
Abstract
Type 2 diabetes (T2D) has many cardiovascular complications, including a thrombotic propensity. Many such chronic, inflammatory diseases are accompanied (and may be exacerbated, and possibly even largely caused) by amyloid fibril formation. Recognising that there are few strong genetic associations underpinning T2D, but that amyloidogenesis of amylin is closely involved, we have been seeking to understand what might trigger the disease. Serum levels of bacterial lipopolysaccharide are raised in T2D, and we recently showed that fibrin(ogen) polymerisation during blood clotting can be affected strongly by LPS. The selectivity was indicated by the regularisation of clotting by lipopolysaccharide-binding protein (LBP). Since coagulopathies are a hallmark of T2D, we wondered whether they might too be caused by LPS (and reversed by LBP). We show here, using SEM and confocal microscopy, that platelet-poor-plasma from subjects with T2D had a much greater propensity for hypercoagulability and for amyloidogenesis, and that these could both be reversed by LBP. These data imply that coagulopathies are an important feature of T2D, and may be driven by ‘hidden’ LPS. Given the prevalence of amyloid formation in the sequelae of diabetes, this opens up novel strategies for both the prevention and treatment of T2D.
Similar content being viewed by others


Introduction
There is an increasing recognition that many chronic, inflammatory diseases are accompanied (and may be exacerbated, and possibly even largely caused) by amyloid fibril formation1,2,3,4,5,6. An amyloid fibril (protein) is defined as: “a protein that is deposited as insoluble fibrils, mainly in the extracellular spaces of organs and tissues as a result of sequential changes in protein folding that result in a condition known as amyloidosis”7. The nature and location of the amyloid typically reflects the disease, such that Parkinson’s is accompanied by amyloid forms of α-synuclein in the substantia nigra pars compacta8,9,10, Alzheimer’s by fibrillar forms of Aβ1-40/42 in many parts of the brain11,12,13, and type 2 diabetes – a disease with few genome-wide associations14 – by amylin fibrils in the insulin-producing cells of the pancreas15,16,17,18,19,20,21,22,23,24,25.
One characteristic of these kinds of fibril is their diameter, which is typically in the range 10–20 nm. By contrast, another type of protein fibre is represented by the fibrin produced during blood clotting. Under normal conditions, the fibres forming the clot have an appearance in a scanning electron microscope like cooked spaghetti, and the fibres have diameters of the order of 80–110 nm26,27,28,29,30. We and others have observed that this diameter can be decreased to something closer to 40 nm in various vascular and inflammatory diseases, including stroke27 and diabetes31,32,33. This is accompanied by many other changes in overall clot morphology and properties (e.g. for diabetes31, 33,34,35,36,37,38,39).
Fibrin is produced by the polymerisation of the protein fibrinogen under the action of thrombin, that removes two fibrinopeptides, thereby allowing the fibrinogen to self-assemble via a ‘knobs and holes’ mechanism into fibres (e.g. refs 40,41,42,43,44), but with no particularly marked changes in secondary structure40, 45,46,47,48. A final, transglutaminase-based crosslinking step49 ensures the stability of the clot.
One hallmark of amyloid proteins is the formation of a rich β-sheet structure, perpendicular to the fibres with a characteristic spacing (observable in X-ray reflections) of 4.7–4.8 Å (e.g. refs 4, 50,51,52,53, while another is the strong fluorescence observable (when excited ca 440 nm) when thioflavin T is bound to them (e.g. refs 54,55,56,57,58,59). Normal amyloidosis is rather slow, so needs some kind of ‘trigger’.
Although fibrin too is an insoluble fibre, fibrinogen has generally not been considered as amyloidogenic, nor fibrin as an amyloid protein (although it can become so in the presence of a rare mutation in the fibrinogen a chain60,61,62,63). However, as part of an extensive study of anomalous blood clotting (e.g. refs 29, 30, 33, 64,65,66,67,68), we recently found48, 68,69,70 that the anomalous clotting was in fact amyloid in nature. The fact that it could be caused by tiny amounts of lipopolysaccharide was seen as consistent with a role for dormant bacteria in the aetiology of such diseases71,72,73,74,75,76,77,78. A particularly strong pointer was the fact that the amyloid formation was abolished when the LPS was administered together with a stoichiometric excess of human lipopolysaccharide binding protein (LBP)68.
There are also strong indications that both iron dysregulation64, 79, 80 and coagulopathies30, 67 accompany a variety of inflammatory diseases. Certainly, too, the amount of LPS is raised in diabetics81,82,83, and this can contribute to insulin resistance84. Thus, the question then arose as to whether the extent of fibrin-type amyloid in blood varies between type 2 diabetics and suitably matched controls, and whether the removal of any LPS using LBP affected this in any way. The present paper shows that the answers to both these questions are strongly in the affirmative.
Materials and Methods
Ethical statement
This study was approved by the Ethical Committee of the University of Pretoria (South Africa): ethics clearance number: 298/2016. A written form of informed consent was obtained from all donors (available on request). The methods were carried out in accordance with the approved guidelines. Blood was collected and methods were carried out in accordance with the relevant guidelines of the ethics committee. We adhered strictly to the Declaration of Helsinki.
Sample population
26 healthy individuals and 25 type 2 diabetes (T2D) individuals were included in the study. We have discussed the changes in clots of healthy individuals in the presence of LPS and LBP extensively in a recent publication68. The present healthy population therefore confirms repeatability of our previous results. Exclusion criteria for the healthy population were known inflammatory conditions such as asthma, human immunodeficiency virus (HIV) or tuberculosis, and risk factors associated with metabolic syndrome (including obesity/high BMI), smoking, and if female, being on contraceptive or hormone replacement treatment. This population did not take any anti-inflammatory medication. As our healthy population were aged between 22 and 91 with BMI < 24.9, we classify them as apparently healthy. We specifically chose not to include controls with increased BMI levels, as literature suggests that an increased BMI is associated with increased levels of inflammation, which may indeed be linked to amyloid formation; furthermore, obesity is also associated with increased endotoxin levels. As there are several levels of increased BMI, from only overweight to morbidly obese, the effect of BMI on clot structure, warrants a separate study.
T2D individuals were voluntarily recruited by their medical practitioner, from a Diabetic Clinic at the Steve Biko academic hospital, Pretoria, South Africa. Demographic data including age, gender, glucose level at time of sample collection, hemoglobin A1c (HbA1c) measures and medication used for typical comorbidities of T2D were recorded. Inclusion criteria consisted of both male and female participants, aged between 40 and 81 (oldest individual), with a diagnosis of T2D for more than 3 months prior to screening and without any signs of infection. Diabetic individuals were diagnosed per the Society for Endocrinology, Metabolism and Diabetes of South Africa (SEMSDA) guidelines. These guidelines follow the American Diabetes Association (ADA) criteria to define T2D. See Table 1 for medication usage. All the T2D individuals had a history of cardiovascular disease, and although a full history of co-morbidities including both macrovascular and microvascular co-morbidities are available in their patient records, we did not record the specific detail for the purposes of this study. All T2D had BMI’s of >25. Exclusion criteria for the T2D were conditions such as previously diagnosed cancer, asthma, human immunodeficiency virus (HIV) or tuberculosis, smoking, and if female, being on contraceptive or hormone replacement treatment. Ethical clearance was obtained from the Human Ethics Committee of the University of Pretoria. Whole blood of the participants was obtained in citrate tubes and platelet poor plasma (PPP) was used for confocal and SEM experiments.
LPS and LPS-binding protein
The LPS used was from E. coli O111:B4 (Sigma, L2630). A final LPS exposure concentration of 0.2 ng.L−1 and a final LPS-binding protein (LBP) exposure concentration of 2 ng.L−1 LBP was used.
Airyscan and scanning electron microscopy: healthy plasma samples: addition of LPS and LBP with thrombin to plasma
PPP was also incubated for 10 minutes, with either LPS (LPS exposure concentration: 0.2 ng.L−1) or LBP (final exposure concentration of 2 ng.L−1). Also, samples were prepared where PPP was incubated with LPS (10 minutes), followed by addition of LBP (incubated for 10 minutes). Adding thrombin to either naïve PPP or PPP with the various added products, in the ratio 1:2, created extensive fibrin networks. For Airyscan preparation, before the thrombin addition, we added Thioflavin T (ThT) at a final concentration of 5 μM to 200 μL of various prepared PPP samples (incubated for one minute, and protected from light). For Airyscan samples, a coverslip was placed over the prepared clot, while the clots were washed, fixed in 4% formaldehyde and prepared for SEM according to known SEM preparation methods. Samples were viewed using a Zeiss LSM 510 META confocal microscope with a Plan-Apochromat 163 and 100×/1.4 Oil DIC objective. Excitation was at 488 nm and emitted light was measured at 505–550 nm. A Zeiss cross beam electron microscope was used to study fibrin fibres.
Airyscan and scanning electron microscopy: type 2 diabetes samples: addition of LBP with thrombin to plasma
Extensive fibrin networks were created by adding thrombin to naïve T2D PPP in the ratio 1:2. PPP was also incubated for 10 minutes with LBP (final exposure concentration of 2 ng.L−1). SEM and Airyscan samples were prepared and viewed as for healthy samples.
Statistical analysis
The non-parametric Mann–Whitney U test (between controls and T2D samples) and the parametric T-test was performed (within samples) using the STATSDIRECT software.
Results
Table 1 shows demographics for the healthy and the T2D groups, including HbA1c levels of T2D and their medication usage. The healthy individuals were on no medication and did not suffer from any inflammatory condition. Our diabetes population contains patients that are mostly on a plethora of medications for cardiovascular co-morbidities e.g. hyperlipidaemia, hypertension and they have high HbA1c levels. We used the definition and the diagnostic criteria used in defining “diabetes” according to the Society for Endocrinology, Metabolism and Diabetes of South Africa (SEMSDA) guidelines. These guidelines follow the American Diabetes Association (ADA) criteria to define T2D (https://diabetes.medicinematters.com/guidelines/diagnosis/what-is-new-in-the-2017-american-diabetes-association-standards-/11932360).
Airyscan results are shown in Figs 1, 2, 3 and 4 for the healthy and T2D population.

Representative micrographs of PPP clots prepared from blood of two healthy individuals. (A and B) Micrographs were taken with a Zeiss LSM 510 META confocal microscope with a Plan-Apochromat 63x/1.4 Oil DIC objective. PPP was exposed to thioflavin T (ThT) (5 μM exposure concentration) followed by adding thrombin to create extensive fibrin clots.

(A and B) Low magnification micrographs of healthy PPP with added 0.2 ng.L−1 LPS Micrographs were taken with a Zeiss LSM 510 META confocal microscope with a Plan-Apochromat 63x/1.4 Oil DIC objective. All PPP was exposed to thioflavin T (ThT) (5 μM) followed by adding thrombin to create extensive fibrin clots. Areas of increased fluorescence are seen in the great majority of the clot area.

(A) A representative micrograph showing healthy PPP with added 0.2 ng.L−1 LPS (10 minutes exposure) followed by 2 ng.L−1 LPS-binding protein and thrombin. PPP with LPS and LBP was then exposed to thioflavin T (ThT) (5 μM exposure concentration) followed by adding thrombin to create extensive clot. Micrographs were taken with a Zeiss LSM 510 META confocal microscope with a Plan-Apochromatt 63x/1.4 Oil DIC objective. Nearly no fluorescence is visible.

(A and C) Micrographs of PPP with added thrombin to form extensive fibrin fibres from 2 individuals with type 2 diabetes; (B and D) PPP from same individuals, but exposed to 2 ng.L−1 LPS-binding protein followed by addition of thrombin. Thioflavin T (ThT) (5 μM) was added before thrombin. Micrographs were taken with a Zeiss LSM 510 META confocal microscope with a Plan-Apochromat 63x/1.4 Oil DIC objective. LBP dramatically reduced the fluorescence seen in samples from patients with T2D.
Airyscan super-resolution microscopy
Healthy plasma with added LPS and LBP (Figs 1, 2 and 3).
In healthy PPP, in the presence of ThT, little to no fluorescence was noted, although (as in Fig. 1) there were occasional very small patches of fluorescence. However, when LPS had been added to the mixture prior to thrombin, fluorescence was greatly enhanced, suggesting increased binding of ThT to β-sheet-rich areas on the fibrin(ogen) (Fig. 2). Previously, we concluded that LPS binding causes the fibrinogen to polymerise into a form with a greatly increased amount of ß-sheet (in the presence of thrombin), reflecting amyloid formation68. This results in a strong fluorescence observable (when excited ca 440 nm) in the presence of ThT (see e.g. refs 56, 57). Here we also showed that even if we add LBP after LPS exposure, less ThT binding (fluorescence) is observed (Fig. 3). This suggests that LBP protects the fibres from LPS damage by binding to the pre-added LPS in the PPP, before clot formation is induced with thrombin.
Airyscan super-resolution microscopy
Naïve type 2 diabetes with and without added LBP (Fig. 4).
Extensive fluorescence was noted in all our T2D samples after incubation with ThT (examples in Fig. 4A,C). However, when PPPs from T2D patients was incubated with LBP, very little to no fluorescence was observed (Fig. 4B,D). This suggests that the added LBP binds to something, removing its effect, and here we suggest (i) that it is LPS that is present in the naïve T2D, which is known, and (ii) that it is this presence of the LPS that is in part responsible for the anomalous clotting in T2D plasma.
We also looked at the clot structure of plasma from healthy and T2D individuals, with added thrombin, using scanning electron microscopy (SEM). Previously we reported, and we confirm here, that LPS causes healthy plasma to from denser and more hypercoagulable clots (confirmed by using PPP form healthy individuals and purified fibrinogen, using thromboelastography, confocal microscopy, SEM and we also tested whether LPS would bind to fibrinogen directly, using isothermal calorimetry). In this paper, we also showed that added LBP has the capability to reverse the damage of the LPS. We suggested that LPS causes anomalous clotting that is actually amyloid protein48, 68.
Here, Fig. 5A shows a representative airyscan micrograph of a typical clot from a healthy individual and Fig. 5B shows a clot where LBP was incubated with plasma before clot formation. In our hands, LBP on its own does not change clot structure significantly. Figure 5C shows a clot formed when plasma had been pre-incubated with LPS, while Fig. 5D shows a clot that was formed with plasma, also pre-incubated with LPS, but followed by addition of LBP, and both with added thrombin. Raw data, extensive SOPs for airyscan and SEM, including micrographs can be accessed at https://1drv.ms/f/s!AgoCOmY3bkKHhHp9zfDsIR_0g9_f and on EP’s researchgate profile, https://www.researchgate.net/profile/Etheresia_Pretorius.

(A) Representative clot structure from a healthy individual; (B) same PPP sample with added LBP; (C) same PPP sample with added LPS; (D) same PPP sample with added LPS, followed by LBP. All clots were created by adding thrombin to PPP.
SEM micrographs show that clots created from T2D PPP (Figs 6 and 7) have a structure similar to those from healthy PPP with added LPS. However, when LBP is added to PPP followed by thrombin, the structure is reverted to that of healthy PPP with added thrombin: see 5 k machine magnification micrographs of clots from T2D (Fig. 6A to D) and with added LBP (Fig. 6E to H). Figure 7A to F show micrographs with higher SEM magnifications (35 k machine magnification). Consequently, in this paper, we show that LBP added to naïve T2D plasma, followed by the addition of thrombin, led to a fibrin fibre structure similar to that of healthy PPP. The fluorescence (Airyscan) results were therefore confirmed by SEM.

(A to D) PPP with added thrombin from 4 naïve type 2 diabetes samples (5 k machine magnification); (E to H) Same samples (5 k machine magnification) with added LBP (incubated for 10 minutes).

(A to C) PPP with added thrombin from 3 naïve type 2 diabetes samples (35 k machine magnification); (D to F) same samples (35 k machine magnification), with added LBP (incubated for 10 minutes).
For a schematic model of a ‘healthy’ clot structure (i.e. a clot formed from the PPP of a healthy individual), and that found in T2D, see Fig. 8A,B. A typical clot is created by adding thrombin to PPP, and this is spread out onto a glass coverslip. The coagulum (clot) is then processed by fixation and dehydration. Such processed clots consist of three layers. These layers vary in thickness and structure depending upon whether the plasma was from a healthy individual or an individual with inflammation. A clot from a healthy individual consists of a bottom layer, which is caused by contact activation of the coagulum and the glass substrate. The second layer is the “true” clot, consisting of a fibrin fibre network of elongated fibres. Sometimes, the contact activation layer is visible through the spaghetti-like fibres. Depending on the preparation procedures, a third, top layer may form on the spaghetti-like fibres. This layer typically washes away during the fixing and dehydration process. However, it may sometimes be visible as patches of coagulum, similar to that of the contact activation layer at the bottom (see Fig. 8A). In T2D, the bottom contact activation layer is not easily distinguishable from the middle “true” clot area, and sparse spaghetti-like fibres are fused into the contact activation layer. This fused layer of matted fibrin may consist of thin branching individual fibres, with very small openings between the individual fibres, giving the impression of a matted plate. In previous publications, we have also referred to this complex and hypercoagulable clot structure as “dense matted deposits” (DMDs) (e.g. refs 48, 65, 68, 85). In T2D, a top netted layer is frequently observed. It mostly does not wash away during the various fixing and dehydration steps, and it forms nearly instantaneously when mixing thrombin with plasma to create the clot. This is due to the hypercoagulable nature of the plasma (see Fig. 8B). During confocal clot preparation, a wet clot is studied. The PPP and thrombin are mixed on a microscope slide and covered after 30 second with a coverslip. Although contact activation happens directly on the glass microscope slide, we therefore look at the fibrous part, which is similar to the middle “true clot” layers of the SEM clots in clots from healthy individuals.

(A) Model of a fibrin clot formed from plasma from a healthy individual. (B) Model of a clot made using PPP taken from an individual with type 2 diabetes. A typical clot consists of 3 layers, fast coagulating top layer, middle fibrous layer and bottom, contact activation layer. The thickness and presence of these 3 layers vary between clots from healthy individuals and inflammatory clots.
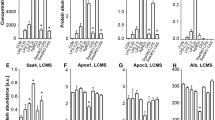
Although Airyscan and SEM analysis are both typically used only as qualitative methods, we aimed to quantify the changes in clot structure. By inspection (see figures above) the control samples without LPS showed much more variance between light and dark pixels, while the samples forming dense matted deposits were more uniformly grey. It thus seemed logical to use this to seek to quantify and discriminate the images. We therefore used the coefficient of variation (CV) as our metric. Thus, while recognising that the pixel intensity data were not normally distributed, we used ImageJ to calculate the mean and standard deviation of the intensity of the pixels in the images of the clot, using the histogram function, followed by the calculation of the coefficient of variation (i.e. SD/mean) of the intensity of the clot structure. Figure 9A to D show examples of representative histograms of the 8-bit intensity for a typical SEM and confocal clot with and without LBP of a patient with T2D. Within-sample analysis was done with the paired T-test and between-samples analysis was done using the Mann-Whitney test. Table 2 shows statistical analysis of the difference in the coefficients of variation (CV) of the pixel intensities in the images of the different clots studied using SEM and Airyscan technology and Fig. 10 shows boxplots of the data.

(A and B) Representative histograms of the 8-bit intensity for a typical SEM clot from PPP of an individual with type 2 diabetes (T2D) (A) and after addition of LBP (B). (C and D) Representative histograms of the 8-bit intensity for a typical Airyscan clot from PPP of an individual with T2D (C) and after addition of LBP.

Boxplots of the distribution of the coefficients of variation (CV) of the pixel intensities of the SEM clot images from the different sample classes analysed (median CV for each group is above boxplots). Note that that the CVs of the clot structures of PPP from naïve T2D and that of healthy individuals with added LPS are both 0.41.
As shown in our stored raw data, our controls included 7 young controls (≤30 years). We used the paired t-test to compare SEM data from our younger group to our older group. No significant difference was seen between them (P = 0.1388), suggesting that, in this population, age did not affect clot structure.
Discussion
Notwithstanding the difficulty of defining its ‘concentration’ in serum73, as most of it is bound to macromolecules, it is known that in type 1 diabetes the serum load of endotoxin (LPS) is raised relative to that of controls86, and that this is associated with disease progression87. There can also be an association between LPS levels in the serum and T2D81,82,83, though the direction of causality (if any) is seemingly not known. It is also known that T2D is accompanied by various kinds of coagulopathies (with both hypercoagulability, e.g. refs 88,89,90,91,92,93,94,95 and hypofibrinolysis31, 36, 37, 39, 89, 92, 94, 96,97,98,99,100,101 being observable.
A question that can arise is why we did not measure the amount of LPS in the plasma. LPS is typically not a molecularly defined substance, and its activity is sometimes reported in ‘endotoxin units’ (EU); this is based on a standard taken from an E. coli O55:B5 strain; an approximate relationship is that 1 ng endotoxin ~10 EU73, 102. The only assay currently available is the Limulus amoebocyte lysate assay; however, its use in the estimation of LPS in blood is not considered especially reliable103,104,105,106,107.
Certainly, some of the fibrin(ogen) changes we see in this paper may be due to glycation of fibrinogen by the raised glucose levels38, 108,109,110,111,112, as well as the increased levels of fibrinogen itself (e.g. refs 35, 112,113,114,115). However, because we had recently shown the extreme sensitivity of the form of the fibrin structure to tiny amounts of (free) LPS, including the formation of amyloid48, 68, it was of interest to enquire as to whether diabetic subjects were more likely to produce an amyloid form of clot.
Although we have seen anomalous clotting structures in diabetes before32, 33, 116, this is the first time that we have shown them to be amyloid in character. This is entirely consistent with the fact that at least some of the amyloidogenesis must be catalysed by fibrinogen-bound LPS, given too, as we show here, that it could be removed, and the amyloidogenesis largely reversed, by LBP. Interestingly, fibrin deposition has also previously been noted in term placentas from diabetic mothers117.
As well as for islets16, 18,19,20, 118,119,120,121,122, amyloid deposition in diabetes is known in kidney123,124,125, as is the deposition of serum amyloid A126, 127 (which may have prognostic value128). We also note the various comorbidities between T2D and CVD129,130,131,132; one interpretation is a common cause. In our T2D sample, most of the individuals suffer from hypertension and increased cholesterol and take anticoagulant medication. These co-morbidities are also common in CVD, and T2D is well known as a major cardiovascular risk factor133. Both T2D and CVD are known to have vascular pathology134, and this is directly associated with coagulation pathology116. We also looked to see it there are any relationships been our newly determined blood parameters and e.g. fasting glucose, but we could not discern any.
Endotoxin levels in T2D have previously been found to be, in part, the cause for hypercoagulation and that T2D patients treated with e.g. the anti-glycemic agent, rosiglitazone showed significantly lower endotoxin levels81. Our sample did not use this medication. Besides medication for glucose control, our T2D group also includes individuals on medication for dyslipidaemia and hypertension, while some are taking anticoagulants (see Table 1). Anticoagulants (e.g. aspirin/disprin) increase fibrin clot porosity and susceptibility to lysis135, 136 (i.e. the exact opposite effect from that noted here with LPS/LBP; Pretorius et al. 2013, 2015); they also have an antiplatelet effect136,137,138,139 and are known to reduce the risk of thrombosis140,141,142,143,144. Such products are also inhibitors of both cyclooxygenase COX-1 and COX-2145. We could not find evidence in the literature that LBP acts as an anti-coagulant.
Concluding remarks
T2D is accompanied by long-term inflammation, and this inflammation is mediated in part by increased fibrinogen levels, as well as a changed cytokine profile that is driven, at least in part, by dysregulated glucose and insulin function. The origin of this inflammation is mostly unclear and remains unresolved in diabetes. It is known that gut dysbioses and atopobioses71 (colloquially referred to as ‘leaky gut’) are a well-known contributor to the pathogenesis of many metabolic diseases, including obesity146, T1D147, 148, T2D146, 149, 150, and CVD151. We have previously suggested that there is a fundamental link between gut dysbioses, the presence of a (dormant) blood microbiome and the presence of the highly inflammatory LPS71,72,73. As discussed in the introduction and shown recently, LPS is known to cause hypercoagulability and anomalous blood clotting, even at very low concentrations48, 68. We have also shown that LPS may directly bind to fibrinogen monomers68. Anomalous clotting is well-known in T2D31, 33,34,35,36,37,38,39, and here we show that LBP added to PPP from individuals with T2D changes fibrin structure to resemble that of healthy fibrin. We confirmed our results with healthy PPP, adding LPS and mopping it up again with added LBP. It is known that LBP is an acutely induced plasma protein that binds avidly to LPS aggregates, and delivers them to CD1473, 152, 153. Our results therefore suggest that, at least in part, the anomalous blood clotting seen in T2D is due to the presence of the potent inflammagen LPS, that can be removed by LBP. A future study could additionally investigate the effect of glucose addition to healthy PPP, with and without the addition of LPS. This will give insights regarding amyloidogenesis and the effects of glucose on such a process. It now seems no coincidence that most of the more serious sequelae of diabetes are accompanied by amyloid formation (e.g. retinopathy154,155,156, nephropathy123, 157,158,159,160, neuropathy161, and cardiovascular problems162, 163). It is well-known that all of these macro- and microvascular complications that form part of the T2D comorbidities cause hypercoagulation and are associated with an upregulated inflammatory profile. A follow-up study should investigate the specific influences of comorbidities like a history of macrovascular (cardiovascular diseases such as coronary artery disease, ischemic stroke, and peripheral artery disease), and microvascular (retinopathy, nephropathy, and neuropathy). This will give further insight with regards to the exact role that these comorbidities play in hypercoagulation.
Thus, the recognition from the present work that amyloid formation is a strong function of the presence of cryptic LPS in type 2 diabetes might have important applications in both the treatment of inflammation in T2D, and the progression of the condition generally. The fundamental question that now remains is how to translate these results into clinical practice.
References
Chiti, F. & Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75, 333–366 (2006).
Herczenik, E. & Gebbink, M. F. B. G. Molecular and cellular aspects of protein misfolding and disease. FASEB J 22, 2115–2133, doi:10.1096/fj.07-099671 (2008).
Rambaran, R. N. & Serpell, L. C. Amyloid fibrils: abnormal protein assembly. Prion 2, 112–117 (2008).
Eisenberg, D. & Jucker, M. The amyloid state of proteins in human diseases. Cell 148, 1188–1203, doi:10.1016/j.cell.2012.02.022 (2012).
Knowles, T. P. J., Vendruscolo, M. & Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15, 384–396, doi:10.1038/nrm3810 (2014).
Tipping, K. W., van Oosten-Hawle, P., Hewitt, E. W. & Radford, S. E. Amyloid Fibres: Inert End-Stage Aggregates or Key Players in Disease? Trends Biochem Sci 40, 719–727, doi:10.1016/j.tibs.2015.10.002 (2015).
Sipe, J. D. et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 21, 221–224, doi:10.3109/13506129.2014.964858 (2014).
Olanow, C. W. & Brundin, P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov Disord 28, 31–40, doi:10.1002/mds.25373 (2013).
Uversky, V. N., Li, J. & Fink, A. L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure. J Biol Chem 276, 44284–44296 (2001).
Vilar, M. et al. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci U S A 105, 8637–8642, doi:10.1073/pnas.0712179105 (2008).
Fändrich, M., Meinhardt, J. & Grigorieff, N. Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion 3, 89–93 (2009).
Petkova, A. T. et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science 307, 262–265, doi:10.1126/science.1105850 (2005).
Serpell, L. C. Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502, 16–30 (2000).
Fuchsberger, C. et al. The genetic architecture of type 2 diabetes. Nature 536, 41–47, doi:10.1038/nature18642 (2016).
Cooper, G. J. S. et al. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA 84, 8628–8632 (1987).
Cooper, G. J. S. et al. Amylin Found in Amyloid Deposits in Human Type-2 Diabetes-Mellitus May Be a Hormone That Regulates Glycogen-Metabolism in Skeletal-Muscle. Proc Natl Acad Sci 85, 7763–7766, doi:10.1073/pnas.85.20.7763 (1988).
Lorenzo, A., Razzaboni, B., Weir, G. C. & Yankner, B. A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 368, 756–760, doi:10.1038/368756a0 (1994).
Jaikaran, E. T. A. S. & Clark, A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta 1537, 179–203 (2001).
Loomes, K. M. Survival of an islet beta-cell in type-2 diabetes: curbing the effects of amyloid cytotoxicity. Islets 3, 38–39 (2011).
Höppener, J. W. M. & Lips, C. J. M. Role of islet amyloid in type 2 diabetes mellitus. Int J Biochem Cell Biol 38, 726–736, doi:10.1016/j.biocel.2005.12.009 (2006).
Pillay, K. & Govender, P. Amylin uncovered: a review on the polypeptide responsible for type II diabetes. Biomed Res Int 2013, 826706, doi:10.1155/2013/826706 (2013).
Zhang, S. et al. The pathogenic mechanism of diabetes varies with the degree of overexpression and oligomerization of human amylin in the pancreatic islet beta cells. FASEB J 28, 5083–5096, doi:10.1096/fj.14-251744 (2014).
Gurlo, T. et al. beta Cell-specific increased expression of calpastatin prevents diabetes induced by islet amyloid polypeptide toxicity. JCI Insight 1, e89590, doi:10.1172/jci.insight.89590 (2016).
Janson, J., Ashley, R. H., Harrison, D., McIntyre, S. & Butler, P. C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48, 491–498 (1999).
Daval, M. et al. The effect of curcumin on human islet amyloid polypeptide misfolding and toxicity. Amyloid 17, 118–128, doi:10.3109/13506129.2010.530008 (2010).
Campbell, R. A., Aleman, M., Gray, L. D., Falvo, M. R. & Wolberg, A. S. Flow profoundly influences fibrin network structure: implications for fibrin formation and clot stability in haemostasis. Thromb Haemost 104, 1281–1284, doi:10.1160/TH10-07-0442 (2010).
Pretorius, E., Steyn, H., Engelbrecht, M., Swanepoel, A. C. & Oberholzer, H. M. Differences in fibrin fiber diameters in healthy individuals and thromboembolic ischemic stroke patients. Blood Coagul Fibrinolysis 22, 696–700, doi:10.1097/MBC.0b013e32834bdb32 (2011).
Weigandt, K. M. et al. Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys J 103, 2399–2407, doi:10.1016/j.bpj.2012.10.036 (2012).
Bester, J., Soma, P., Kell, D. B. & Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget Gerontology 6, 35284–35303 (2015).
Kell, D. B. & Pretorius, E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr Biol 7, 24–52, doi:10.1039/c4ib00173g (2015).
Alzahrani, S. H. & Ajjan, R. A. Coagulation and fibrinolysis in diabetes. Diabet Vasc Dis Res 7, 260–273, doi:10.1177/1479164110383723 (2010).
Pretorius, E., Oberholzer, H. M., van der Spuy, W. J., Swanepoel, A. C. & Soma, P. Qualitative scanning electron microscopy analysis of fibrin networks and platelet abnormalities in diabetes. Blood Coagul Fibrinol 22, 463–467, doi:10.1097/MBC.0b013e3283468a0d (2011).
Pretorius, E. et al. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc Diabetol 13, 30 (2015).
Jörneskog, G. et al. Altered properties of the fibrin gel structure in patients with IDDM. Diabetologia 39, 1519–1523 (1996).
Dunn, E. J. & Ariëns, R. A. S. Fibrinogen and fibrin clot structure in diabetes. Herz 29, 470–479, doi:10.1007/s00059-004-2607-z (2004).
Dunn, E. J., Ariëns, R. A. S. & Grant, P. J. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 48, 1198–1206, doi:10.1007/s00125-005-1742-2 (2005).
Dunn, E. J., Philippou, H., Ariëns, R. A. S. & Grant, P. J. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia 49, 1071–1080, doi:10.1007/s00125-006-0197-4 (2006).
Pieters, M. et al. The effect of glycaemic control on fibrin network structure of type 2 diabetic subjects. Thromb Haemost 96, 623–629 (2006).
Alzahrani, S. H. et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: associations with cardiometabolic and vascular markers. J Clin Endocrinol Metab 97, E2282–2287, doi:10.1210/jc.2012-2128 (2012).
Weisel, J. W. Fibrinogen and fibrin. Adv Protein Chem 70, 247–299, doi:10.1016/s0065-3233(05)70008-5 (2005).
Wolberg, A. S. Thrombin generation and fibrin clot structure. Blood Rev 21, 131–142, doi:10.1016/j.blre.2006.11.001 (2007).
C L Corte, A. L., Philippou, H. & Ariëns, R. A. S. Role of fibrin structure in thrombosis and vascular disease. Adv Protein Chem Struct Biol 83, 75–127, doi:10.1016/B978-0-12-381262-9.00003-3 (2011).
Undas, A. & Ariëns, R. A. S. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol 31, e88–99, doi:10.1161/ATVBAHA.111.230631 (2011).
Wolberg, A. S. Determinants of fibrin formation, structure, and function. Curr Opin Hematol 19, 349–356, doi:10.1097/MOH.0b013e32835673c2 (2012).
Averett, L. E. et al. Complexity of "A-a" knob-hole fibrin interaction revealed by atomic force spectroscopy. Langmuir 24, 4979–4988, doi:10.1021/la703264x (2008).
Yermolenko, I. S., Lishko, V. K., Ugarova, T. P. & Magonov, S. N. High-resolution visualization of fibrinogen molecules and fibrin fibers with atomic force microscopy. Biomacromolecules 12, 370–379, doi:10.1021/bm101122g (2011).
Protopopova, A. D. et al. Visualization of fibrinogen alphaC regions and their arrangement during fibrin network formation by high-resolution AFM. J Thromb Haemost 13, 570–579, doi:10.1111/jth.12785 (2015).
Kell, D. B. & Pretorius, E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Progr Biophys Mol Biol 123, 16–41, doi:10.1016/j.pbiomolbio.2016.08.006 (2017).
Dickneite, G. et al. Coagulation factor XIII: a multifunctional transglutaminase with clinical potential in a range of conditions. Thromb Haemost 113, 686–697, doi:10.1160/TH14-07-0625 (2015).
Maji, S. K., Wang, L., Greenwald, J. & Riek, R. Structure-activity relationship of amyloid fibrils. FEBS Lett 583, 2610–2617, doi:10.1016/j.febslet.2009.07.003 (2009).
Morris, K. L. & Serpell, L. C. X-ray fibre diffraction studies of amyloid fibrils. Methods Mol Biol 849, 121–135, doi:10.1007/978-1-61779-551-0_9 (2012).
Tycko, R. & Wickner, R. B. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res 46, 1487–1496, doi:10.1021/ar300282r (2013).
Langkilde, A. E., Morris, K. L., Serpell, L. C., Svergun, D. I. & Vestergaard, B. The architecture of amyloid-like peptide fibrils revealed by X-ray scattering, diffraction and electron microscopy. Acta Crystallogr D Biol Crystallogr 71, 882–895, doi:10.1107/S1399004715001674 (2015).
LeVine, H. 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol 309, 274–284 (1999).
Biancalana, M., Makabe, K., Koide, A. & Koide, S. Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J Mol Biol 385, 1052–1063, doi:10.1016/j.jmb.2008.11.006 (2009).
Biancalana, M. & Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta 1804, 1405–1412, doi:10.1016/j.bbapap.2010.04.001 (2010).
Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J Chem Biol 3, 1–18, doi:10.1007/s12154-009-0027-5 (2010).
Sulatskaya, A. I., Kuznetsova, I. M. & Turoverov, K. K. Interaction of thioflavin T with amyloid fibrils: stoichiometry and affinity of dye binding, absorption spectra of bound dye. J Phys Chem B 115, 11519–11524, doi:10.1021/jp207118x (2011).
Freire, S., de Araujo, M. H., Al-Soufi, W. & Novo, M. Photophysical study of Thioflavin T as fluorescence marker of amyloid fibrils. Dyes and Pigments 110, 97–105, doi:10.1016/j.dyepig.2014.05.004 (2014).
Serpell, L. C., Benson, M., Liepnieks, J. J. & Fraser, P. E. Structural analyses of fibrinogen amyloid fibrils. Amyloid 14, 199–203, doi:10.1080/13506120701461111 (2007).
Picken, M. M. Fibrinogen amyloidosis: the clot thickens! Blood 115, 2985–2986, doi:10.1182/blood-2009-12-236810 (2010).
Stangou, A. J. et al. Hereditary fibrinogen A alpha-chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 115, 2998–3007, doi:10.1182/blood-2009-06-223792 (2010).
Haidinger, M. et al. Hereditary amyloidosis caused by R554L fibrinogen Aalpha-chain mutation in a Spanish family and review of the literature. Amyloid 20, 72–79, doi:10.3109/13506129.2013.781998 (2013).
Kell, D. B. & Pretorius, E. Serum ferritin is an important disease marker, and is mainly a leakage product from damaged cells. Metallomics 6, 748–773, doi:10.1039/C3MT00347G (2014).
Pretorius, E., Vermeulen, N., Bester, J., Lipinski, B. & Kell, D. B. A novel method for assessing the role of iron and its functional chelation in fibrin fibril formation: the use of scanning electron microscopy. Toxicol Mech Methods 23, 352–359, doi:10.3109/15376516.2012.762082 (2013).
Pretorius, E. et al. Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS One 9, e85271 (2014).
Pretorius, E. & Kell, D. B. Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integrative Biol 6, 486–510 (2014).
Pretorius, E., Mbotwe, S., Bester, J., Robinson, C. J. & Kell, D. B. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J R Soc Interface 123, 20160539, doi:10.1098/rsif.2016.0539 (2016).
Pretorius, E., Mbotwe, S., Bester, J., Robinson, C. & Kell, D. B. Acute induction of anomalous blood clotting by highly substoichiometric levels of bacterial lipopolysaccharide (LPS). bioRxiv, 2016-053538v053531, doi:10.1101/053538 (2016).
Kell, D. B. & Pretorius, E. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. bioRxiv preprint. bioRxiv, 054734, doi:10.1101/054734 (2016).
Potgieter, M., Bester, J., Kell, D. B. & Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev 39, 567–591, doi:10.1093/femsre/fuv013 (2015).
Kell, D. B., Potgieter, M. & Pretorius, E. Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Research 4, 179, doi:10.12688/f1000research.6709.1 (2015).
Kell, D. B. & Pretorius, E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol 7, 1339–1377, doi:10.1039/C5IB00158G (2015).
Itzhaki, R. F. et al. Microbes and Alzheimer's Disease. J Alzheimers Dis 51, 979–984, doi:10.3233/JAD-160152 (2016).
Kell, D. B. & Kenny, L. C. A dormant microbial component in the development of pre-eclampsia. BioRxiv preprint. bioRxiv, 057356 (2016).
Kell, D. B. & Pretorius, E. To what extent are the terminal stages of sepsis, septic shock, SIRS, and multiple organ dysfunction syndrome actually driven by a toxic prion/amyloid form of fibrin? bioRxiv preprint. bioRxiv, 057851, doi:10.1101/057851 (2016).
Pretorius, E., Akeredolu, O.-O., Soma, P. & Kell, D. B. Major involvement of bacterial components in rheumatoid arthritis and its accompanying oxidative stress, systemic inflammation and hypercoagulability. Exp Biol Med, in press (2016).
Pretorius, E., Bester, J. & Kell, D. B. A bacterial component to Alzheimer-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J Alzheimers Dis 53, 1237–1256 (2016).
Kell, D. B. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics 2, 2 (2008).
Kell, D. B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol 577, 825–889, doi:10.1007/s00204-010-0577-x (2010).
Al-Attas, O. S. et al. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol 8, 20, doi:10.1186/1475-2840-8-20 (2009).
Hawkesworth, S. et al. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr Diabetes 3, e83, doi:10.1038/nutd.2013.24 (2013).
Zaman, G. S. & Zaman, F. Relationship between postprandial endotoxemia in nonobese postmenopausal women and diabetic nonobese postmenopausal women. J Nat Sci Biol Med 6, 89–93, doi:10.4103/0976-9668.149098 (2015).
Liang, H., Hussey, S. E., Sanchez-Avila, A., Tantiwong, P. & Musi, N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS One 8, e63983, doi:10.1371/journal.pone.0063983 (2013).
Lipinski, B., Pretorius, E., Oberholzer, H. M. & Van Der Spuy, W. J. Iron enhances generation of fibrin fibers in human blood: Implications for pathogenesis of stroke. Microsc Res Tech 75, 1185–1190, doi:10.1002/jemt.22047 (2012).
Lassenius, M. I. et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 34, 1809–1815, doi:10.2337/dc10-2197 (2011).
Nymark, M. et al. Serum lipopolysaccharide activity is associated with the progression of kidney disease in finnish patients with type 1 diabetes. Diabetes Care 32, 1689–1693, doi:10.2337/dc09-0467 (2009).
Carr, M. E. Diabetes mellitus: a hypercoagulable state. J Diabet Complic 15, 44–54 (2001).
Banga, J. D. Coagulation and fibrinolysis in diabetes. Semin Vasc Med 2, 75–86, doi:10.1055/s-2002-23098 (2002).
Thor, M., Yu, A. & Swedenborg, J. Markers of inflammation and hypercoagulability in diabetic and nondiabetic patients with lower extremity ischemia. Thromb Res 105, 379–383 (2002).
Aras, R., Sowers, J. R. & Arora, R. The proinflammatory and hypercoagulable state of diabetes mellitus. Rev Cardiovasc Med 6, 84–97 (2005).
Grant, P. J. Diabetes mellitus as a prothrombotic condition. J Intern Med 262, 157–172, doi:10.1111/j.1365-2796.2007.01824.x (2007).
Tripodi, A. et al. Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis 31, 165–172, doi:10.1007/s11239-010-0506-0 (2011).
Ye, Y., Perez-Polo, J. R., Aguilar, D. & Birnbaum, Y. The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Res Cardiol 106, 925–952, doi:10.1007/s00395-011-0216-6 (2011).
Beijers, H. J. B. H. et al. Impaired glucose metabolism and type 2 diabetes are associated with hypercoagulability: potential role of central adiposity and low-grade inflammation–the Hoorn Study. Thromb Res 129, 557–562, doi:10.1016/j.thromres.2011.07.033 (2012).
Cucuianu, M., Fekete, T., Marcusiu, C., Mosler, R. & Dutu, A. Fibrinolysis in diabetes mellitus. Role of overweight and hypertriglyceridemia. Medecine interne 22, 171–177 (1984).
Yano, Y. et al. Increased plasma thrombin-activatable fibrinolysis inhibitor levels in normotensive type 2 diabetic patients with microalbuminuria. The Journal of clinical endocrinology and metabolism 88, 736–741, doi:10.1210/jc.2002-020691 (2003).
Hess, K. et al. A novel mechanism for hypofibrinolysis in diabetes: the role of complement C3. Diabetologia 55, 1103–1113, doi:10.1007/s00125-011-2301-7 (2012).
Walus-Miarka, M. et al. Altered fibrin-clot properties are associated with retinopathy in type 2 diabetes mellitus. Diabetes & Metabolism 38, 462–465, doi:10.1016/j.diabet.2012.03.007 (2012).
Bochenek, M., Zalewski, J., Sadowski, J. & Undas, A. Type 2 diabetes as a modifier of fibrin clot properties in patients with coronary artery disease. J Thromb Thrombolysis 35, 264–270, doi:10.1007/s11239-012-0821-8 (2013).
Konieczynska, M., Fil, K., Bazanek, M. & Undas, A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb Haemost 111, 685–693, doi:10.1160/th13-07-0566 (2014).
Pearson, F. C. et al. Comparison of several control standard endotoxins to the National Reference Standard Endotoxin–an HIMA collaborative study. Appl Environ Microbiol 50, 91–93 (1985).
Andrä, J. et al. Biophysical characterization of the interaction of Limulus polyphemus endotoxin neutralizing protein with lipopolysaccharide. Eur J Biochem 271, 2037–2046, doi:10.1111/j.1432-1033.2004.04134.x (2004).
Ketchum, P. A. & Novitsky, T. J. Assay of endotoxin by limulus amebocyte lysate. Methods Mol Med 36, 3–12, doi:10.1385/1-59259-216-3:3 (2000).
Mattsby-Baltzer, I., Lindgren, K., Lindholm, B. & Edebo, L. Endotoxin shedding by enterobacteria: free and cell-bound endotoxin differ in Limulus activity. Infect Immun 59, 689–695 (1991).
Novitsky, T. J. Limitations of the Limulus amebocyte lysate test in demonstrating circulating lipopolysaccharides. Ann N Y Acad Sci 851, 416–421 (1998).
Novitsky, T. J. Biomedical Applications of Limulus Amebocyte Lysate. Biology and Conservation of Horseshoe Crabs, 315–329, doi:10.1007/978-0-387-89959-6_20 (2009).
Brownlee, M., Vlassara, H. & Cerami, A. Nonenzymatic glycosylation reduces the susceptibility of fibrin to degradation by plasmin. Diabetes 32, 680–684 (1983).
Pieters, M. et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes - a purified fibrinogen model. Thromb Haemost 99, 691–700, doi:10.1160/TH07-11-0699 (2008).
Howard, S. C., Algra, A. & Rothwell, P. M. Effect of age and glycaemic control on the association between fibrinogen and risk of acute coronary events after transient ischaemic attack or stroke. Cerebrovasc Dis 25, 136–143, doi:10.1159/000112324 (2008).
Svensson, J. et al. Acetylation and glycation of fibrinogen in vitro occur at specific lysine residues in a concentration dependent manner: a mass spectrometric and isotope labeling study. Biochem Biophys Res Commun 421, 335–342, doi:10.1016/j.bbrc.2012.03.154 (2012).
Fan, N. K., Keegan, P. M., Platt, M. O. & Averett, R. D. Experimental and imaging techniques for examining fibrin clot structures in normal and diseased states. J Vis Exp, e52019, doi:10.3791/52019 (2015).
Bembde, A. S. A study of plasma fibrinogen level in type-2 diabetes mellitus and its relation to glycemic control. Indian J Hematol Blood Transfus 28, 105–108, doi:10.1007/s12288-011-0116-9 (2012).
McBane, R. D. 2nd, Hardison, R. M. & Sobel, B. E. Comparison of plasminogen activator inhibitor-1, tissue type plasminogen activator antigen, fibrinogen, and D-dimer levels in various age decades in patients with type 2 diabetes mellitus and stable coronary artery disease (from the BARI 2D trial). Am J Cardiol 105, 17–24, doi:10.1016/j.amjcard.2009.08.643 (2010).
Ariëns, R. A. S. Fibrin(ogen) and thrombotic disease. J Thromb Haemost 11(Suppl 1), 294–305, doi:10.1111/jth.12229 (2013).
Soma, P. & Pretorius, E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc Diabetol 14, 96, doi:10.1186/s12933-015-0261-9 (2015).
Mayhew, T. M. & Sampson, C. Maternal diabetes mellitus is associated with altered deposition of fibrin-type fibrinoid at the villous surface in term placentae. Placenta 24, 524–531 (2003).
Cooper, G. J. S. et al. Amylin and the amylin gene: structure, function and relationship to islet amyloid and to diabetes mellitus. Biochim Biophys Acta 1014, 247–258 (1989).
Marzban, L., Park, K. & Verchere, C. B. Islet amyloid polypeptide and type 2 diabetes. Exp Gerontol 38, 347–351 (2003).
Miklossy, J. et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol Aging 31, 1503–1515, doi:10.1016/j.neurobiolaging.2008.08.019 (2010).
Westermark, P., Andersson, A. & Westermark, G. T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91, 795–826, doi:10.1152/physrev.00042.2009 (2011).
Westermark, G. T. & Westermark, P. Islet amyloid polypeptide and diabetes. Curr Protein Pept Sci 14, 330–337 (2013).
Díez, R., Madero, M., Gamba, G., Soriano, J. & Soto, V. Renal AA Amyloidosis in Patients with Type 2 Diabetes Mellitus. Nephron Extra 4, 119–126, doi:10.1159/000363625 (2014).
Anderberg, R. J. et al. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab Invest 95, 250–262, doi:10.1038/labinvest.2014.163 (2015).
Dieter, B. P. et al. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J Diabetes Complications, doi:10.1016/j.jdiacomp.2016.07.018 (2016).
Kumon, Y., Suehiro, T., Itahara, T., Ikeda, Y. & Hashimoto, K. Serum amyloid A protein in patients with non-insulin-dependent diabetes mellitus. Clin Biochem 27, 469–473 (1994).
Eren, M. A. et al. Association of serum amyloid A with subclinical atherosclerosis in women with gestational diabetes. Gynecol Endocrinol 28, 1010–1013, doi:10.3109/09513590.2012.705371 (2012).
Marzi, C. et al. Acute-phase serum amyloid A protein and its implication in the development of type 2 diabetes in the KORA S4/F4 study. Diabetes Care 36, 1321–1326, doi:10.2337/dc12-1514 (2013).
Reusch, J. E. Diabetes, microvascular complications, and cardiovascular complications: what is it about glucose? J Clin Invest 112, 986–988 (2003).
Targher, G. et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 30, 1212–1218 (2007).
Singh, G. M. et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: a pooled analysis. PloS one 8, e65174, doi:10.1371/journal.pone.0065174 (2013).
Basuli, D., Stevens, R. G., Torti, F. M. & Torti, S. V. Epidemiological associations between iron and cardiovascular disease and diabetes. Front Pharmacol 5, 117, doi:10.3389/fphar.2014.00117 (2014).
Bertoni, A. G., Kramer, H., Watson, K. & Post, W. S. Diabetes and Clinical and Subclinical CVD. Glob Heart 11, 337–342, doi:10.1016/j.gheart.2016.07.005 (2016).
Di Pino, A., Urbano, F., Piro, S., Purrello, F. & Rabuazzo, A. M. Update on pre-diabetes: Focus on diagnostic criteria and cardiovascular risk. World J Diabetes 7, 423–432, doi:10.4239/wjd.v7.i18.423 (2016).
Undas, A., Celinska-Lowenhoff, M., Lowenhoff, T. & Szczeklik, A. Statins, fenofibrate, and quinapril increase clot permeability and enhance fibrinolysis in patients with coronary artery disease. J Thromb Haemost 4, 1029–1036, doi:10.1111/j.1538-7836.2006.01882.x (2006).
Undas, A., Brummel-Ziedins, K. E. & Mann, K. G. Antithrombotic properties of aspirin and resistance to aspirin: beyond strictly antiplatelet actions. Blood 109, 2285–2292 (2007).
Knapp, M., Lisowska, A., Knapp, P. & Baranowski, M. Dose-dependent effect of aspirin on the level of sphingolipids in human blood. Adv Med Sci 58, 274–281, doi:10.2478/ams-2013-0021 (2013).
Manolis, A. S., Manolis, T. A., Papadimitriou, P., Koulouris, S. & Melita, H. Combined antiplatelet therapy: still a sweeping combination in cardiology. Cardiovasc Hematol Agents Med Chem 11, 136–167 (2013).
Mehta, S. R. Aspirin for prevention and treatment of cardiovascular disease. Ann Intern Med 150, 414–416 (2009).
Angiolillo, D. J. & Ferreiro, J. L. Antiplatelet and anticoagulant therapy for atherothrombotic disease: the role of current and emerging agents. Am J Cardiovasc Drugs 13, 233–250, doi:10.1007/s40256-013-0022-7 (2013).
Elblbesy, M. A., Hereba, A. R. & Shawki, M. M. Effects of aspirin on rheological properties of erythrocytes in vitro. Int J Biomed Sci 8, 188–193 (2012).
Gasparyan, A. Y., Ayvazyan, L., Pretorius, E. & Kitas, G. D. Platelets in rheumatic diseases: friend or foe? Curr Pharm Des 20, 552–566 (2014).
Santos, M. T. et al. Residual platelet thromboxane A2 and prothrombotic effects of erythrocytes are important determinants of aspirin resistance in patients with vascular disease. J Thromb Haemost 6, 615–621, doi:10.1111/j.1538-7836.2008.02915.x (2008).
Sostres, C., Gargallo, C. J. & Lanas, A. Aspirin, cyclooxygenase inhibition and colorectal cancer. World J Gastrointest Pharmacol Ther 5, 40–49, doi:10.4292/wjgpt.v5.i1.40 (2014).
Berk, M. et al. Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Med 11, 74, doi:10.1186/1741-7015-11-74 (2013).
Slyepchenko, A. et al. Intestinal dysbiosis, gut hyperpermeability and bacterial translocation: missing links between depression, obesity and type 2 diabetes? Curr Pharm Des (2016).
Li, X. & Atkinson, M. A. The role for gut permeability in the pathogenesis of type 1 diabetes - a solid or leaky concept? Pediatr Diabetes 16, 485–492, doi:10.1111/pedi.12305 (2015).
Vaarala, O., Atkinson, M. A. & Neu, J. The "perfect storm" for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 57, 2555–2562, doi:10.2337/db08-0331 (2008).
de Kort, S., Keszthelyi, D. & Masclee, A. A. Leaky gut and diabetes mellitus: what is the link? Obes Rev 12, 449–458, doi:10.1111/j.1467-789X.2010.00845.x (2011).
Delzenne, N. M., Cani, P. D., Everard, A., Neyrinck, A. M. & Bindels, L. B. Gut microorganisms as promising targets for the management of type 2 diabetes. Diabetologia 58, 2206–2217, doi:10.1007/s00125-015-3712-7 (2015).
Schicho, R., Marsche, G. & Storr, M. Cardiovascular complications in inflammatory bowel disease. Curr Drug Targets 16, 181–188 (2015).
Kitchens, R. L. & Thompson, P. A. Impact of sepsis-induced changes in plasma on LPS interactions with monocytes and plasma lipoproteins: roles of soluble CD14, LBP, and acute phase lipoproteins. J Endotoxin Res 9, 113–118, doi:10.1179/096805103125001504 (2003).
Kopp, F., Kupsch, S. & Schromm, A. B. Lipopolysaccharide-binding protein is bound and internalized by host cells and colocalizes with LPS in the cytoplasm: Implications for a role of LBP in intracellular LPS-signaling. Biochim Biophys Acta 1863, 660–672, doi:10.1016/j.bbamcr.2016.01.015 (2016).
Kam, J. H., Lenassi, E. & Jeffery, G. Viewing Ageing Eyes: Diverse Sites of Amyloid Beta Accumulation in the Ageing Mouse Retina and the Up-Regulation of Macrophages. Plos One 5, doi:10.1371/journal.pone.0013127 (2010).
Ma, Y., Tao, Y., Lu, Q. & Jiang, Y. R. Intraocular expression of serum amyloid a and interleukin-6 in proliferative diabetic retinopathy. Am J Ophthalmol 152, 678–685 e672, doi:10.1016/j.ajo.2011.03.007 (2011).
Yoneda, S. et al. Vitreous fluid levels of beta-amyloid((1-42)) and tau in patients with retinal diseases. Jpn J Ophthalmol 49, 106–108, doi:10.1007/s10384-004-0156-x (2005).
Gong, W. et al. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int 72, 213–218, doi:10.1038/sj.ki.5002305 (2007).
Stettler, C. et al. Serum amyloid A, C-reactive protein, and retinal microvascular changes in hypertensive diabetic and nondiabetic individuals: an Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) substudy. Diabetes Care 32, 1098–1100, doi:10.2337/dc08-2137 (2009).
Khalighi, M. A., Dean Wallace, W. & Palma-Diaz, M. F. Amyloid nephropathy. Clin Kidney J 7, 97–106, doi:10.1093/ckj/sfu021 (2014).
Huang, F. et al. Valsartan inhibits amylin-induced podocyte damage. Microvasc Res 106, 101–109, doi:10.1016/j.mvr.2016.04.007 (2016).
Shin, S. C. & Robinson-Papp, J. Amyloid Neuropathies. Mount Sinai Journal of Medicine 79, 733–748, doi:10.1002/msj.21352 (2012).
Guan, J., Mishra, S., Falk, R. H. & Liao, R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol 302, H544–552, doi:10.1152/ajpheart.00815.2011 (2012).
Perfetto, F. et al. Cardiac amyloidosis: the heart of the matter. Intern Emerg Med 8, 191–203, doi:10.1007/s11739-011-0647-y (2013).
Acknowledgements
We thank the Biotechnology and Biological Sciences Research Council (grant BB/L025752/1) as well as the National Research Foundation (NRF) and the Medical Research Council of South Africa (MRC) for supporting this collaboration. The authors thank Dr Prashilla Soma: Diabetes Clinician. Biotechnology and Biological Sciences Research Council (grant BB/L025752/1). National Research Foundation (NRF) of South Africa (91548: Competitive Program) and Medical Research Council (MRC) of South Africa (Self-Initiated Research Program: A0X331). Grant holder: E Pretorius.
Author information
Authors and Affiliations
Contributions
E.P. is the study leader, analysed all samples, prepared all figures, co-wrote paper; S.M.: technical assistance with SEM preparation; D.B.K. is the study co-leader, and co-wrote and edited the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pretorius, E., Mbotwe, S. & Kell, D.B. Lipopolysaccharide-binding protein (LBP) reverses the amyloid state of fibrin seen in plasma of type 2 diabetics with cardiovascular co-morbidities. Sci Rep 7, 9680 (2017). https://doi.org/10.1038/s41598-017-09860-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09860-4
This article is cited by
-
Proteomics of fibrin amyloid microclots in long COVID/post-acute sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system
Cardiovascular Diabetology (2022)
-
Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC)
Cardiovascular Diabetology (2022)
-
Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin
Cardiovascular Diabetology (2021)
-
Colorectal cancer is associated with increased circulating lipopolysaccharide, inflammation and hypercoagulability
Scientific Reports (2020)
-
Is Porphyromonas gingivalis involved in Parkinson’s disease?
European Journal of Clinical Microbiology & Infectious Diseases (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
