural Science Foundation of China (31470322, 31430011 and 31501030) and Foundation of He’nan Educational Committee (15A180066).
Author information
Authors and Affiliations
Contributions
Z.H.G., D.Z.L. and X.Q.W. designed the research, X.Q.W. and L.Z. collected data, X.Q.W., L.Z. and H.F.Z. analyzed the data, and X.Q.W. and X.Y.Y. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Ye, X., Zhao, L. et al. Genome-wide RAD sequencing data provide unprecedented resolution of the phylogeny of temperate bamboos (Poaceae: Bambusoideae). Sci Rep 7, 11546 (2017). https://doi.org/10.1038/s41598-017-11367-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11367-x
This article is cited by
-
Phylogenomics of Palythoa (Hexacorallia: Zoantharia): probing species boundaries in a globally distributed genus
Coral Reefs (2022)
-
Phylogenetic relationships and migration patterns of the Fargesia spathacea complex inferred from genomic data
Plant Systematics and Evolution (2022)
-
Traditional System Versus DNA Barcoding in Identification of Bamboo Species: A Systematic Review
Molecular Biotechnology (2021)
-
Phylogeny of Fargesia (Poaceae: Bambusoideae) and infrageneric adaptive divergence inferred from three cpDNA and nrITS sequence data
Plant Systematics and Evolution (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
