
Abstract
Eukaryotic N-glycosylation pathways are dependent of N-acetylglucosaminyltransferase I (GnTI), a key glycosyltransferase opening the door to the formation of complex-type N-glycans by transferring a N-acetylglucosamine residue onto the Man5GlcNAc2 intermediate. In contrast, glycans N-linked to Chlamydomonas reinhardtii proteins arise from a GnTI-independent Golgi processing of oligomannosides giving rise to Man5GlcNAc2 substituted eventually with one or two xylose(s). Here, complementation of C. reinhardtii with heterologous GnTI was investigated by expression of GnTI cDNAs originated from Arabidopsis and the diatom Phaeodactylum tricornutum. No modification of the N-glycans was observed in the GnTI transformed cells. Consequently, the structure of the Man5GlcNAc2 synthesized by C. reinhardtii was reinvestigated. Mass spectrometry analyses combined with enzyme sequencing showed that C. reinhardtii proteins carry linear Man5GlcNAc2 instead of the branched structure usually found in eukaryotes. Moreover, characterization of the lipid-linked oligosaccharide precursor demonstrated that C. reinhardtii exhibit a Glc3Man5GlcNAc2 dolichol pyrophosphate precursor. We propose that this precursor is then trimmed into a linear Man5GlcNAc2 that is not substrate for GnTI. Furthermore, cells expressing GnTI exhibited an altered phenotype with large vacuoles, increase of ROS production and accumulation of starch granules, suggesting the activation of stress responses likely due to the perturbation of the Golgi apparatus.
Similar content being viewed by others

Introduction
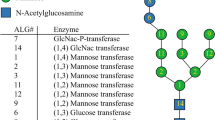
N-linked glycosylation is an extensive eukaryotic post-translational modification of secreted proteins consisting of the covalent attachment of an oligosaccharide onto asparagine residues belonging to the consensus sequence Asn-X-Ser/Thr/Cys, where X represents any amino acid except proline1,2,3,4. The process starts in the endoplasmic reticulum (ER) with the biosynthesis of a lipid-linked oligosaccharide (LLO) precursor which involves the action of a conserved set of enzymes named Asparagine-Linked Glycosylation (ALG). First steps of the LLO synthesis occur on the cytosolic face of the ER membrane where N-acetylglucosamine (GlcNAc) and mannose (Man) residues are added step by step onto a membrane-anchor dolichol pyrophosphate (Dol-PP) to form a Man5GlcNAc2-PP-Dol. Then, the Man5GlcNAc2-PP-Dol is flipped into the lumen of the ER5, 6 where its extension occurs by the addition of several Man and glucose (Glc) residues into a complete oligosaccharide precursor Glc3Man9GlcNAc2-PP-Dol7. This LLO is thereafter transferred by the oligosaccharyltransferase (OST) complex onto the asparagine of the N-glycosylation consensus sequence of the nascent polypeptides8. The N-glycan is then trimmed by the action of α-glucosidases I and II, and eventually an ER-mannosidase into Man8-9GlcNAc2 oligomannoside. During these ER events, interactions between the glycoprotein and ER-resident chaperones ensure the glycoprotein folding9, 10. Correctly folded glycoproteins then enter the Golgi apparatus where their N-glycans are processed through the action of an α-mannosidase I into Man5GlcNAc2. Subsequently, N-acetylglucosaminyltransferase I (GnTI), α-mannosidase II and finally N-acetylglucosaminyltransferase II (GnTII) give rise to the canonical GlcNAc2Man3GlcNAc2 core common to mammals, insects and land plants11, 12. This core undergoes further maturation into organism-specific complex-type N-glycans that are involved in several physiological functions like cell-cell interaction, intracellular communication and signaling13, 14.
The structures of N-linked glycans of the green microalgae C. reinhardtii were recently characterized15. Predominant N-glycans carried by C. reinhardtii endogenous proteins are oligomannosides ranging from Man2GlcNAc2 to Man5GlcNAc2. In addition, mature N-glycans were identified as oligomannosides Man4GlcNAc2 and Man5GlcNAc2 presenting methylation on mannose residues and substituted by one or two xylose residues15. The absence of terminal GlcNAc residues on these N-linked glycans suggested that mature N-glycans in C. reinhardtii are derived from GnTI-independent Golgi events11, 15. In silico analysis of the C. reinhardtii genome allowed the identification of numerous candidate genes encoding enzymes of the protein N-glycosylation pathway, such as the ALG, OST subunits, glycosidases and glycosyltransferases involved either in the ER or Golgi N-glycan biosynthesis steps11, 15. However, in agreement with biochemical analyses, no GnTI candidate is predicted in C. reinhardtii genome suggesting that the N-glycosylation pathway in this organism is different from that found in land plants, insects, mammals12, 16 and diatoms for which a functional GnTI has been recently characterized17.
Considering the difference in the N-glycosylation pathways between C. reinhardtii and land plants, as well as between C. reinhardtii and diatoms, we have undertaken the expression of either a diatom or a plant GnTI in C. reinhardtii to investigate the effects of such a complementation on the physiology and N-glycan pathway in C. reinhardtii. Our findings demonstrate that the expression of GnTI induces stress responses in C. reinhardtii and reveal a truncated ER N-glycosylation pathway in C. reinhardtii.
Results
The commonly used cw92 laboratory strain was transformed with the codon optimized sequences encoding for the catalytically active GnTI from Arabidopsis (AtGnTI) or from the diatom Phaeodactylum tricornutum (PtGnTI). Both have been shown to be able to process N-linked glycans. AtGnTI (At4g38240) encodes for a Golgi enzyme that is responsible for the transfer of a terminal GlcNAc residue onto Man5GlcNAc2 N-glycan18, 19. PtGnTI has been shown to restore in vivo the biosynthesis of complex-type N-glycans in CHO Lec1 mutant that lacks endogenous GnTI activity17. These sequences fused to a tag sequence encoding for a V5-epitope were used for nuclear expression in C. reinhardtii 20,21,22. RT-PCR analyses show that both plant and diatom GnTI were expressed in C. reinhardtii transformed lines (Fig. 1 and Fig. S1). Further experiments were focused on two AtGnTI and four PtGnTI expressing lines, all of which exhibit the highest transcription levels (Fig. 1a). Furthermore, using anti-V5 epitope antibodies, a signal around 56 kDa was immunodetected on a western blot in the microsomal fraction isolated from the transgenic lines but not in that of the non-transformed cells (Fig. 1b and Fig. S2).

(a) RT-PCR analysis of GnTI transcription level in cw92 cells, cell lines transformed with AtGnTI-V5 (lines AtGnTI#3 and AtGnTI#5) or PtGnTI-V5 (lines PtGnTI#1, PtGnTI#6, PtGnTI#8, PtGnTI#10 and PtGnTI#11) using AtGnTI (1) or PtGnTI (2) specific primers. Actin (lower panel) was used as an RT-PCR control. (b) Immunodetection of recombinant GnTI in the microsomal fraction isolated from cw92 cells and AtGnTI#3 lines respectively. The immunodetection was performed using an anti-V5 antibody as a primary antibody. A protein extract from CHO cells expressing PtGnTI-V5 (+) was used as a positive control17. Full images of the agarose gel and the Western blot are presented in Figs S1 and S2.
The phenotypic analysis of C. reinhardtii transformed cell lines revealed that the expression of AtGnTI or PtGnTI was correlated to a modification of the cell size. Indeed, measurements of longitudinal cell diameters showed that cells expressing AtGnTI or PtGnTI were statistically enlarged as compared to the cw92 cells and cells transformed with the empty vector (Fig. 2a). Despite the size difference, the growth rates of transformed cells were similar to those of cw92 cells indicating that the swelling observed in transformed lines did not alter cell growth (Fig. 2b).

(a) Measurement of the cell diameters of C. reinhardtii cells expressing AtGnTI or PtGnTI as compared to the cw92 cells and cells transformed with an empty vector (Kruskal-Wallis test with n > 200 and p-value fixed at 0.05; stars indicate the significant level of the test). (b) Growth rate of cw92 and transformed cell lines grown in TAP medium. (c,d) Ultrastructure of cw92 cells (c) and transformed cell line AtGnTI#3 (d) by Transmission Electron Microscopy (TEM). chl: chloroplast, fl: flagella, G: Golgi apparatus, m: mitochondrion, n: nucleus, P: pyrenoid, s: starch granules, cv: contractile vacuoles, lv: large vesicles. (e) ROS levels in transformed cell lines determined through the oxidation measurement of CMH spin probe by electron paramagnetic resonance spectroscopy. The ROS level (arbitrary units/12 × 104 cells hour−1) in each transformed cell line was normalized against ROS level measured in cw92 cells. After normalization, a statistical test was performed between GnTI expressing cell lines and cells transformed with the empty vector using Ordinary One-Way ANOVA with n = 3 and p-value fixed at 0.05.
To examine whether the ultrastructure of swollen cells is altered, cells were high pressure frozen and analyzed by thin-section electron microscopy. To further characterize the cell swelling, the ultrastructure of the transformed lines was analyzed by Transmission Electron Microscopy (TEM). As illustrated in Fig. 2c,d, GnTI expressing cells exhibited a large number of starch granules. In addition, large vesicles appeared in the GnT I expressing lines as compared to the cw92 and cells transformed with the empty vector. In order to check if the increase in size and the starch accumulation were associated to a stress phenotype in transformed cells, we measured the production of reactive oxygen species (ROS) in the different lines using electron paramagnetic resonance (EPR) spectroscopy. Figure 2e shows that ROS levels were significantly higher in the cells expressing AtGnTI or PtGnTI than in the cw92 cells, except for PtGnTI#1 line, in which ROS content was not significantly increased.
Protein N-glycosylation in cw92 and transformed cells were then investigated by mass spectrometry profiling. N-linked glycans were released from total protein extracts using PNGase F, labeled with 2-aminobenzamide (2AB) and analyzed by liquid chromatography coupled to electrospray ionization mass spectrometer (LC-ESI-MS). Although the expression of GnTI induced altered phenotypes in C. reinhardtii, the comparison of the N-glycan structures between cw92 cells and the transformed lines did not reveal any modification of the N-glycan profiles (Fig. S3). A specific search for mono- and discharged ions corresponding to N-glycans exhibiting extra terminal GlcNAc residue was unsuccessful. This suggested that the expression of AtGnTI or PtGnTI did not affect, in a detectable manner, the N-glycosylation of endogenous proteins in C reinhardtii.
Different scenarios may explain the inability of exogenous GnTI to affect the protein N-glycan profiles in C. reinhardtii. This may result from a very weak protein expression of active GnTI in the Golgi apparatus or its mislocalization in this organelle. Absence of the appropriate substrates in the Golgi apparatus could also explain the GnTI inactivity. This concerns the nucleotide-sugar (UDP-GlcNAc) or the oligomannoside Man5GlcNAc2 N-linked to secreted proteins. Man5GlcNAc2 oligomannoside was previously identified in C. reinhardtii cw92 proteins and was assigned to a branched Man5GlcNAc2 by analogy with data published regarding the Golgi N-glycan processing in eukaryotes15. The non-effect of GnTI on the N-glycan profiles from C. reinhardtii raised questions regarding the structure of this Man5GlcNAc2, which therefore needs to be reinvestigated. Here, we made use of Ion Mobility Spectrometry–Mass Spectrometry (IMS-MS), a reliable analytical method which allows the separation of isomers including oligosaccharide isomers23,24,25. In this analysis, the ion mobility of Man5GlcNAc2 coupled to 2AB oligosaccharide (sodium adduct) prepared from C. reinhardtii proteins was compared to the ion mobility of branched sodiated 2AB labeled Man5GlcNAc2 obtained from the bovine ribonuclease B. As illustrated in Fig. 3, Man5GlcNAc2-2AB from C. reinhardtii and from bovine ribonuclease B exhibited different ion mobilities (drift time of 10.12 ms and 11.28 ms, respectively) which suggested that they possess distinct structures. This data indicated that Man5GlcNAc2 in C. reinhardtii is different from the branched Man5GlcNAc2 that results from the trimming of oligomannosides Man8-9GlcNAc2 in the ER and the Golgi apparatus in plants and mammals.

(a–c) Ion mobility spectra of Man5GlcNAc2-2AB derivatives prepared from two independent preparations of C. reinhardtii proteins (a,b) and bovine ribonuclease B (c) (M + Na+ adducts).
The structure of C. reinhardtii Man5GlcNAc2 isomer was further investigated using electrospray ionization-multistage tandem mass spectrometry (ESI-MSn) (with n = 2, n = 3 and n = 4) taking advantage of specific ion transitions reported in ref. 26 (Table 1). Fragmentation patterns of the doubly charged [M + 2Na]2+ of permethylated Man5GlcNAc2-2AB (m/z 882) prepared from proteins of PtGnTI#1 transformed line and from bovine ribonuclease B were compared (Fig. 4). The MS3 “m/z 882 → m/z 1302 → product ions” analysis of PtGnTI#1 N-glycans revealed the presence of discriminant product ions m/z 880 and m/z 676 (Fig. 4a, middle panel) that are not detected for the permethylated Man5GlcNAc2-2AB released from bovine ribonuclease B (Fig. 4b, middle panel). Moreover, the MS4 “m/z 882 → m/z 1302 → m/z 880 → product ions’’ analysis showed that m/z 880, which arose from the neutral loss of two terminal mannose residues from m/z 1302, fragments into m/z 676 (Fig. 4a, lower panel). Such a fragmentation pattern does not correspond to that of a branched Man5GlcNAc2 isomer, but to the one of a linear Man5GlcNAc2 structure26 (Table 1). Similar fragmentation patterns were observed for permethylated Man5GlcNAc2-2AB prepared from cw92 cells and AtGnTI cells (Fig. S4). In contrast, the fragmentation pattern deduced from the MS4 analysis of the m/z 882 for Man5GlcNAc2-2AB isolated from bovine ribonuclease B revealed the specific fragmentations m/z 882 → m/z 1302 → m/z 866 → m/z 648, as it is expected for the branched isomer of Man5GlcNAc2-2AB (Fig. 4b)26 (Table 1). In conclusion, the comparison of ESI-MSn fragmentation patterns suggested that C. reinhardtii proteins carry linear Man5GlcNAc2 (Fig. 5d) instead of the conventional branched isomer (Fig. 5c).

ESI-MSn spectra with n = 2 (upper panel), n = 3 (middle panel) and n = 4 (lower panel) of permethylated Man5GlcNAc2-2AB derivative (m/z 882 corresponding to [M + 2Na]2+ precursor ion) isolated from PtGnTI C. reinhardtii proteins (a) and from Ribonuclease B (b) On each panel, the precursor ion selected for the fragmentation analysis is shown with a diamond and its fragmentation pattern is proposed according to one of the possible structures. 2AB: 2-aminobenzamide; Black square: GlcNAc; grey circle: Man.

N-glycan biosynthesis in land plants and mammals (a) and proposed pathway in C. reinhardtii (b). Asn: asparagine residue of the N-glycosylation site (Asn-X-S/T/C). PP-Dol: dolichol pyrophosphate; Black square: GlcNAc; grey circle: Man; Star: xylose and Me: methyl substituent. Detailed structure of Man5GlcNAc2 in land plants and mammals (c) and in C. reinhardtii (d).
According to the N-glycan biosynthesis pathway described in land plants and mammals, a linear Man5GlcNAc2 is meant to exhibit terminal α(1,2/6)-Man residues instead of α(1,3/6)-Man. For further confirmation of the C. reinhardtii Man5GlcNAc2 topology, the N-glycan mixture was submitted to exoglycosidase degradations using either Jack bean mannosidase, a non-specific α-mannosidase and Aspergillus saitoi mannosidase, an exoglycosidase specific for α(1,2)-mannose residues27. Man5GlcNAc2-2AB from C. reinhardtii was efficiently converted into ManGlcNAc2-2AB by Jack bean mannosidase after removal of the four α-mannose units. In contrast, Man3GlcNAc2-2AB was the major end product after treatment with Aspergillus saitoi α(1,2)-mannosidase that results from the removal of two terminal α(1,2)-mannose residues from the linear trimannoside arm (Fig. S5). Taken together, both mass spectrometry and enzyme sequencing analyses demonstrated that C. reinhardtii glycoproteins carry a non-canonical linear Man5GlcNAc2, as depicted in Fig. 5d.
The occurrence of a linear Man5GlcNAc2 oligosaccharide onto C. reinhardtii gycoproteins may result either from the trimming of Man8-9GlcNAc2 in the Golgi apparatus by the action of α-mannosidases or from a truncated biosynthesis of the LLO in the ER (Fig. 5). The latter hypothesis was investigated by isolation and analysis of C. reinhardtii LLO. LLO were isolated from cw92 microsomal preparations of C. reinhardtii using a methanol/chloroform extraction procedure according to a protocol adapted from27, 28. The oligosaccharide was hydrolyzed from the PP-dolichol anchor by mild acidic cleavage and then permethylated as previously reported27, 29. A predominant ion at m/z 2192.7 was observed using MALDI-TOF-MS and could be assigned to a permethylated oligosaccharide containing 8 hexoses and 2 HexNAc residues (Hex8HexNAc2) (Fig. S6). A minor ion at m/z 1987.6 could be assigned to Hex7HexNAc2 (Fig. S6). The sequence of this oligosaccharide was analyzed by ESI-MSn and compared to the one isolated from LLO of the YG170 (alg3) yeast mutant (Fig. 6). Indeed, this strain lacks α1,3-mannosyltransferase ALG3 activity and accumulates Glc3Man5GlcNAc2 LLO in the ER30, 31. Both ESI-MSn data exhibited similar fragmentation patterns that are consistent with a linear arrangement in the oligosaccharide. Taken together, these analyses demonstrated that C. reinhardtii accumulates a predominant linear truncated Glc3Man5GlcNAc2 LLO in the ER (Fig. 5b).

ESI-MSn spectra with n = 2 (upper panel), n = 3 (middle panel + lower panel (a)) and n = 4 (lower panel (b)) of permethylated LLO derivative (m/z 1107 corresponding to [M + 2Na]2+ precursor ion) from lipid-linked oligosaccharides of cw92 C. reinhardtii cells (a) and the YG170 yeast mutant cells (b). On each panel, the precursor ion selected for the fragmentation analysis is shown with a diamond and its fragmentation pattern is proposed. Black square: GlcNAc; grey circle: Man; black circle: Glc.
Discussion
In previous work, we characterized N-glycan structures linked to endogenous proteins in C. reinhardtii and showed they were mostly oligomannosides and for 30% novel mature structures containing xylose residues and methylated mannoses on Man4-5GlcNAc2 15. These structures resulted from a Golgi GnTI-independent processing of oligomannosides as the bioinformatics analysis of the genome has revealed that it lacks GnTI. Furthermore, no N-glycan harboring terminal GlcNAc residues has been identified in the whole glycan population11, 15. This situation contrasts with most eukaryotic organisms in which the GnTI is a key Golgi enzyme in the N-glycosylation pathway opening the door to the biosynthesis of structurally diverse mature N-glycans12. In this canonical pathway, the addition of a first GlcNAc by GnTI is required for the sequential activity of a large repertoire of other specific transferases giving rise to complex N-glycans involved in numerous biological processes, such as intracellular communication and signaling13, 14. Inactivation of GnTI in those organisms induces strong developmental phenotypes. For instance, GnTI-null embryos of mouse die at about 10 days after fertilization indicating that mature N-glycans are required for morphogenesis in mammals32, 33. Also, inactivation of GnTI reduces the viability in worm and fly34, 35. In plants, GnT I mutants exhibit a stress phenotype, thus suggesting a role for mature N-glycans in specific physiological processes36, 37. In rice, gntI mutants, impaired in the N-glycans maturation showed severe developmental defects, resulting in early lethality, associated to reduced sensitivity to cytokinin36.
GnTI genes are predicted in different microalgae genomes. In the diatom P. tricornutum, PtGnTI was demonstrated to encode for an active glycosyltransferase17. Therefore, the capacity of C. reinhardtii to express GnTI from Arabidopsis and from P. tricornutum was analyzed to investigate whether C. reinhardtii N-glycan biosynthesis can be complemented with this key transferase and shift into a GnTI – dependent pathway. Moreover, in a biotech context, the production, in microalgae, of recombinant glycoproteins for therapeutic applications will require the engineering of their endogenous N-glycosylation pathway for the production of biopharmaceuticals exhibiting human-compatible N-glycans. Therefore, implementation of a GnTI - dependent pathway is a prerequisite for any production of glycosylated biopharmaceuticals in C. reinhardtii.
Transgenic lines expressing the Arabidopsis or diatom GnTI were obtained and GnTI protein was immunodetected as expected in the microsomal fraction. However, further experiments are required to confirm the GnTI localization within the Golgi apparatus. Mass spectrometry analyses of N-glycan profiles from proteins secreted in transgenic lines did not show any modification of the N-glycan population by comparison with the cw92 cells. Among the different scenarios that may explain this result, the absence of UDP-GlcNAc nucleotide sugar in the Golgi apparatus was first considered. The transport of the cytosolic nucleotide sugars across the Golgi membrane is performed by Nucleotide Sugar Transporters (NSTs). Searching for NST orthologues in the C. reinhardtii genome allowed the identification of 23 candidate genes of which the deduced amino-acid sequences harbor the characteristic Triose Phosphate Translocator (TPT) domain (Pfam 03151) present in NSTs (Mathieu-Rivet et al., in press). However, as most of the characterized NSTs has been shown to transport at least two distinct substrates, determination of their specificity for nucleotide sugars based on sequence homologies with other Golgi NSTs remains difficult without additional biochemical evidence38. Therefore, further experimental work is needed to determine whether a specific Golgi UDP-GlcNAc transporter exists in C. reinhardtii. In relation to this, we cannot ruled out that the cytosolic abundance of UDP-GlcNAc may not be sufficient to supply the Golgi apparatus which would consequently limit GnTI in C. reinhardtii.
Inability of GnTI to affect the endogenous N-glycan population in C. reinhardtii transformed lines may also result from the absence of the appropriate glycan substrate. Man5GlcNAc2 oligomannoside was previously identified in C. reinhardtii 15 and considered as being the branched isomer substrate for GnTI by analogy with mammals and plants N-glycan pathways. Its structure was reinvestigated by mass spectrometry and enzyme sequencing and compared to a branched Man5GlcNAc2 standard from mammals26. Ion mobilities determined by IMS-MS and fragmentation patterns resulting from ESI-MSn clearly show that C. reinhardtii and mammalian oligomannosides differ in their shape and sequence. Moreover, the fragmentation pattern determined by MSn and enzyme digestion with an α(1,2)-mannosidase are consistent with the presence of a linear Man5GlcNAc2 on C. reinhardtii proteins as depicted in Fig. 5d. This oligosaccharide may result from a truncated LLO biosynthesis occurring in the ER (Fig. 5). Indeed, ALG3, ALG9 and ALG12 candidates are not predicted in the C. reinhardtii genome15, 39, 40. These ER enzymes are involved in the completion of the biosynthesis of the LLO precursor Man9GlcNAc2-PP-Dol prior to its glucosylation (Fig. 5a). As a consequence, the absence of ALG3, ALG9 and ALG12 activities results in the secretion, from ER, of proteins carrying a linear Man5GlcNAc2 instead of Man8-9GlcNAc2 (Fig. 5b). In contrast, in land plants and mammals, branched Man5GlcNAc2 is obtained by the trimming of mannose residues of Man8-9GlcNAc2 oligomannosides by Golgi α-mannosidases (Fig. 5a). However, linear Man5GlcNAc2 oligomannoside could also result from the trimming of Man8-9GlcNAc2 by Golgi α-mannosidases predicted in C. reinhardtii genome11, 15 but having different glycan specificities as compared to those of homologous enzymes involved in mammalian and plant N-glycan pathways. To discriminate between the two possibilities, C. reinhardtii LLO was isolated, characterized by mass spectrometry analysis and compared with the one extracted from the ALG3 deficient yeast mutant31. These analyses demonstrated that this microalga accumulates a linear Glc3Man5GlcNAc2 (Fig. 6 and Fig. S6). Such a truncated ER pathway has been already characterized in some unicellular organisms such as the coccidian parasites Toxoplasma and Cryptosporidium41,42,43,44,45. In conclusion, data obtained in this study revealed a truncated ER N-glycan pathway in C. reinhardtii and required the reevaluation of the previously published N-glycan pathway15. In this reassessed N-glycan processing, Man5GlcNAc2 results from the deglucosylation of Glc3Man5GlcNAc2 precursor in the ER and its methylation and xylosylation in the Golgi apparatus (Fig. 5). Location of xylose residues on Man5GlcNAc2 was reinvestigated by ESI-MSn (Fig. S7). This analysis confirmed the presence of the first xylose residue onto the β-Man15. In addition, the second xylose could be positioned either on the α(1, 3)-linked Man (I) or the first α(1, 2)-linked Man (II). MS4 experiments are more consistent with the structure I depicted in Fig. 5.
Surprisingly, cells expressing GnTI exhibited an altered phenotype although no modification of the N-glycans has been observed. The presence of large vacuoles as well as the increase of ROS production observed in transformed cell lines suggested that transformation with GnTI induced the activation of stress responses such as autophagy and oxidative stress46, 47. Previously, Pérez-Martin and collaborators showed that ER stress caused by tunicamycin or DTT triggers autophagy48. It can be hypothesized that AtGnTI or PtGnTI may localize in the ER of C. reinhardtii. Indeed, a previous study published by Schoberer and coworkers in 2009 demonstrated that overexpressed GnTI in tobacco recruit the ER protein Sar1p resulting in a GnTI-Sar1p association in the ER membranes49.
In addition, chlamydomonas cells expressing AtGnTI or PtGnTI accumulate starch. In BY2 cells and chlamydomonas noctigama, Hummel and collaborators50 reported in 2010 that the disassembly of the Golgi apparatus using the secretion inhibitor Brefeldin A (BFA) caused the accumulation of plastid starch. This starch increase was also observed when the CopII-mediated ER to Golgi transport was inhibited in tobacco plants expressing a dominant negative version of the small GTPase Sar1p51. Moreover, it was proposed that disruption of the Golgi apparatus resulting in the loss of secretory activity may cause the redirection of free carbohydrates to the plastids where they would be converted into starch51.
Therefore, we postulate here that the heterologous expression of GnTI may affect Golgi organization or the endosomal system, which would consequently disturb the biosynthesis of glycans or glycoconjugates and the cell biology of C. reinhardtii. Additional work and phenotypic studies of transformants expressing a non-functional GnTI would allow the confirmation of this hypothesis.
Methods
Strains and growth conditions
CC-503 cw92 strain, later called cw92 cells, was obtained from the Chlamydomonas Culture Collection at Duke University (Durham, NC, USA) and grown in batch cultures at 25 °C, illuminated with 150 μmol m−2 s−1 using TAP medium redirection of free carbohydrates to the plastids where they would be converted into starch52.
Vector construction and transformation
The sequences encoding for AtGnTI (At4g38240) and PtGnTI (gi: 307604450) fused to the V5 epitope17 were codon optimized and synthesized by Sloning Biotechnology. Optimized sequences were cloned as a XbaI/NdeI fragment in the pSL18 vector53. pSL18 (empty vector), pSL18-AtGnTI or pSL18-PtGnTI construct was introduced using the glass beads method54. To detect the transgene in genomic DNA, screening was performed by PCR using V5-reverse GGAGTCCAGGCCCAGCAGG and with AtGnTI-forward GCCCTAAGTGGCCAAGGC or PtGnT1-forward CCAGTCCAAGTGGCCGGGC as primers.
RT-PCR analysis
Total RNA were isolated from fresh cell pellets (1.5 × 106 cells), using TRIzol (Invitrogen). gDNA contamination was removed by a Turbo DNase treatment (Thermo Fisher Scientific). Reverse transcription was performed on 2 µg of RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The transcription level of AtGnTI and PtGnTI transgenes was analyzed by PCR using the V5-reverse primer, with either AtGnTI-forward or PtGnT1-forward primers. Each PCR contained 2 µL of diluted cDNA (1/10), 0.8 mM of each primer, 0.25 mM dNTPs and 0.6 U GoTaq polymerase (Promega) and was performed in the reaction buffer provided by the manufacturer, according the following program: 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 62 °C for 30 s, 72 °C for 30 s and a final step of 72 °C for 5 min. The expression of the actin gene was monitored as a control using CrActin-forward CGCTGGAGAAGACCTACGAG and CrActin-reverse GGAGTTGAAGGTGGTGTCGT as primers.
Microsomal preparations and Western-blot analysis
1.4 × 107 of C. reinhardtii cells were collected (2,500 g for 5 min) and washed with a 20 mM potassium phosphate buffer at pH 7.4. All the following preparation steps were carried out at 4 °C. The cell pellet was broken with 2 mL of protease inhibitor cocktail 25X (Roche) dissolved in 10 mM potassium phosphate buffer (pH 7.4) using the FastPrep-24™ 5G15. Samples were then spun (300 g for 3 min) in order to remove intact cells and debris. The supernatant was collected and centrifuged (20,000 g for 30 min) in order to eliminate pigments and chloroplasts. Finally, the supernatant was ultracentrifuged (100,000 g for 1 h) to pellet the microsomal fraction. Immunodetection using anti-V5 antibodies has been performed as described in ref. 17.
N-glycans preparation and derivatization
Total proteins were extracted and deglycosylated using Peptide N-glycosidase F (PNGase F) as described in ref. 15. The released N-glycans were then labeled with 2-aminobenzamide (2AB) according to ref. 55. Excess of reagent was removed using a cartridge D1 from Ludger. Freeze dried samples were resuspended in 10 µL of water. Jack bean mannosidase (Sigma M7944) or Aspergillus saitoi α(1,2)-mannosidase (ProZyme) enzymatic treatments were performed on 4.5 µL of 2AB-labeled N-glycans according to the manufacturer’s instructions.
Permethylation - The 2AB-labeled N-glycans were permethylated29 and cleaned-up according to ref. 56.
LC-ESI-MS
The analyses were performed using the nano-LC1200 system coupled to a QTOF 6550 mass spectrometer equipped with a nanospray source and a LC-Chip Cube interface (Agilent Technologies, les Ulis, France). Briefly, 2AB-labeled N-glycans were enriched and desalted on a 500 nL PGC trap column and separated on a PGC (3-μm particle size) column (150 mm long × 75 μm inner diameter, Agilent Technologies). A 30-min linear gradient (5–75% acetonitrile in 0.1% formic acid) at a flow rate of 400 nL min−1 was used. Separated N-glycans were analyzed with the QTOF analyser. Full autoMS scans from 290 to 1,700 m/z and autoMS/MS from 59 to 1,700 m/z were recorded. In every cycle, a maximum of 5 precursors sorted by charge state (1+ and 2+ preferred) were isolated and fragmented in the collision cell with fixed collision cell energy at 15 eV. Scan speed raise based on precursor abundance (target 5,000 counts/spectrum) and precursors sorted only by abundance. Active exclusion of precursors was enabled and the threshold for precursor selection was set to 1,000 counts. The ESI acquisition parameters in positive mode were: capillary voltage; 1.9 kV, drying gas temperature; 250 °C, gas flow (air); 11 L min−1; fragmentor voltage, 360 V; Skimmer1 voltage, 65 V and OctopoleRFPeak voltage, 750 V.
IM-MS analyses
The ESI-IM-MS experiments were performed using a Waters SYNAPT G2 hybrid quadrupole/HDMS instrument equipped with an ESI LockSpray™source, the MassLynx 4.1 and the DriftScope 2.2 softwares (Waters, Manchester, UK). The SYNAPT HDMS system was calibrated using sodium formate cluster ions (2 mg mL−1) and operated in ‘V’ resolution mode (resolution 20,000 FWHM). The ESI parameters were in positive ion mode: capillary voltage, 3 kV; sample cone voltage, 70 V; source temperature, 90 °C; desolvation temperature, 250 °C; desolvation gas flow (N2), 700 L h−1. The data were acquired using a 50–2000 m/z range with 1 s scan time and 0.02 s interscan delay. Sample solutions were infused into the source at a flow rate of 400 µL h−1 with a syringe pump (Cole-Palmer, Vernon Hills, Illinois, USA). The IMS conditions were optimized as followed: gas flow (N2), 90 mL min−1; IMS pressure, 3.05 mbar; wave height voltage, 40 V and T-wave velocity, 550 m s−1. The ion mobility spectra were fitted using the Origin 9.0 software (OriginLab).
ESI-MSn analyses
Permethylated 2AB-derivatives of N-glycans were analyzed by ESI-MSn (n = 1 to 4) using a Bruker HCT Ultra ETD II quadrupole ion trap (QIT) mass spectrometer equipped with the Esquire control 6.2 and Data Analysis 4.0 softwares (Bruker Daltonics, Bremen, Germany). The ESI parameters were as followed: capillary and end plate voltages respectively set at −3.5 kV and −3.0 kV in positive ion mode, skimmer and capillary exit voltages set at 40 V and 200, respectively, nebulizer gas (N2), pressure, drying gas (N2) flow rate and drying gas temperature were 10 psi, 7.0 L min−1 and 300 °C, respectively. Helium pressure in the ion trap was 1.2 × 10−5 mbar. The data were acquired using a 200–2200 m/z range, using a scan speed of 8,000 m/z per second. The number of ions entering the trap cell was automatically adjusted by controlling the accumulation time using the ion charge control (ICC) mode (target 200,000) with a maximum accumulation time of 50 ms. The injection low-mass cut-off (LMCO) value was m/z 100 for Man5GlcNAc2 derivatives and 140 for LLO precursor respectively. The values of spectra averages and rolling average were 6 and 2. ESI-MSn experiments were carried out by collision-induced dissociation (CID) using helium as the collision gas, isolation width of 2 m/z unit for the precursor ions and for the intermediate ions using a resonant excitation frequency with an amplitude from 0.8 to 1.0 Vp–p. Sample solutions were infused into the source at a flow rate of 300 µL h−1 by means of a syringe pump (Cole-Palmer, Vernon Hills, IL, USA).
Electron microscopy
High pressure freezing was performed with the freezer HPM100 Leica-microsystems. Prior to freezing, 72 h old C. reinhardtii cells were treated at room temperature 1 hour with 100 mM mannitol as cryoprotectant diluted in fresh culture medium. Pre-treated C. reinhardtii cells were transferred into an aluminium cryocapsule covered by soy lecithin dissolved at 100 mM in chloroform. Excess medium was absorbed by filter paper. After fixation on the loading device, samples were frozen according to a maximum cooling rate of 20,000 °C s−1 and a pressure of 2,100 bars. Samples were transferred to a freeze substitution automate (AFS2, Leica) pre-cooled to −110 °C. Samples were substituted in anhydrous acetone with 0.5% uranyl acetate and 0.5% osmium tetroxide at −90 °C for 72 h. The temperature was gradually raised to −60 °C using a gradient of +2 °C h−1 and stabilized during 12 h, then gradually raised to −30 °C by using the same gradient during 12 h and gradually raised again to +4 °C. Then, samples were rinsed twice at room temperature with anhydrous acetone. Infiltration was then processed in acetone-Spurr’s resin (Spurr 25%, 8 h at +4 °C; Spurr 50%, 16 h at +4 °C; Spurr 75%, 8 h at room temperature; Spurr 100%, 16 h at room temperature; Spurr 100%, 24 h at room temperature). The Spurr’s resin was finally polymerised at +50 °C during 24 h. Ultrathin sections (70 nm; ultracut UC6, Leica) were collected onto carbon-formvar-coated nickel grids. A classical staining using 0.5% uranyl acetate and 0.5% lead citrate was done before sections were observed in a Philips, FEI Tecnai 12 Biotwin transmission electron microscope operating at 80 kV, with ES500W Erlangshen CCD camera (Gatan).
Size measurements
Cells (1.5 × 107) were collected (4,500 g for 5 min). The cell pellet was incubated in TAP medium containing 4% of paraformaldehyde (PAF) (v/v) during 30 min, under shaking, at room temperature. Then, cells were washed four times and finally resuspended in 200 µL of TAP. 20 µL of fixed cells were spotted between slide and coverslip (Superfrost™Plus, Thermo Scientific) and were observed with a Leica DMI-6000B inverted microscope at magnification 400. Cell size was determined using fixed cells by measuring the cell longitudinal diameter with Image J software. The measurements were done in triplicates on a minimum of 200 cells (n > 200).
Reactive oxygen species measurement
Reactive oxygen species (ROS) production was evaluated by electron paramagnetic resonance (EPR) spectroscopy. Cells were incubated at room temperature in the dark for 60 min in Krebs-HEPES buffer containing 5 µM diethyl dithiocarbamate, 25 µM deferoxamine, and the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl pyrrolidine hydrochloride (CMH; 500 µM; Noxygen, Elzach, Germany). Spectra of the oxidized product of CMH (CM.) were recorded from frozen samples with a X-band spectrometer (MS-400; Magnetech, Berlin, Germany) with the following acquisition parameters: microwave power, 1 mW; modulation amplitude, 5 G; sweep time, 60 s; and 1 scans. After correction of the baseline, the total amplitude of the signal was measured and expressed in arbitrary units produced per 12 × 104 cells for 60 min. After EPR analyses, total chlorophyll was quantified for each sample according to ref. 57. The quantity of total chlorophyll was used to normalize the signal intensity evaluated by EPR for each sample. The results were statistically analyzed using GraphPad Prism® software. The ROS level in each transformed cell line was normalized against ROS level measured in cw92 cells. To determine the significant level, a statistical test was performed between the GnTI transformed cell lines and the lines transformed with the empty vector using Ordinary One-Way ANOVA with n = 3 and p-value fixed at 0.05.
LLO preparation
The LLO extraction was performed on microsomal fractions from C. reinhardtii according to a protocol adapted from27, 28. Released oligosaccharides were then lyophilized and resuspended in 500 µL of water and purified on a carbograph column (Hypersep Hypercarb, Thermo Scientific) using the manufacturer’s instructions.
References
Austen, B. M. & Marshall, R. D. Acid dissociation constants of glycopeptides. Biochem. J. 163, 31–38 (1977).
Gavel, Y. & von Heijne, G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433–442 (1990).
Gil, G.-C., Velander, W. H. & Van Cott, K. E. N-glycosylation microheterogeneity and site occupancy of an Asn-X-Cys sequon in plasma-derived and recombinant protein C. PROTEOMICS 9, 2555–2567 (2009).
Matsui, T. et al. N-glycosylation at noncanonical Asn-X-Cys sequences in plant cells. Glycobiology 21, 994–999 (2011).
Helenius, J. et al. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 415, 447–450 (2002).
Helenius, J. & Aebi, M. Transmembrane movement of dolichol linked carbohydrates during N-glycoprotein biosynthesis in the endoplasmic reticulum. - PubMed – NCBI, Available at: http://www.ncbi.nlm.nih.gov/pubmed/12137737. (Accessed: 28th March 2016) (2002).
Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1833, 2430–2437 (2013).
Mohorko, E., Glockshuber, R. & Aebi, M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 34, 869–878 (2011).
Williams, D. B. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 119, 615–623 (2006).
Määttänen, P., Gehring, K., Bergeron, J. J. M. & Thomas, D. Y. Protein quality control in the ER: The recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511 (2010).
Mathieu-Rivet, E. et al. Protein N-glycosylation in eukaryotic microalgae and its impact on the production of nuclear expressed biopharmaceuticals. Frontiers in plant science, section Plant Physiol. 5, 359 (2014).
Strasser, R. Plant protein glycosylation. Glycobiology cww023 (2016).
Varki, A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3, 97–130 (1993).
Gagneux, P. & Varki, A. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology 9, 747–755 (1999).
Mathieu-Rivet, E. et al. Exploring the N-glycosylation pathway in Chlamydomonas reinhardtii unravels novel complex structures. Mol. Cell. Proteomics 12, 3160–3183 (2013).
Lerouge, P. et al. in Protein Trafficking in Plant Cells 31–48 (Springer, 1998).
Baïet, B. et al. N-Glycans of Phaeodactylum tricornutum Diatom and Functional Characterization of Its N-Acetylglucosaminyltransferase I Enzyme. J. Biol. Chem. 286, 6152–6164 (2011).
Strasser, R. et al. Molecular cloning and characterization of cDNA coding for beta1, 2N-acetylglucosaminyltransferase I (GlcNAc-TI) from Nicotiana tabacum. Glycobiology 9, 779–785 (1999).
Wenderoth, I. & von Schaewen, A. Isolation and characterization of plant N-acetyl glucosaminyltransferase I (GntI) cDNA sequences. Functional analyses in the Arabidopsis cgl mutant and in antisense plants. Plant Physiol. 123, 1097–1108 (2000).
Fuhrmann, M. et al. Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mol. Biol. 55, 869–881 (2004).
Fuhrmann, M., Oertel, W. & Hegemann, P. A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in Chlamydomonas reinhardtii†. Plant J. 19, 353–361 (1999).
Shao, N. & Bock, R. A codon-optimized luciferase from Gaussia princeps facilitates the in vivo monitoring of gene expression in the model alga Chlamydomonas reinhardtii. Curr. Genet. 53, 381–388 (2008).
Pagel, K. & Harvey, D. J. Ion mobility-mass spectrometry of complex carbohydrates: collision cross sections of sodiated N-linked glycans. Anal. Chem. 85, 5138–5145 (2013).
Struwe, W. B., Pagel, K., Benesch, J. L. P., Harvey, D. J. & Campbell, M. P. GlycoMob: an ion mobility-mass spectrometry collision cross section database for glycomics. Glycoconj. J. 33, 399–404 (2016).
Zhu, F., Lee, S., Valentine, S. J., Reilly, J. P. & Clemmer, D. E. Mannose7 glycan isomer characterization by IMS-MS/MS analysis. J. Am. Soc. Mass Spectrom. 23, 2158–2166 (2012).
Prien, J. M., Ashline, D. J., Lapadula, A. J., Zhang, H. & Reinhold, V. N. The High Mannose Glycans from Bovine Ribonuclease B Isomer Characterization by Ion Trap MS. J. Am. Soc. Mass Spectrom. 20, 539–556 (2009).
Garénaux, E. et al. The Dual Origin of Toxoplasma gondii N -Glycans†. Biochemistry (Mosc.) 47, 12270–12276 (2008).
Pabst, M. et al. Isomeric analysis of oligomannosidic N-glycans and their dolichol-linked precursors. Glycobiology 22, 389–399 (2012).
Ciucanu, I. & Kerek, F. A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res 131, 209–217 (1998).
Huffaker, T. C. & Robbins, P. W. Yeast mutants deficient in protein glycosylation. Proc. Natl. Acad. Sci. 80, 7466–7470 (1983).
Zufferey, R. et al. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J. 14, 4949–4960 (1995).
Ioffe, E. & Stanley, P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. 91, 728–732 (1994).
Metzler, M. et al. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 13, 2056–2065 (1994).
Sarkar, M. et al. Null Mutations in Drosophila N-Acetylglucosaminyltransferase I Produce Defects in Locomotion and a Reduced Life Span. J. Biol. Chem. 281, 12776–12785 (2006).
Schachter, H. Mgat1-dependent N-glycans are essential for the normal development of both vertebrate and invertebrate metazoans. Semin. Cell Dev. Biol. 21, 609–615 (2010).
Fanata, W. I. D., Lee, K. H. & Son, B. H. N-glycan maturation is crucial for cytokinin-mediated development and cellulose synthesis in Oryza sativa. Plant J. 73, 966–979 (2013).
Kang, J. S. et al. Salt tolerance of Arabidopsis thaliana requires maturation of N-glycosylated proteins in the Golgi apparatus. Proc. Natl. Acad. Sci. 105, 5933–5938 (2008).
Orellana, A., Moraga, C., Araya, M. & Moreno, A. Overview of Nucleotide Sugar Transporter Gene Family Functions Across Multiple Species. J. Mol. Biol. 428, 3150–3165 (2016).
Gomord, V. et al. Plant-specific glycosylation patterns in the context of therapeutic protein production: PMP-specific glycosylation patterns. Plant Biotechnol. J. 8, 564–587 (2010).
Levy-Ontman, O. et al. Genes Involved in the Endoplasmic Reticulum N-Glycosylation Pathway of the Red Microalga Porphyridium sp.: A Bioinformatic Study. Int. J. Mol. Sci. 15, 2305–2326 (2014).
Fauquenoy, S. et al. Proteomics and Glycomics Analyses of N-Glycosylated Structures Involved in Toxoplasma gondii-Host Cell Interactions. Mol. Cell. Proteomics 7, 891–910 (2008).
Haserick, J. R., Leon, D. R., Samuelson, J. & Costello, C. E. Asparagine-Linked Glycans of Cryptosporidium parvum Contain a Single Long Arm, Are Barely Processed in the Endoplasmic Reticulum (ER) or Golgi, and Show a Strong Bias for Sites with Threonine. Mol. Cell. Proteomics 16, S42–S53 (2017).
Luk, F. C. Y., Johnson, T. M. & Beckers, C. J. N-linked glycosylation of proteins in the protozoan parasite Toxoplasma gondii. Mol. Biochem. Parasitol. 157, 169–178 (2008).
Samuelson, J. et al. The diversity of dolichol-linked precursors to Asn-linked glycans likely results from secondary loss of sets of glycosyltransferases. Proc. Natl. Acad. Sci. USA 102, 1548–1553 (2005).
Samuelson, J. & Robbins, P. W. Effects of N-glycan precursor length diversity on quality control of protein folding and on protein glycosylation. Semin. Cell Dev. Biol. 41, 121–128 (2015).
Pérez-Pérez, M. E., Couso, I. & Crespo, J. L. Carotenoid deficiency triggers autophagy in the model green alga Chlamydomonas reinhardtii. Autophagy 8, 376–388 (2012).
Pérez-Pérez, M. E. & Crespo, J. L. Autophagy in the model alga Chlamydomonas reinhardtii. Autophagy 6, 562–563 (2010).
Pérez-Martín, M., Pérez-Pérez, M. E., Lemaire, S. D. & Crespo, J. L. Oxidative stress contributes to autophagy induction in response to endoplasmic reticulum stress in Chlamydomonas reinhardtii. Plant Physiol. 166, 997–1008 (2014).
Schoberer, J. et al. Arginine/Lysine Residues in the Cytoplasmic Tail Promote ER Export of Plant Glycosylation Enzymes. Traffic 10, 101–115 (2009).
Hummel, E., Osterrieder, A., Robinson, D. G. & Hawes, C. Inhibition of Golgi function causes plastid starch accumulation. J. Exp. Bot. 61, 2603–2614 (2010).
Hummel, E., Schmickl, R., Hinz, G., Hillmer, S. & Robinson, D. G. Brefeldin A action and recovery in Chlamydomonas are rapid and involve fusion and fission of Golgi cisternae. Plant Biol. Stuttg. Ger. 9, 489–501 (2007).
Harris, E. H. The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use. Academic Press (2009).
Depège, N., Bellafiore, S. & Rochaix, J.-D. Role of Chloroplast Protein Kinase Stt7 in LHCII Phosphorylation and State Transition in Chlamydomonas. Science 299, 1572–1575 (2003).
Kindle, K. L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. 87, 1228–1232 (1990).
Séveno, M. et al. Plant N-glycan profiling of minute amounts of material. Anal. Biochem. 379, 66–72 (2008).
North, S. J. et al. Glycomics Profiling of Chinese Hamster Ovary Cell Glycosylation Mutants Reveals N-Glycans of a Novel Size and Complexity. J. Biol. Chem. 285, 5759–5775 (2010).
Arnon, D. I. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in beta vulgaris. Plant Physiol. 24, 1–15 (1949).
Acknowledgements
The authors acknowledge the University of Rouen Normandie, the Région Normandie, the Institut Universitaire de France (I.U.F.), the European regional development fund (ERDF) No. 31708 and the Labex SynOrg (ANR-11-LABX-0029) for financial support. They thank Prof Dr. Markus Aebi, Institute of Microbiology, Department of Biology, ETH Zürich, Switzerland for the kind gift of the YG170 yeast strain, Candice Prevost, Rodolphe Dumontier and Dr Abderrakib Zahid for technical support.
Author information
Authors and Affiliations
Contributions
P.L.; M.B., E.M.R. planned and designed of the research; G.V., P.L.L., J.V., C.P., C.L.B., M.L.W.B., P.C., S.B., performed experiments; G.V., P.L.L., J.V., C.L.B., I.R.J. and V.R. collected and analyzed data, G.V., P.L.L., J.V., M.B., P.L., C.A., E.M.R. interpreted data and A.D., P.L., M.B., E.M.R., G.V., P.L.L., C.L.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vanier, G., Lucas, PL., Loutelier-Bourhis, C. et al. Heterologous expression of the N-acetylglucosaminyltransferase I dictates a reinvestigation of the N-glycosylation pathway in Chlamydomonas reinhardtii . Sci Rep 7, 10156 (2017). https://doi.org/10.1038/s41598-017-10698-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10698-z
This article is cited by
-
Golgi fucosyltransferase 1 reveals its important role in α-1,4-fucose modification of N-glycan in CRISPR/Cas9 diatom Phaeodactylum tricornutum
Microbial Cell Factories (2023)
-
Algal glycobiotechnology: omics approaches for strain improvement
Microbial Cell Factories (2021)
-
N-glycans of the microalga Chlorella vulgaris are of the oligomannosidic type but highly methylated
Scientific Reports (2019)
-
Current state-of-the-art in plant-based antibody production systems
Biotechnology Letters (2019)
-
User-friendly extraction and multistage tandem mass spectrometry based analysis of lipid-linked oligosaccharides in microalgae
Plant Methods (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
