
Abstract
Murine double minute 2 (MDM2) is an E3-ubiquitin ligase critical for various biological functions. Previous data have revealed an indispensable role of MDM2 in kidney homeostasis. However, its role in glomerular mesangial cell (GMC) proliferation and extracellular matrix (ECM) accumulation during hyperglycemia condition remains unclear. In our present study, we found that MDM2 protein level was significantly upregulated in high glucose-treated GMCs, while knocking down MDM2 by siRNA could attenuate high glucose-induced ECM accumulation and GMCs proliferation. Unexpectedly, Nutlin-3a, a MDM2-p53 interaction blocker, had no benefit in protecting diabetic mice from renal impairment in vivo and in alleviating high glucose-induced ECM accumulation in vitro. Intriguingly, we found that Notch1 signaling activation was obviously attenuated by MDM2 depletion in GMCs with high glucose exposure. However, Numb, a substrate of MDM2 which suppresses Notch1 signaling, was found not to be involved in the MDM2 and Notch1 association. Moreover, our findings demonstrated that MDM2 interacted with Notch1 intracellular domain (NICD1) independent of Numb and regulated the ubiquitination status of NICD1. Collectively, our data propose a pivotal role of MDM2 in high glucose-induced GMC proliferation and ECM accumulation, via modulating the activation of Notch1 signaling pathway in an ubiquitination-dependent way.
Similar content being viewed by others


Introduction
Diabetic kidney disease (DKD) is one of the most serious microvascular complications of both type 1 and type 2 diabetic mellitus, resulting in end stage renal disease (ESRD)1, 2. Unfortunately, there has been no satisfying intervention strategy for DKD until now, especially in its early stage. Glomerular mesangial cell (GMC) proliferation and excessive extracellular matrix (ECM) deposition in mesangium are hallmark pathological features in the onset and progression of DKD, which lead to glomerulosclerosis and deterioration of renal function3, 4. However, the mechanisms mediating GMC proliferation and ECM accumulation under hyperglycemia condition have not been fully clarified.
Murine double minute-2 (MDM2) is an important E3 ubiquitin ligase which promotes the nuclear exportation, ubiquitination and proteasomal degradation process of p535,6,7. In this regard, MDM2 is considered as an onco-protein by accelerating tumor growth and promoting cancer progression in a p53-dependent manner8,9,10; hence, small molecule inhibitors targeting MDM2-p53 interaction, such as Nutlin-3a, have been developed for the treatment of various types of cancer and are under clinical trial presently11,12,13. Furthermore, MDM2 also correlates to various physiological and pathological processes independent of p53. For example, MDM2 can exert its oncogenic profile via activating Akt signaling14, and facilitate sterile inflammation through NF-κB pathway15.
MDM2 is widely presented in kidney resident cells including glomerular endothelial cells (GECs), podocytes, parietal epithelial cells (PECs) and tubular epithelial cells (TECs)16, 17. Physiologically, MDM2 is indispensable for resting podocytes and TECs in maintaining cell homeostasis and survival by protecting them from p53-related cell death. Knocking down MDM2 results in functional and structural abnormalities of podocytes and TECs18, 19. Meanwhile, MDM2 dysregulation is also implicated in the pathological events of various kidney diseases. For example, a German group has demonstrated that MDM2 plays a dual role in post-ischemic acute tubular injury, as it promotes renal sterile inflammation in a NF-κB-dependent manner and drives tubular cell regeneration by blocking p53-mediated cell cycle arrest and apoptosis16. Additionally, we recently found MDM2 upregulation is implicated in renal fibroblast activation, leading to tubulointerstitial fibrosis (TIF)20. In glomerular injury disease, MDM2 promotes PEC proliferation and intraglomerular inflammation during anti-GBM serum induced-crescentic glomerulonephritis17, and also leads to podocyte loss by facilitating cell entering mitosis (i.e., mitotic catastrophe) in adriamycin nephropathy21. However, whether MDM2 contributes to the pathogenesis of DKD is rarely reported, thus we are interested in exploring its role in GMC proliferation and ECM accumulation, which are the critical components of DKD.
In our present study, we tested the hypothesis that MDM2 could drive GMC proliferation and ECM accumulation under hyperglycemia condition, and then explored its related downstream signaling pathways. Our findings demonstrate that MDM2 plays a crucial role in high glucose-induced GMC proliferation and ECM accumulation, through modulating the activation status of Notch1 signaling pathway in an ubiquitination-dependent manner, independent of p53.
Results
High glucose stimulates cell proliferation and ECM accumulation and induces the upregulation of MDM2 level in GMCs
To determine the influence of high glucose on GMC proliferation and ECM protein accumulation, HBZY-1 cells were exposed to high glucose media for indicated times. In line with previously reports22, 23, the cell proliferation was significantly induced by high glucose, and the expression of collagen III and fibronectin (indicators for ECM accumulation) also increased (Fig. 1A–C). Next, we detected MDM2 expression in cultured GMCs and we found the protein abundance of MDM2 was upregulated in a similar time-dependent manner (Fig. 1D,E), the influence of osmotic pressure on MDM2 expression was rule out using Mannitol (data not presented). These data suggest that MDM2 may be correlated to GMC proliferation and ECM accumulation under hyperglycemia situation.

Cell proliferation and collagen III, fibronectin accumulation along with upregulated MDM2 expression in high glucose-cultured GMCs. (A) BrdU summarized data showing that high glucose induces HBZY-1 cell proliferation from 6 h to 24 h n = 7. (B) Representative western blot images and (C) summarized data showing the collagen III and fibronectin protein level in HBZY-1 treated with high glucose for indicated time n ≥ 3. (D) Representative western blot images and (E) summarized data showing MDM2 protein level in HBZY-1 treated with high glucose for indicated time n = 6. Ctrl: control; HG: high glucose. * P < 0.05 vs. Ctrl.
MDM2 contributes to high glucose-induced ECM accumulation and cell proliferation in GMCs
To evaluate the role of MDM2 in mediating GMC proliferation and ECM accumulation, we designed three different sequence siRNA oligonucleotides targeting MDM2 gene and the most efficient one (002) was utilized to knockdown MDM2 expression in the following experiments. The transfection efficiency of this siRNA was approximately 50% under normal or high glucose condition, and we found MDM2 inhibition dramatically decreased the high glucose-induced upregulation of collagen III and fibronectin (Fig. 2A–D). In addition, we investigated the influence of MDM2 silencing on GMC proliferation. As shown in Fig. 2E, high glucose-induced GMC proliferation was also largely reversed by MDM2 depletion, indicating that MDM2 plays a critical role in GMC dysfunction under hyperglycemia condition.

Genetic depletion of MDM2 attenuates high glucose-induced collagen III, fibronectin accumulation and cell proliferation in GMCs. (A) Representative western blot images and (B) summarized data showing the interfere effect of three different sequence siRNA oligonucleotides targeting MDM2 n = 3. (C) Representative western blot images and (D) summarized data showing that depletion MDM2 by siRNA partially attenuates collagen III and fibronectin accumulation in high glucose-stimulated HBZY-1 cells n = 6. (E) BrdU summarized data showing that high glucose-induced cell proliferation is inhibited by MDM2 genetic depletion n = 6. Scra: scramble shRNA; siMDM2: MDM2 siRNA. * P < 0.05 vs. Ctrl + Scra; # P < 0.05 vs. HG + Scra.
Nutlin-3a cannot alleviate renal impairment of diabetic mice and high glucose-induced ECM accumulation in vivo and in vitro
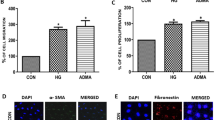
Next, we explored the possible mechanisms involved in MDM2-associated GMC dysfunction. p53 is a classic downstream target of MDM2 and Nutlin-3a is a cis-imidazoline compound that inhibits MDM2-mediated p53 degradation, which has been reported to play a beneficial role in variety of kidney diseases17, 21, 24. Thus we treated the STZ-induced diabetic mice with Nutlin-3a to investigate the p53-dependent role of MDM2. First, by immunohistochemistry we found the intensity of p53 was markedly increased in the glomeruli of mice with Nutlin-3a treatment (Fig. 3A,B), indicating the efficient blocking of MDM2-p53 signaling. However, there was no significant difference between vehicle and Nutlin-3a treated diabetic mice in the physical and biochemical parameters including serum glucose, body weight, kidney weight/body weight, serum albumin, serum creatinine, blood urea nitrogen, urine total protein and urine albumin to creatinine ratio (Fig. 3C–J). Meanwhile, no alleviation of pathological changes in glomeruli was observed in Nutlin-3a treated mice (Fig. 3K,L). Collectively, these findings suggest a p53-independent effect of MDM2 in glomerular impairment under diabetic status in vivo.

Nutlin-3a cannot ameliorate the renal impairment in STZ-induced diabetic mice. (A) Representative photomicrograph of p53 density in the glomeruli of mice in each group (original magnification 200x). (B) Quantification of p53-positive cells/glomerular cross section for each group n = 6. (C–J) Summarized data showing the serum glucose, body weight, Kw/Bw, serum albumin, serum creatinine, blood urea nitrogen, urine total protein, and UACR of the experimental mice in our study n ≥ 7. (K) Representative PAS staining images of glomeruli (original magnification 200x). (L) Summarized data showing the glomerular damage index (GDI) of the three groups of mice. n ≥ 7. Vehl: vehicle; Kw: kidney weight; Bw: bodyweight; UACR: urine albumin to creatinine ratio. * P < 0.05 vs. Ctrl mice; ** P < 0.01 vs. Ctrl mice; *** P < 0.0001 vs. Ctrl mice; # P > 0.05 vs. Ctrl mice; ### P > 0.05 vs. diabetic mice + Vehl; & P > 0.05 vs. diabetic mice + Vehl.
To confirm the role of Nutlin-3a in ECM accumulation in vitro, HBZY-1 cells were treated with Nutlin-3a before exposed to high glucose media. Nutlin-3a treatment markedly upregulated the expression of p53 in cultured GMCs (Fig. 4A,B). However, as shown in Fig. 4C,D, Nutlin-3a treatment couldn’t prevent the upregulation of collagen III and fibronectin induced by high glucose in HBZY-1 cells. According to these data, there must be other ways independent of p53 to explain MDM2-mediated GMC dysfunction under hyperglycemia condition.

Nutlin-3a cannot reverse high glucose-induced collagen III and fibronectin accumulation in vitro. (A) Representative western blot images and (B) summarized data showing that the elevation expression of p53 in Nutlin-3a-treated HBZY-1 cells n = 4. # P < 0.05 vs. HG + Vehl. (C) Representative western blot images and (D) summarized data showing that Nutlin-3a treatment has no benefit on high glucose-induced collagen III and fibronectin accumulation in cultured HBZY-1 cells n = 7. * P < 0.05 vs. Ctrl; & P > 0.05 vs. HG + Vehl.
Aberrant activation of Notch1 signaling pathway induced by high glucose in GMCs is largely attenuated by MDM2 depletion
Notch1 signaling pathway activation has been reported in glomeruli of diabetic kidney and high glucose-stimulated GMCs25, which is well-recognized playing a pathogenic part in GMC proliferation and ECM accumulation. In our study, we confirmed Notch1 signaling is activated in HBZY-1 cells exposed to high glucose, characterized by upregulated Notch1 intracellular domain (NICD1) and its downstream target gene Hes1 and Hey1 (Fig. 5A,B,E,F). MDM2 has been reported to activate Notch1 signaling pathway during tumorigenesis26, thus we wondered whether MDM2 promotes GMC dysfunction via Notch1 signaling activation in GMCs with high glucose treatment. By knocking down MDM2, the high glucose-enhanced expression of NICD1, Hes1 and Hey1 were significantly reduced (Fig. 5C,D,G,H), indicating that the high glucose-induced activation of Notch1 signaling pathway is attenuated by MDM2 depletion.

Genetic deletion of MDM2 attenuates aberrant Notch1 signaling in high glucose-treated GMCs. (A) Representative western blot images and (B) summarized data showing the protein level of NICD1 and Hes1 in HBZY-1 cells exposed to high glucose for indicated times n ≥ 5. * P < 0.05 vs. Ctrl. (C) Representative western blot images and (D) summarized data showing that the genetic deletion of MDM2 by siRNA effectively suppresses the protein level of NICD1 and Hes1 n = 5. * P < 0.05 vs. Ctrl + Scra; # P < 0.05 vs. HG + Scra. (E) Representative western blot images and (F) summarized data showing the protein level of Hey1 in HBZY-1 cells exposed to high glucose for indicated times n = 6. * P < 0.05 vs. Ctrl. (G) Representative western blot images and (H) summarized data showing that the genetic deletion of MDM2 by siRNA effectively suppresses the protein level of Hey1 n = 3. * P < 0.05 vs. Ctrl + Scra; # P < 0.05 vs. HG + Scra.
Numb expression is upregulated and cannot be affected by MDM2 inhibition in GMCs exposed to high glucose
We further moved on to investigate how MDM2 regulates Notch1 signaling pathway in high glucose-cultured GMCs. Numb is another important ubiquitination target of MDM227 and also a negative regulator of Notch1 signaling pathway28. MDM2 indirectly activates Notch1 signaling pathway by disrupting Numb-Notch1 complex. However, the expression of Numb and whether Numb is a mediator of MDM2-Notch1 interaction in GMCs with high glucose exposure are not known. In our study, the protein abundance of Numb was increased in high glucose-cultured HBZY-1 cells in a time-dependent manner (Fig. 6A,B), and the depletion of MDM2 had no significant influence on the expression of Numb (Fig. 6C,D), suggesting that Numb may not be involved in the association between MDM2 and Notch1 during GMCs dysfunction induced by high glucose.

Numb expression is upregulated by high glucose simulation in GMCs and not affected by MDM2 inhibition. (A) Representative western blot images and (B) summarized data showing the Numb protein level in HBZY-1 with high glucose treatment at indicated time points n = 6. * P < 0.05 vs. Ctrl. (C) Representative western blot images and (D) summarized data showing that the abundance of Numb is not influenced by MDM2 inhibition n = 6. * P < 0.05 vs. Ctrl + Scra; & P > 0.05 vs. HG + Scra.
MDM2 regulates the ubiquitination status of NICD1
As MDM2 modulates the activation of Notch1 signaling pathway while Numb is not implicated, we explored other possible mechanisms. It has been reported that MDM2 also modulates Notch1 signaling pathway via direct ubiquitination modification of NICD1, leading to activation of Notch1 signaling pathway instead of degradation26, but whether similar regulation contributes to the pathogenesis of DKD is not clarified. In our study, by using immunoprecipitation method, we found that NICD1 could be pulled down by MDM2 primary antibody in high glucose-cultured GMCs, without Numb binding (Fig. 7A). More importantly, the ubiquitination status of NICD1 was regulated by the level of MDM2 when knocking down MDM2 by siRNA (Fig. 7B,C), suggesting an ubiquitination modification role of MDM2 in GMCs under hyperglycemia condition.

MDM2 binds with NICD1 in GMCs exposed to high glucose and regulates the ubiquination status of NICD1. (A) Cell lysates of high glucose-cultured HBZY-1cells were incubated with MDM2 primary antibody, and NICD1 was pulled down by MDM2 primary antibody without Numb binding. Input: whole lysate; IP: immunoprecipitation; IgG: mouse IgG. (B) Cell lysates of HBZY-1 cells transfected with MDM2 siRNA were incubated with Notch1 primary antibody, and ubiquitination was detected by immunoblotting with anti-ubiquitin antibody. Ub-NICD1 represents ubiquitinated NICD1. (C) Summarized data showing that the ubiquitination NICD1 is regulated by MDM2 inhibition n = 4. * P < 0.05 vs. Ctrl + Scra; & P < 0.05 vs. HG + Scra.
Discussion
In the current study, we explored the role and mechanism of MDM2 in GMC proliferation and ECM accumulation under hyperglycemia condition. Here, we showed that MDM2 is upregulated in high glucose-cultured GMCs, and genetic ablation of MDM2 attenuates cell proliferation and ECM accumulation induced by high glucose. However, neither p53 nor Numb is involved in these processes mediated by MDM2 aforementioned. Instead, our findings suggest that MDM2-mediated Notch1 activation is one of the potential mechanisms responsible for high glucose-induced GMC proliferation and ECM accumulation.
MDM2 is a well-known onco-protein that suppresses p53-dependent cell cycle arrest, which also exerts numerous p53-independent functions by interacting with other substrates such as Numb, NF-κB, and so on. Accumulating studies have documented a strong correlation between MDM2 and physiologic homeostasis of kidney18, 19 as well as pathological events during diseased status17, 24. MDM2 is proposed to be a survival factor for resting podocytes, and also essential for the lifespan and the maintenance of renal function in mice18, 19. Meanwhile, the same research group identified MDM2 as a contributor to podocyte mitotic catastrophe in adriamycin nephropathy21. Moreover, MDM2 is involved in anti-GBM serum induced-crescentic glomerulonephritis by triggering the aberrant cell cycle entry of PECs and inflammatory cell infiltration17. However, whether MDM2 is implicated in modulating GMC function during hyperglycemia is largely unknown. Our data first revealed that high glucose upregulated the protein level of MDM2 in a time-dependent manner, accompanied with an elevation of collagen III and fibronectin accumulation, as well as cell proliferation in vitro. These data drive us to explore whether MDM2 is a mediator of high glucose-induced GMC dysfunction. Indeed, inhibiting MDM2 by genetic strategy confirmed the pivotal role of MDM2 in mediating high glucose-induced cell proliferation and ECM accumulation in GMCs, which are two crucial events leading to glomerulosclerosis in DKD.
Then we explored the underlying mechanisms. It has been reported that inhibition of MDM2-p53 interaction using Nutlin-3a attenuates kidney disorders in various diseased situations. For example, Nutlin-3a treatment prevents adriamycin-induced podocyte detachment and alleviates glomerular pathological change21. Moreover, Nutlin-3a treatment ameliorates kidney injury in MRL-Faslpr mice24 and abrogates anti-GBM serum induced crescentic glomerulonephritis17. Thus we examined the intervention effect of Nutlin-3a in DKD animal model. Unexpectedly, Nutlin-3a treatment failed to improve biochemical parameters of STZ-induced diabetic mice. Also, there was no significant alleviation of pathological changes in the glomeruli of these mice, especially mesangial lesions. Corresponding to these in vivo data, Nutlin-3a couldn’t alleviate collagen III and fibronectin accumulation in cultured GMCs exposed to high glucose in vitro. It is worth mentioning that, in a recent published report, inhibition of MDM2 with Nutlin-3a in normal C57BL/6 mice leads to damage of glomerular filtration barrier and renal function, meanwhile a 4-week Nutlin-3a treatment also shows no renal protection effect in db/db diabetic mice29. Thus, we concluded that the benefit of MDM2 ablation described above is obtained via a p53-independent way.
Notch1 is a single trans-membrane protein that contributes to the formation and development of kidney, and expresses less in adult kidney30, 31. The release of active NICD1 by γ-secretase is critical for the subsequent transcriptional activation of downstream Notch1-targeted genes, such as Hes1/Hey132, 33. Abnormal activation of Notch1 signaling pathway is implicated in diabetic-related podocyte injury and GMC dysfunction25, 34, 35, while podocyte-specific deletion of Notch1 protects mice from diabetic-associated podocyte damage and depletion34, 35. In accordance with these findings, inhibition of Notch1 pathway by blocking the release of NICD1 using γ-secretase inhibitor also alleviates albuminuria and glomerulosclerosis in rat DKD model36. Thus, getting a better understanding of Notch1 signaling regulation during hyperglycemia is of great importance. The connection between MDM2 and Notch signaling pathway including Notch1 and Notch4 has been observed during tumorigenesis26, 37, 38. But it is rarely known if there is a link between MDM2 and Notch1 in high glucose-induced GMC dysfunction. In this regard, we first confirmed the activation of Notch1 signaling pathway in high glucose-cultured GMCs, and further revealed that this process is largely modulated by MDM2, if not all. Our data indicated that Notch1 plays an important role in the pathogenetic process associated with MDM2 in mesangial cells, especially under high glucose conditions.
To further clarify the mechanism of Notch1 activation, we tested the expression of Numb, a protein that suppresses Notch1 signaling pathway by binding with NICD1, preventing its nuclear localization and recruiting ubiquitination, and leading to degradation of NICD128, 39. It is widely accepted that MDM2 promotes aberrant activation of Notch1 signaling via ubiquitinating and degrading of Numb27, 40, 41. However, the expression change of Numb and its potential role in mediating the effect of MDM2 in high glucose-cultured GMCs is unclear. In the present study, we found that high glucose induced the expression of Numb protein in a time-dependent manner, while knockdown of MDM2 had no effect on Numb expression. Therefore, Numb may not be involved in MDM2-regulated Notch1 activation, at least in GMCs. MDM2 is suggested to modulate Notch1 signaling pathway through other mechanisms.
Ubiquitination is one type of posttranslational modifications, which is important for regulating protein activity and stability. Usually, protein ubiquitination is considered as a proteasome-dependent degradation fate of protein. However, it is also reported that protein ubiquitination plays a role in the activation of signal transduction pathways, such as Akt hyperactivation42 in cancer and activation of IKK (IκB kinase) in NF-κB pathway43. MDM2 has been reported to directly interact with NICD1, which leads to the ubiquitination and the transcriptional activation of Notch1 signaling pathway in breast cancer cells26. In our study, it was found that MDM2 interacted with NICD1 without Numb binding. More importantly, the ubiquitination status of NICD1 was regulated by MDM2, which parallels with the activation status of Notch1 signaling pathway described above. Thus, ubiquitination modification-related Notch1 activation may be a reasonable explanation for the relationship between MDM2 and Notch1 signaling pathway in GMCs exposed to high glucose.
In summary, we demonstrated that MDM2 is essential in mediating high glucose-induced GMC proliferation and ECM accumulation, through the activation of Notch1 via ubiquitination modification of NICD1. Our current study reveals a novel mechanism involved in GMCs dysfunction under hyperglycemia conditions and might provide a potential target for the early intervention of DKD.
Research Materials and Methods
Animals
The animal experiments were performed according to the guidelines for use and care of laboratory animals of National Institutes of Health, and approved by the Animal Care and Use Committee of Tongji Medical College. 7–8 weeks old C57BL/6 mice weighing 21–24 g were obtained from Charles River (Beijing, China), and raised in a pathogen-free environment with a 12-hour light and dark cycle, and unrestricted access to food and water. To create the diabetic model, the mice were starved for 12 h and intraperitoneally injected with a single dose of streptozotocin (STZ, 150 mg/kg body weight, BOSTER, Wuhan, China), while control mice received only citrate buffer. Blood glucose was monitored weekly by glucometer readings. Only the mice with stable serum glucose levels over 16.7 mmol/l were included in the following experiment. The diabetic mice were divided into two groups: one group treated with 60% DMSO (vehicle) and the other with Nutlin-3a (20 mg/kg body weight, Biochempartner, Shanghai, China) every day for 12 weeks. Then urine samples, blood samples and kidney tissues were collected.
Serum, urine samples detection
The blood harvested from the mice were clotted for 2 h at room temperature, and then the blood and urine samples were centrifuged for 20 min at 1000 rpm to get the supernatants. The samples were aliquoted and stored in −80 °C until detected. The serum glucose, albumin, creatinine, blood urea nitrogen, urine total protein and urine creatinine were measured by Auto-Chemistry Analyzer. Urine albumin was detected by Elisa kit (Elabscience, Wuhan, China) according to the manufacturer’s instructions.
Immunohistochemistry
Kidney tissues were fixed in 4% neutral buffered formalin, dehydrated in graded sucrose, and embedded in paraffin. Immunohistochemical staining of p53 was performed using a standard biotin-streptavidin-peroxidase method. Briefly, 4 μm thick sections were deparaffinized and rehydrated. Endogenous peroxidase was blocked by 5% BSA for 30 min at room temperature, then the sections were incubated with the primary antibody for p53 (rabbit, 1:100; Protein Tech Group, Chicago, IL, USA) overnight at 4 °C. Then, sections were washed in PBS and incubated with biotinylated goat anti-rabbit antibody and HRP-labeled streptavidin (Beyotime, Jiangsu, China), each for 20 min. At last, peroxidase activity was visualized by diaminobenzidine, and sections were counterstained with hematoxylin and observed under light microscope and 20 glomeruli were randomly chosen for counting the p53-positive cells as described previously44.
Morphological examination
Periodic acid-schiff (PAS) staining of 4 μm paraffin sections was used to examine the morphometric change in the glomeruli. 50 randomly chosen glomeruli of each kidney section were calculated to get the score of glomerular damage index (GDI) as described previously45.
Cell culture, treatment, RNA interference
A rat GMCs line (HBZY-1) was routinely cultured in MEM/EBSS media containing 10% fetal bovine serum, 100 U/ml penicillin and 100 U/ml streptomycin in 37 °C incubator with 5% CO2. The cells were exposed to media with 30 mmol/l final glucose concentration for indicated time.
For chemical intervention, the cells were pretreated with 5 μmol/l Nutlin-3a for 1 h, and then exposed to high glucose media for 12 h before harvested.
For RNA interference, short-interfering RNA (siRNA) target on rat MDM2 was obtained from Ribobio (Guangzhou, China). Cells reaching an appropriate confluence were transfected with MDM2 siRNA for 48 h and the scramble siRNA was used as a negative control. The transfection procedure was carried out according to the manufacturer’s instructions. Cells were exposed to high glucose media for 12 h before harvested.
Cell proliferation
BrdU assay was used to measure cell proliferation. The BrdU incorporation was carried out according to the manufacturer’s instructions of commercial kit (Abcam, Cambridge, MA, USA). Briefly, HBZY-1 cells were seeded into 96-weel plate at a proper density, after indicated treatment, BrdU solution was added into culture media and cultured for another 2 h in the incubator, and then cells were fixed. BrdU incorporation was detected by BrdU detector antibody and peroxidase substrate. The absorbance was read at 450 nm.
Western blotting analysis
The total protein of cultured cells was extracted by RIPA lysis buffer containing protease inhibitors. BCA detection method was used to determine the protein concentration of cell lysates. Then the cell lysates were supplied with SDS protein loading buffer and boiled in 98 °C for 5 minutes, leading to protein denaturation. Western blotting analysis was carried out briefly as follows: the denatured proteins were separated by SDS-PAGE and transferred to PVDF membranes. After blocking step, the membranes were incubated with indicated primary antibodies overnight at 4 °C. These antibodies were used as follows: MDM2 (mouse, 1:400); Hes1 (mouse, 1:1,000); β-actin (1:10,000) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Collagen III (rabbit, 1:1,000); Fibronectin (rabbit, 1:1,000), p53 (rabbit, 1:1,000), Hey1 (rabbit, 1:1,000), Ubiquitin (rabbit, 1:1,000) were obtained from Protein Tech Group (Chicago, IL, USA), Notch1 (rabbit, 1:500) was obtained from Abcam (Cambridge, MA, USA), Numb (rabbit, 1:1,000) was obtained from Cell Signal Transduction (Danvers, MA, USA). The next day, the membranes were washed three times with Tris-buffered saline with 0.1% Tween 20 (TBST, pH 7.2–7.6) before incubating with corresponding HRP-conjugated secondary antibodies with a 1:20,000 dilution for 1 h at room temperature. The ECL reagents were used to detect immunoreactive bands with film. The relative intensity of target bands were quantified by Image J software and normalized by the intensity of β-actin.
Immunoprecipitation
The cell lysates were prepared just as described above. Then equal amounts (0.5–1 mg) of total protein samples were supplied with 1 μg primary antibody and 30 μl Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA), then incubated overnight at 4 °C on a rotating device. The primary antibody used for immunoprecipitation in our present study including MDM2 (mouse, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Notch1 (rabbit, Sigma, St Louis, MO, USA). The following day, the immunoprecipitates were collected by centrifuging at 2,500 rpm for 5 min at 4 °C. The pellets were washed 3 times with 1.0 ml PBS on a rotating device for 5 min each time. After the final wash, the pellets were resuspended in SDS sample buffer, then boiled in 98 °C for 5 min. The immunoprecipitated proteins were detected by western blotting analysis.
Statistical Analysis
All of the result data in this study were expressed as mean ± SEM. Significant difference were analyzed using two-tailed T test with GraphPad Software. P < 0.05 was considered as of statistical significance.
Data availability
The authors declare the data availability.
References
Hadjadj, S. et al. Death, end-stage renal disease and renal function decline in patients with diabetic nephropathy in French cohorts of type 1 and type 2 diabetes. Diabetologia 59, 208–216 (2016).
Panduru, N. M. et al. Urinary adiponectin is an independent predictor of progression to end-stage renal disease in patients with type 1 diabetes and diabetic nephropathy. Diabetes care 38, 883–890 (2015).
Park, M. J. et al. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: the possible role in the development of diabetic nephropathy. J Biol Chem 289, 13519–13530 (2014).
Wang, J. et al. Tetrahydrobiopterin contributes to the proliferation of mesangial cells and accumulation of extracellular matrix in early-stage diabetic nephropathy. J Pharm Pharmacol 69, 182–190 (2017).
Yu, J. et al. Inhibition of cell proliferation, migration and invasion by a glioma-targeted fusion protein combining the p53 C terminus and MDM2-binding domain. Cell Prolif 49, 79–89 (2016).
Raza, S. et al. The cholesterol metabolite 27-hydroxycholesterol regulates p53 activity and increases cell proliferation via MDM2 in breast cancer cells. Mol Cell Biochem 410, 187–195 (2015).
Hashimoto, H., Sue, Y., Saga, Y., Tokumitsu, M. & Yachiku, S. Roles of p53 and MDM2 in tumor proliferation and determination of the prognosis of transitional cell carcinoma of the renal pelvis and ureter. Int J Urol 7, 457–463 (2000).
Florenes, V. A. et al. MDM2 gene amplification and transcript levels in human sarcomas: relationship to TP53 gene status. J Natl Cancer Inst 86, 1297–1302 (1994).
Turbin, D. A. et al. MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod Pathol 19, 69–74 (2006).
Bueso-Ramos, C. E. et al. The human MDM-2 oncogene is overexpressed in leukemias. Blood 82, 2617–2623 (1993).
Wang, B., Fang, L., Zhao, H., Xiang, T. & Wang, D. MDM2 inhibitor Nutlin-3a suppresses proliferation and promotes apoptosis in osteosarcoma cells. Acta Biochim Biophys Sin 44, 685–691 (2012).
Trino, S. et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: a new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget 7, 12951–12961 (2016).
Zajkowicz, A. et al. Nutlin-3a, an MDM2 antagonist and p53 activator, helps to preserve the replicative potential of cancer cells treated with a genotoxic dose of resveratrol. Mol Biol Rep 40, 5013–5026 (2013).
Singh, S. et al. Human oncoprotein MDM2 activates the Akt signaling pathway through an interaction with the repressor element-1 silencing transcription factor conferring a survival advantage to cancer cells. Cell Death Differ 20, 558–566 (2013).
Thomasova, D., Mulay, S. R., Bruns, H. & Anders, H. J. p53-independent roles of MDM2 in NF-kappaB signaling: implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia 14, 1097–1101 (2012).
Mulay, S. R., Thomasova, D., Ryu, M. & Anders, H. J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int 81, 1199–1211 (2012).
Mulay, S. R. et al. Murine Double Minute-2 Inhibition Ameliorates Established Crescentic Glomerulonephritis. Am J Pathol 186, 1442–1453 (2016).
Thomasova, D. et al. MDM2 prevents spontaneous tubular epithelial cell death and acute kidney injury. Cell Death Dis 7, e2482 (2016).
Thomasova, D. et al. Murine Double Minute-2 Prevents p53-Overactivation-Related Cell Death (Podoptosis) of Podocytes. J Am Soc Nephrol 26, 1513–1523 (2015).
Ye, C. et al. MDM2 mediates fibroblast activation and renal tubulointerstitial fibrosis via a p53-independent pathway. Am J Physiol Renal Physiol 312, F760–F768 (2017).
Mulay, S. R. et al. Podocyte loss involves MDM2-driven mitotic catastrophe. J Pathol 230, 322–335 (2013).
Wolf, G., Sharma, K., Chen, Y., Ericksen, M. & Ziyadeh, F. N. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int 42, 647–656 (1992).
Liu, F. et al. The effect of FoxO1 on the proliferation of rat mesangial cells under high glucose conditions. Nephrol Dial Transplant 29, 1879–1887 (2014).
Allam, R., Sayyed, S. G., Kulkarni, O. P., Lichtnekert, J. & Anders, H. J. Mdm2 promotes systemic lupus erythematosus and lupus nephritis. J Am Soc Nephrol 22, 2016–2027 (2011).
Liu, L. et al. Notch Signaling Molecules Activate TGF- beta in Rat Mesangial Cells under High Glucose Conditions. J Diabetes Res 2013, 979702 (2013).
Pettersson, S. et al. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem J 450, 523–536 (2013).
Yogosawa, S., Miyauchi, Y., Honda, R., Tanaka, H. & Yasuda, H. Mammalian Numb is a target protein of Mdm2, ubiquitin ligase. Biochem Biophys Res Commun 302, 869–872 (2003).
McGill, M. A. & McGlade, C. J. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 278, 23196–23203 (2003).
Saito, R. et al. Systems biology analysis reveals role of MDM2 in diabetic nephropathy. JCI insight 1, e87877 (2016).
Hilton, M. J. et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med 14, 306–314 (2008).
Lasagni, L. et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem cells 28, 1674–1685 (2010).
Bray, S. J. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7, 678–689 (2006).
Bray, S. & Bernard, F. Notch targets and their regulation. Curr Top Dev Biol 92, 253–275 (2010).
Sweetwyne, M. T. et al. Notch1 and Notch2 in Podocytes Play Differential Roles During Diabetic Nephropathy Development. Diabetes 64, 4099–4111 (2015).
Gao, F. et al. Valsartan ameliorates podocyte loss in diabetic mice through the Notch pathway. Int J Mol Med 37, 1328–1336 (2016).
Lin, C. L. et al. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes 59, 1915–1925 (2010).
Liu, Y. et al. MDM2-MOF-H4K16ac axis contributes to tumorigenesis induced by Notch. FEBS J 281, 3315–3324 (2014).
Sun, Y. et al. Trp53 regulates Notch 4 signaling through Mdm2. J Cell Sci 124, 1067–1076 (2011).
Gulino, A., Di Marcotullio, L. & Screpanti, I. The multiple functions of Numb. Exp Cell Res 316, 900–906 (2010).
Pece, S. et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167, 215–221 (2004).
Sczaniecka, M. et al. MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J Biol Chem 287, 14052–14068 (2012).
Yang, W. L. et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 (2009).
Chen, Z. J. Ubiquitination in signaling to and activation of IKK. Immunol Rev 246, 95–106 (2012).
Steffes, M. W. et al. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int 59, 2104–2113 (2001).
Zhang, C. et al. Protection of podocytes from hyperhomocysteinemia-induced injury by deletion of the gp91phox gene. Free Radic Biol Med 48, 1109–1117 (2010).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81522010, No. 81470964, No. 81570671, No. 81570657, and No. 81400720), a grant from Wuhan Science and Technology Bureau (No. 2015060101010039), a Doctoral Fund from the Ministry of Education of China (No. 20130142110064), and the Academic Frontier Youth Team Program of Huazhong University of Science and Technology (2016).
Author information
Authors and Affiliations
Contributions
C.Z. conception and design of research; C.-T.L., H.T., C.Y., C.-Q.Y., J.Z., C.-Y.Z. and W.X. performed experiments; C.-T.L., H.S., H.T. and C.Y. analyzed data; C.Z., H.S. and C.-T.L. interpreted results of experiments; C.Z., C.-T.L., and H.S. prepared figures; C.Z. and C.-T.L. drafted manuscript; C.Z. and H.S. edited and revised manuscript; C.Z. approved final version of manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, CT., Tang, H., Ye, C. et al. MDM2 Contributes to High Glucose-Induced Glomerular Mesangial Cell Proliferation and Extracellular Matrix Accumulation via Notch1. Sci Rep 7, 10393 (2017). https://doi.org/10.1038/s41598-017-10927-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10927-5
This article is cited by
-
Asparaginyl endopeptidase protects against podocyte injury in diabetic nephropathy through cleaving cofilin-1
Cell Death & Disease (2022)
-
PHLPP1/Nrf2–Mdm2 axis induces renal apoptosis via influencing nucleo-cytoplasmic shuttling of FoxO1 during diabetic nephropathy
Molecular and Cellular Biochemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
