
Abstract
Tigers have lost 93% of their historical range worldwide. India plays a vital role in the conservation of tigers since nearly 60% of all wild tigers are currently found here. However, as protected areas are small (<300 km2 on average), with only a few individuals in each, many of them may not be independently viable. It is thus important to identify and conserve genetically connected populations, as well as to maintain connectivity within them. We collected samples from wild tigers (Panthera tigris tigris) across India and used genome-wide SNPs to infer genetic connectivity. We genotyped 10,184 SNPs from 38 individuals across 17 protected areas and identified three genetically distinct clusters (corresponding to northwest, southern and central India). The northwest cluster was isolated with low variation and high relatedness. The geographically large central cluster included tigers from central, northeastern and northern India, and had the highest variation. Most genetic diversity (62%) was shared among clusters, while unique variation was highest in the central cluster (8.5%) and lowest in the northwestern one (2%). We did not detect signatures of differential selection or local adaptation. We highlight that the northwest population requires conservation attention to ensure persistence of these tigers.
Similar content being viewed by others

Introduction
Genetic variation and its partitioning across populations helps inform evolutionary history, population connectivity, and demographic fluctuations1, 2. Such information is particularly important for endangered species, where conservation strategy and management planning depends on accurate detection of species status, population structure and connectivity, inbreeding, adaptive variation, admixture, and population demographic history2.
Until recently, highly polymorphic microsatellites were the markers of choice for conservation genetic studies3. Rapid development of next generation sequencing (NGS) has facilitated survey of genome-wide variation. Such data has helped identify African savannah (Loxodonta africana) and forest elephants (Loxodonta cyclotis) as separate species4, detect cryptic population structure in chimpanzees (Pan troglodytes)5, reveal local adaptation in giant panda (Ailuropoda melanoleuca) populations6 and quantify genetic diversity in Tasmanian devils7 (Sarcophilus harrisii). Genome-wide Single Nucleotide Polymorhphisms (SNPs) have been developed for monitoring populations in the wild, including bears8 (Ursus arctos), wolves9 (Canis lupus) and river otters10 (Lontra canadensis). Several thousands of SNPs provide higher statistical power in comparison to examining a limited number of microsatellites, resulting in more robust population genetic inference11, 12.
Many large carnivores are currently threatened, with around 61% of them designated as ‘threatened’13 by the IUCN redlist. A majority of them have suffered range reduction and fragmentation, with surviving individuals restricted to small isolated populations that tend to have low genetic variation and a high probability of extinction14. Identifying such populations can help devise conservation strategies to avoid inbreeding depression, aid in population recovery and plan assisted gene flow for genetic rescue. Such strategies have been implemented effectively in carnivores like the Florida panther15 (Puma concolor coryi). However, genetic rescue could be counter-productive if populations are locally adapted16. Conservation strategy must consider the relative risks of inbreeding and outbreeding depression before genetic rescue is implemented17.
Tigers have lost an estimated 93% of their historical range18. Of the nine tiger subspecies, four are extinct (P. tigris sondaica, P. Tigris balica, P. tigris virgata, and P. tigris amoyensis). The remaining five subspecies have experienced intense population bottlenecks (e.g. 90% decline in the Indo-Chinese tigers)19, 20 in historical times and prolonged reduction in distribution21. Tiger conservation and recovery is a global priority as exemplified by several international efforts (e.g. Global Tiger Forum). Recent surveys suggest an increase in tiger numbers22 indicating the success of such initiatives.
The Indian subcontinent is home to more than 60% of all tigers and harbors over half the global genetic diversity of the species23. Securing the future of Indian tigers is critical for survival of the species. From a time when tigers were widespread24, today tigers survive in small (median = 19, mean = 35)25, often isolated populations, many of which may not be viable on their own. Tiger monitoring is conducted at the scale of protected areas or geographically defined landscapes (e.g., central India) with clustered protected areas. Data on historical occupancy of Indian tigers24 reveals relatively continuous distributions. This implies that tiger genetic clusters may have wider geographical extents than currently monitored landscape units, as supported by microsatellite data20. However, tiger monitoring surveys indicate that tiger populations within some geographic landscapes have become isolated and habitat connectivity between others is tenuous25. Additionally, these landscapes represent different biogeographic zones, such as the Semi-Arid or the Western Ghats, with distinctive habitats26 such as grasslands, subtropical moist forests, mangroves and tropical dry forests27. Given their wide distribution, tigers may be categorized as generalist species. However, tiger populations living in different habitats may potentially be locally adapted; in such cases genetic rescue may not be considered a valid conservation strategy17. Therefore, understanding the spatial extent of genetic clusters as determined by genetic connectivity and local adaptation is essential for conservation.
In this paper, we collect genome-wide SNP data from wild tigers to better understand how protected area-based populations in India are segregated into genetic clusters. We investigate the following questions: (i) what are the genetic clusters for tigers in India? (ii) how is genetic variation distributed among these genetic clusters? (iii) are there any signatures of differential local adaptation between these clusters?
Results
Sequencing Data, SNP calling, and Filtration
The total number of retained reads per sample varied from 4,869 to 12,882,482, with an average of 4,118,660 (as seen in the output of process_radtags command from the program Stacks28). After alignment, the program Stacks was used to assess the depth at unique loci obtained per individual. The number of unique loci (as called by Stacks) obtained per sample (Table S2) was also highly variable, ranging from 1 to 693,476, with an average of 268,964. The raw vcf file (after calling SNPs in Freebayes29) had 1,527,595 loci. This vcf file was passed through several filters, as mentioned in the methods. In the final dataset, only individuals with less than 25% missing data (although the average percentage of missing data was much lower at 4.2%) and SNPs with less than 5% missing data were retained. Details of samples removed are presented in Table S3.
Genetic Differentiation
Genetic differentiation was assessed using 10,184 SNPs from 38 samples. Tigers from across India (Fig. 1) were partitioned into three major clusters in all analyses. Further sub-structuring within one cluster was indicated by most analyses. The three clusters identified by Admixture (best support for K = 3) and PCA (Fig. 2a,c), geographically coincided with individuals in the northwest, central and southern India. The first cluster, designated as the ‘NW’ cluster, was solely composed of individuals from a single protected area in the northwest region - Ranthambore (Fig. 1). The third cluster, designated ‘SI’, was comprised of all samples from southern India (Fig. 1). The remaining individuals formed a single cluster designated as ‘C’. Within this cluster, northeast samples (Arunachal, Kaziranga and Morigaon), Bandhavgarh and Simlipal derived between 12 and 21% of their ancestry from the other two clusters.

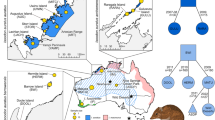
Map depicting tiger reserves in India. The total number of samples obtained for the study was 54 from across 21 locations (Table S1). Sample size from each location is denoted by ‘N’. Of these samples however, only 38 were used for the final analyses. The map was generated using the ArcGIS software (Desktop), an ESRI product version 10.2. (http://www.esri.com/software/arcgis/arcgis-for-desktop). The forest cover map, published in the State of Forest report-2013 (http://fsi.nic.in/cover_2013/sfr_forest_cover.pdf), was purchased from the Forest Survey of India, and the Protected Area boundaries from the Wildlife Institute of India, Dehradun.

Genetic clusters inferred using multiple methods. (a) Genetic clusters inferred at different values of K (2–5) in Admixture. Each vertical bar indicates a single individual, with the Y axis depicting the proportion derived from each cluster. The optimal suggested value of number of clusters was K = 3. The cluster names above indicate clusters detected at K = 3, and K = 5. (b) An illustration to depict the geographical extant of the three clusters inferred at K = 3. The figure was generated in QGIS version 2.0.1. (https://qgis.org/downloads/). The polygons have been added just for illustrative purposes. (c) Principal components analysis depicting the first and second principal components. The percentage of variation explained is given in brackets. (d) A phylogenetic network constructed in SplitsTree4. Sample names have been coloured for the purpose of representation only.
Cluster C was further subdivided into ‘N’ (comprising individuals from the North), ‘NE’ (northeast individuals from Kaziranga, Morigaon and Arunachal) and ‘CI’ (remaining individuals from central India), supported to varying degrees by different analyses. The phylogenetic network (Fig. 2d) showed two further splits in the C cluster separating out N and NE, although these were shallower splits in comparison to SI and NW. Network analysis also supported the presence of hierarchical structure (Fig. 3a). At a low threshold of genetic similarity (0.16), three clusters were inferred, consistent with our clustering analyses. However, two individuals from central India were assigned to the NW cluster (albeit with very low affinity – Fig. S11). Community detection at higher thresholds (0.163, 0.165), identified four clusters, splitting C into a north- northeast (N-NE) and a CI cluster. A further increase in threshold (0.167) separated individuals from the northeast, Bandhavgarh and Simlipal into a fifth cluster. Admixture results for higher values of K (Fig. 2a) also supported this inference. Estimates of ancestry coefficients (range: 0.00001–0.99998, at K = 3) are provided in supplementary materials (Table S6).

Networks plotted at four thresholds of genetic similarity. (a) Each panel (i–iv) in the figure depicts the threshold of genetic similarity used to connect individuals – 0.160–0.167, With increasing thresholds, dissimilar populations break off into separate modules. NW and SI separate into modules at a low threshold. Differentiation within C is evident at higher thresholds. Individuals are geographically placed. (b) Test of significance of modularity at each genetic similarity threshold. 5000 permutations were done to generate the distribution of the permuted data. The red line indicates the observed modularity.
Summary Statistics
Theta (H) was highest for C (0.48), intermediate for SI (0.35) and lowest for NW (0.22) (Table 1). Mean heterozygosity (He, Table 1) followed a similar trend. Inbreeding coefficients (Fig. S5) were higher in NW (mean = 0.48) compared to other clusters. Pair-wise relatedness estimates (Supplementary Fig. S4) revealed two samples from Ranthambore were identical (pair-wise relatedness = 0.9) and hence one of these individuals was removed from all analyses. Overall, relatedness was high in NW as well (range = 0–0.5, mean = 0.27) compared to those in other protected areas, e.g. Kanha (0) and Wayanad (range = 0–0.45, mean = 0.19), and other genetic clusters (C: range = 0–0.65, mean = 0.08).
Pair-wise FST, estimated assuming three clusters (NW, C, SI), was highest for the NW–SI comparison (0.35, Table 2). When assuming five genetic clusters (NW, N, NE, CI and SI), with N, NE and CI combined as a group (Table 3), AMOVA revealed that 65.32% (Vd) of the total variation observed was due to variation within individuals (p value = 0). Variation contributed by differences among groups (Va), although not statistically significant (p value = 0.09), was much lower (9.26%). FIS was higher in CI and NE clusters (Table 4).
Isolation by Distance
A Mantel test revealed a positive correlation between genetic (DPS, based on proportion of shared alleles) and geographic distances (Mantel’s R = 0.62, p-value = 0.001). This positive relationship was observed only at short distances (Fig. S6). The Mantel’s correlogram (Fig. S7) confirmed this positive relationship over distances of less than 500 km (Mantel’s R = 0.79, p-value = 0.001). Further increase in geographic distance did not result in increase in the genetic distance.
Genetic Diversity
For almost all sample sizes, NW had the lowest genetic diversity and C had the highest (Fig. 4a). Much of the diversity (Venn diagram, Fig. 4b) was shared across all groups (~62%) with private variation being much lower (NW - 2%, SI - 4% and C - 8.6%, when considering three clusters). Individually, the NW and C clusters represented the lowest and the highest proportion (~73% and ~91% respectively) of total diversity in Indian tigers. Therefore, the combined diversity of NW and SI accounts for about 91% of the total genetic diversity (Fig. 4c). However, CI and SI together account for 97% of the total diversity.

Private and shared genetic diversity between the genetic clusters. (a) Mean private allelic richness of each cluster (Y axis) at different sample sizes of alleles (X axis). Alleles were sub-sampled for all possible combinations of loci. The error bars represent standard error values. (b) A Venn diagram depicting proportion of private and shared richness in each cluster and their combinations. (c) Using the data from the Venn diagram, a bar plot depicting the proportion of genetic diversity retained for two different combinations of clusters.
Climate and vegetation based differentiation of tiger reserves
The PCA including all points across India showed a gradient of separation of tiger reserves along PC2 – mainly driven by precipitation (Figs S13 and S14). When only points falling within tiger reserves were plotted, the protected areas showed a north to south gradient, while the northeast regions showed separation along a different axis (Fig. S16). Multiple temperature and precipitation variables contributed to the two PC axes (Fig. S15).
Loci under Selection
The Bayescan analysis did not identify any outlier loci (Fig. S8) at a false discovery rate (FDR) of 0.05.
Discussion
In this study, we investigated population differentiation and genetic diversity of Indian tigers using 10,184 SNPs typed for 38 wild individuals from 17 protected areas (PAs). Bayesian clustering, PCA plots and isolation by distance tests establish genetic differentiation within the Indian subcontinent. Our data and analyses reveal that tiger populations in India cluster into three genetic groups that broadly map onto geographic tiger landscapes and represent groups of proximate protected areas. A large proportion of genetic diversity is shared between clusters, while population specific diversity is variable but low. Importantly, from a conservation perspective, our study reveals an isolated tiger population with low genetic diversity.
Our analyses reveal that tigers from the NW cluster (Ranthambore) are genetically isolated. Such genetic isolation is supported by the fact that they form a distinct cluster in structure analysis (Fig. 2a) and have the highest pair-wise FSTs (Table 2) with other genetic clusters. They separate from other individuals in the network analysis, even at low thresholds of genetic similarity (Fig. 2, 0.16). Geographically, Ranthambore is poorly connected to other tiger conservation landscapes30. Historically, tigers extended further northwest of Ramthambore into Pakistan, but went locally extinct in the early 1900s31. Genetic data reveal that northwest tigers and these extinct populations were connected to other tiger populations in the past20. More recently, tiger populations from the two closest PAs to Ranthambore, Panna and Sariska, were extirpated (2009 and 2004 respectively)32, 33. Together, this makes Ranthambore the western-most extant population of wild tigers today.
Our data also suggest that genetic diversity in Ranthambore is much lower in comparison to all other genetic clusters (theta (H) = 0.21), possibly due to small effective size and/or isolation. Low genetic variation contributes to higher extinction risk in wild populations14. Deleterious mutations also accumulate in small populations (e.g. Woolly mammoths (Mammuthus primigenius)34). While we point out that our data contain some related individuals (as measured by the p^ statistic, Fig. S4), we reanalyzed population structure excluding these (supplementary materials). Ranthambore individuals (2 samples) still form a separate cluster at the most likely value of K (Fig. S18). Our results suggest that the Ranthambore population is isolated and that inbreeding depression is a realistic possibility in this population. Future research efforts can further investigate this possibility.
Samples from southern India form a distinct genetic cluster (supported by Admixture, PCA, network analysis, and high pair-wise FST). This is in contrast to a previous microsatellite-based study20 that found no evidence for differentiation within peninsular India, but consistent with a report on tiger monitoring in India25. Our genome-wide SNP data was able to reveal this cryptic population structure35, 36. We caution that our final dataset did not include the only large PA in the Eastern Ghats (Nagarjunsagar Srisailam Tiger Reserve - NSTR, Fig. 1), a population that could mediate connectivity between South and central Indian clusters. Admixture analysis with one sample from NSTR was not conclusive as very few reads from the sample passed quality filtering (Fig. S12).
Northeastern tigers were previously assigned to a common cluster with central India23. In contrast, a recent report that extensively sampled northeast tigers suggests that they form a distinct genetic cluster25. Owing to our limited sampling in this region, our data is unable to resolve the genetic status of northeastern tigers. These individuals show varied genetic affinity in different analyses – part of the central cluster (Admixture analysis), an independent cluster (phylogenetic network) and part of a North – northeast cluster (the network analysis). Further, tigers from this region also have high genetic diversity (high heterozygosity – Table S5 and FIS– Table 4). Northeast tigers could be genetically closer to southeast Asian tigers. Evaluating range-wide genomic diversity for tigers (including samples from southeast Asia) may better resolve the population status of the northeast tigers.
The central genetic cluster harbors the highest diversity. High effective size and/or gene flow with neighbouring clusters could explain this. Our results are also consistent with theoretical37 and empirical studies (based on microsatellite data) that show greater diversity in centrally located populations, including in endangered species, such as the Cross River gorilla38 (Gorilla gorilla diehli) and violets39 (Viola pumila, V. stagnina).
Despite relatively continuous historical tiger distributions, we observe a signal of genetic isolation by distance. Ecological theory predicts that large-bodied carnivores like tigers may move far (~450 km40). On the other hand, a genetic study found support for even higher dispersal distances (~650 km41). Our results support ecological theory, with isolation by distance operating at the scale of 300–400 km (Fig. S6). However, loss of connectivity and genetic differentiation can be observed at short distances, as in the case of Ranthambore, and even in central India, as has been reported in previous studies42, 43. Overall, if functional connectivity (e.g. through corridors) is maintained between PAs within the identified (or between) clusters, further genetic differentiation can be avoided.
The widely distributed North American grey wolf (Canis lupus) occurs over varied environments and reveals signatures of local adaption despite high gene flow44. Tigers occur across a range of vegetation types and climatic gradients across India. However, while we do find high genetic differentiation among the clusters, we do not find evidence of adaptive divergence (based on outlier tests). In summary, the clusters identified do not appear to merit the status of conservation units45. Our analyses suggest that assisted gene flow between genetic clusters could potentially be a viable conservation strategy if genetic rescue is required. We caution that methods such as ddRAD usually sample only about 1–10% of the genome46 and data from whole genomes may be better able to identify signatures of selection.
Population structure can arise through the interplay of multiple factors including population history, selection and connectivity. Disentangling the effects of each of these is often challenging. However, understanding their relative contributions can have important consequences for species survival. For instance, while it is generally understood that small populations are at greater risk of inbreeding and extinction, the duration of time for which the population has been small may have important consequences. Recently bottlenecked populations may be of greater conservation concern than populations that have remained small for a long time47. Further, understanding whether population change has occurred in response to older climatic fluctuations, as opposed to more recent anthropogenic effects is important to consider when deciding endangerment status (IUCN criteria) and planning species recovery45.
In the case of Indian tigers, we find evidence for population structure. While the southern Indian cluster includes several populations, the northwestern cluster is restricted to a single tiger population, Ranathambore. The observed genetic differentiation between Ranathambore and the central cluster could signify the lack of gene flow relatively recently and/or older population divergence. Additionally, differential population size changes through historical time could impact the magnitude of differentiation. We attempted to understand the demographic history of the genetic clusters we identified using a site frequency spectrum-based approach. However, the estimated site frequency spectrum had very few singletons and none of the models fit our data well (supplementary materials, Fig. S19). This could be due to a combination of our small sample size, as well as the fact that tigers from the Indian subcontinent have undergone a very recent bottleneck about 200 years ago23, making it difficult to accurately infer demographic history48. In summary, we cannot explain which parameters (changes in population size, changes in connectivity, or both) are responsible for the northwestern differentiation we observe. Whole genome sequencing and higher sample sizes may allow better detection of low frequency alleles and more accurate reconstruction of demographic history in the future.
Because it is difficult to sample wild tigers invasively, our sampling is opportunistic. As a result, our sample sizes are low and tend to be clustered in space, both of which may impact our results49, 50. It is possible that we may be underestimating the number of genetic clusters due to underrepresentation of populations. Similarly, higher sample sizes in the northeast may resolve admixture signatures51. However, while our sample sizes are small, the number of loci is high, providing greater power to detect population structure50, 52. Future studies that acquire genome-wide data from non-invasive sources such as tiger scat will enhance our ability to sample geographically53, allowing us to detect existing and on-going changes in population structure and connectivity.
Population genomics of wild species is being increasingly used to inform conservation practice, including to make recommendations on reintroduction (e.g. wolves (Canis lupus)54), to set up breeding programs (e.g. Tasmanian devils (Sarchophilus harrisii)7) and to designate management/conservation units55. Our population genomic study of wild Indian tigers finds evidence for a small and isolated population of tigers in northwest India. The long-term persistence of this population may require connectivity with neighboring populations, such as with central India. In summary, our study reveals how genome-wide data and analyses can flag populations that may require urgent conservation attention (such as Ranthambore) and identify strongholds of variation (such as central India). Such inference has significant impacts for on-ground conservation practice.
Methods
Samples and DNA Extraction
Tissue samples of wild tigers are difficult to obtain since capturing them is logistically challenging in India. However, multiple laboratories working on tiger population genetics have access to post-mortem samples and these constitute the bulk of our sampling. In some cases, samples were obtained from captured tigers (e.g. individuals involved in conflict). The list of samples is provided in supplementary Table S1. Geographical locations of samples are shown in Fig. 1. We were able to acquire a total of 54 samples, 50 samples representing 17PAs (out of 49 tiger reserves) and four samples from outside PAs. Samples were preserved in absolute ethanol and stored at −20 °C. DNA extraction was conducted using the spin column method following the manufacturer’s instructions (Qiagen).
Library preparation
Double-digested RAD libraries were prepared as outlined in Peterson et al.56. A combination of two frequent cutters, Nla III and MluC I (NEB), was used to maximize coverage by getting more fragments. DNA concentration of all extracts was measured using Qubit (Invitrogen) and the initial quantity for library preparation was standardized to 200 ng. DNA quality was not uniformly high, especially in samples that had been preserved for a long time. Some samples were processed despite poor quality if they were single representatives of their population and were usually included in the library twice. Extended ligation was carried out for 13 hours, as standardized by Chattopadhyay et al.51. Library quality was assessed based on the bioanalyzer profile. A total of ten libraries was prepared and sent for paired-end sequencing on three lanes of an Illumina HiSeq. 1000 machine. The low diversity issue encountered with ddRAD samples during sequencing was circumvented by the addition of 30% phiX genome into the sequencing run (as suggested by Illumina).
Processing the Raw Data
The sequencing quality of reads was initially assessed using the program FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The data was demultiplexed into individual samples using the program Stacks28. Reads with one mismatch in the index were rescued. Demultiplexed data were filtered to remove reads with poor quality (option –q) and the presence of uncalled bases (option –c), and were also truncated to 80 base pairs (option –t). The read numbers retained per sample varied considerably. The quality-filtered reads were aligned to the reference genome57 using Bowtie258. Both paired and unpaired reads were aligned with default criteria. Technical replicates were merged and Samtools59 was used to process the aligned data. Read group information was added using Picard tools (http://picard.sourceforge.net) to reflect the unique sample identity. Reads were realigned to account for indels using the GATK package60. Since data from samples that had been sequenced twice were combined after alignment, the number of unique loci/regions identified in each sample (instead of raw reads per sample) was used to compare data obtained per sample. Depth and coverage stats were calculated using Stacks and Bedtools61, 62.
SNP Calling and Filtering
Unique regions were identified in the program Stacks for each sample. Samples that had fewer than 100,000 loci were eliminated from further analysis. Our data had low average depth (Supplementary Fig. S1), and therefore Freebayes29 was used to call SNPs, as it considers multiple samples from a population simultaneously to call variants with high confidence. Only reads supported by at least 8x depth of coverage in the population were considered to identify an allele. Additionally, only dinucleotide variants (excluding indels, MNPs and complex variants) were retained. To filter loci with low data, we calculated the missing data per sample using vcftools63. To optimize the tradeoff between the number of samples retained and the number of loci with low missing data, the dataset was filtered multiple times. The vcf file obtained was subjected to filtering based on missing data (<5% per SNP), mapping quality (>= 30) and minor allele frequency (>= 0.05). Thinning of loci within 500 bp of each other was done to remove SNPs that might be in linkage disequilibrium. SNPs that were not in Hardy-Weinberg equilibrium (p < 0.01) were filtered out. Additionally, SNPs were also filtered based on mean depth of coverage across samples. However, as the results obtained with this set of loci (7741 SNPs) were qualitatively similar to those observed with the larger dataset (comparison of datasets based on FST and Admixture results), the smaller dataset was not utilized in the final analysis (data not shown).
Genetic Differentiation
Multiple methods were used to infer tiger genetic clusters. A phylogenetic network was constructed in the program SplitsTree464. A NeighborNet network was computed based on p-distance generated from the input data in the form of fasta sequences. A maximum likelihood based approach implemented in the program Admixture65 was used to identify the number of population clusters supported by the data. The program was run with 10-fold cross-validation for K values ranging from 1 to 6. The best K was inferred based on the value of K that had the least cross-validation error. For a more visual interpretation of population structure, a principal components analysis (PCA) was performed using an R package66. Additionally, a network-based approach was also used to infer hierarchical clustering as implemented in NetStruct67. The program constructs a network by connecting individuals above a genetic similarity threshold. This is followed by community detection to identify groups of individuals much more closely related to each other than to those in other groups – a process analogous to detecting genetic clusters. Based on the observed range of genetic distance, the network was plotted at multiple thresholds, followed by community detection using a fast-greedy algorithm. A permutation test was done to test the significance of the observed network.
Pair-wise FST was calculated between clusters using the Weir and Cockerham estimator in Arlequin68. Summary statistics, including heterozygosity (He) values and theta (H), were computed in Arlequin68. Pair-wise relatedness (p^) and individual inbreeding coefficients (f) were estimated in Plink69. AMOVA68 was used to test for hierarchical structure. Theta (H) was estimated as a proxy for effective size (Ne) of each genetic cluster.
Isolation by Distance
A Mantel test was performed to check for isolation by distance in R66. Genetic (DPS = 1− proportion of shared alleles) and geographic distance were calculated for all pairs of individuals. To test for significance of the observed Mantel’s R a randomization was done with 999 replicates. A Mantel’s correlogram (package vegan) was used to identify the distance classes at which the correlation was significant.
Distribution of Genetic Diversity between Clusters
To see how genetic diversity is distributed across the populations, private allele richness was estimated for each cluster, as well as for combinations of clusters. Since diversity measures can be influenced by sample size, these were standardized across clusters using rarefaction. Private allele richness was estimated for each standardized sample size using the program ADZE70 and plotted. To visualize sharing of diversity, the results were plotted as a Venn diagram by calculating the proportion for total diversity in each (or combination of) cluster(s).
Climate and vegetation based differentiation of tiger reserves
In order to visually inspect whether the available habitats in tiger reserves were differentiated based on climate and/or vegetation, a PCA was run for 19 bioclimatic variables (bioclim), aridity (FAO Global Aridity Index http://www.fao.org/nr/aboutnr/nrl/en/) and land cover classification (GlobCover Land Cover v2.2 - http://geodata.grid.unep.ch/options.php?selectedID = 2054&selectedDatasettype = 16). Regular points were placed on an outline of the India map (grid spacing 0.1) in QGis (v2.0.1). Multiple climate layers (http://www.worldclim.org/bioclim), aridity and vegetation (GlobCover) were used to define the environmental space for the sampled points. The ‘point sampling tool’ was used to extract data for the previously generated points. Points falling within tiger reserves were marked in the attribute table. This table was exported to the software R (v3.3.2) and after centering and scaling the data, a PCA was performed with all points. To closely examine the separation between the tiger reserves, a PCA of only points falling in tiger reserves was performed.
Test for Loci under Selection
A Bayesian approach was used to test for SNPs that might be under selection among the different genetic clusters identified using the software tool Bayescan71. A 5% false discovery rate was used to identify outlier loci. The Bayescan analysis was run with prior odds for the neutral model set to 1000; higher odds lower the possibility of obtaining false positives. It is recommended to set the prior odds high (1000) when using thousands of markers.
Data Accessibility
The sequences are available on GenBank and can be obtained from the SRA database with the accession number SRP114885.
Ethical Approval
Our study does not involve any experiments with live animals. Further, while we use tissue samples, none of these were directly obtained for the purpose of this study. We only used samples from tissue previously collected for other studies/purposes. Therefore, ethical clearance regarding sample collection is not applicable to our study.
References
Allendorf, F. W., Hohenlohe, Pa & Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709 (2010).
Steiner, C. C., Putnam, A. S., Hoeck, P. E. A. & Ryder, O. A. Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 1, 261–281 (2013).
Bruford, M. W. et al. In Molecular Genetic Approaches in Conservation 278–297 (1996).
Rohland, N. et al. Genomic DNA Sequences from Mastodon and Woolly Mammoth Reveal Deep Speciation of Forest and Savanna Elephants. PLoS Biology 8, e1000564 (2010).
Bowden, R. et al. Genomic tools for evolution and conservation in the chimpanzee: Pan troglodytes ellioti is a genetically distinct population. PLoS Genet. 8, 1–10 (2012).
McMahon, B. J., Teeling, E. C. & Höglund, J. How and why should we implement genomics into conservation? Evol. Appl. 7, 999–1007, doi:10.1111/eva.12193 (2014).
Miller, W. et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc. Natl. Acad. Sci. 108, 12348–12353 (2011).
Norman, A. J., Street, N. R. & Spong, G. De novo SNP discovery in the Scandinavian brown bear (Ursus arctos). PLoS One 8, e81012 (2013).
Kraus, R. H. S. et al. A SNP-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on non-invasively collected samples. Mol. Ecol. Res. 15, 295–305, doi:10.1111/1755-0998.12307 (2014).
Stetz, J. B. et al. Discovery of 20,000 RAD–SNPs and development of a 52-SNP array for monitoring river otters. Cons. Gen. Res. 8, 299–302, doi:10.1007/s12686-016-0558-3 (2016).
Jeffries, D. L. et al. Comparing RADseq and microsatellites to infer complex phylogeographic patterns, a real data informed perspective in the Crucian carp, Carassius carassius, L. Mol. Ecol. 25, 2997–3018 (2016).
Benestan, L. et al. RAD genotyping reveals fine-scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Mol. Ecol. 24, 3299–3315 (2015).
Ripple, W. J. et al. Status and ecological effects of the world’s largest carnivores. Science 343, 1241484–1241484 (2014).
Saccheri, I., Kuussaari, M., Kankare, M., Vikman, P. & Hanski, I. Inbreeding and extinction in a butterfly metapopulation. Nature 392, 1996–1999 (1998).
Johnson, W. E. et al. Genetic restoration of the Florida panther. Science 329, 1641–1645 (2010).
Edmands, S. Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 16, 463–475, doi:10.1111/j.1365-294X.2006.03148.x (2007).
Frankham, R. et al. Predicting the probability of Outbreeding Depression. Conserv. Biol. 25, 465–475 (2011).
Dinerstein, E. et al. A USER’ S GUIDE WILD TIGERS: 2005–2015 Setting Priorities for the Conservation and Recovery of Wild Tigers: 2005–2015. (2006).
Luo, S.-J. et al. Phylogeography and genetic ancestry of tigers (Panthera tigris). PLoS Biol. 2, e442 (2004).
Mondol, S., Bruford, M. W. & Ramakrishnan. Demographic loss, genetic structure and the conservation implications for Indian tigers. Proc. R. Soc. B 280, doi:10.1098/rspb.2013.0496 (2013).
Goodrich, J. M. et al. Panthera tigris. The IUCN Red List of Threatened Species 2015. Iucn 8235 (2015).
WWF Tx2 Annual Report. Doubling Wild Tigers (2016).
Mondol, S., Karanth, K. U. & Ramakrishnan, U. Why the Indian subcontinent holds the key to global tiger recovery. PLoS Genet. 5, e1000585 (2009).
Karanth, K. K., Nichols, J. D., Karanth, K. U., Hines, J. E. & Christensen, N. L. The shrinking ark: patterns of large mammal extinctions in India. Proc. R. Soc. B Biol. Sci. 277, 1971–1979 (2010).
Jhala, Y. V., Qureshi, Q. & Gopal, R. (eds). Status of Tigers in India 2014. National Tiger Conservation Authority, New Delhi & The Wildlife Institute of India, Dehradun (2015).
Rodgers, W. A., Panwar, H. S. & Mathur, V. B. Wildlife Protected Area Network in India: A Review (Executive summary) (2002).
Ranganathan, J., Chan, K. Ma, Karanth, K. U. & Smith, J. L. D. Where can tigers persist in the future? A landscape-scale, density-based population model for the Indian subcontinent. Biol. Conserv. 141, 67–77 (2008).
Catchen, J., Hohenlohe, Pa, Bassham, S., Amores, A. & Cresko, Wa Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–40 (2013).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. bioRxiv 1207, 3907 (2013).
Reddy, P. A. et al. Genetic evidence of tiger population structure and migration within an isolated and fragmented landscape in Northwest India. PLoS One 7, e29827 (2012).
Nowell, K. & Jackson, P. Wild cats. Status Survey and Conservation Action Plan. IUCN, Gland, Switzerland, doi:10.1023/A:1008907403806 (1996).
Gopal, R., Qureshi, Q., Bhardwaj, M., Jagadish Singh, R. K. & Jhala, Y. V. Evaluating the status of the Endangered tiger Panthera tigris and its prey in Panna Tiger Reserve, Madhya Pradesh, India. Oryx 44, 383–389 (2010).
Gratwicke, B. Poaching laws are useless without solid enforcement. Nature 445, 147–147 (2007).
Rogers, R. L. & Slatkin, M. Excess of genomic defects in a woolly mammoth on Wrangel island. PLoS Genet. 13, e1006601, doi:10.1371/journal.pgen.1006601 (2017).
Glover, K. A. et al. A comparison of SNP and STR loci for delineating population structure and performing individual genetic assignment. BMC Genetics 11, 2–12 (2010).
Gärke, C. et al. Comparison of SNPs and microsatellites for assessing the genetic structure of chicken populations. Animal Genetics 43, 419–428, doi:10.1111/j.1365-2052.2011.02284.x (2011).
Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188 (2008).
Bergl, R. A., Bradley, B. J., Nsubuga, A. & Vigilant, L. Effects of habitat fragmentation, population size and demographic history on genetic diversity: the Cross River gorilla in a comparative context. Am. J. Primatol. 70, 848–59 (2008).
Jaquiéry, J., Guillaume, F. & Perrin, N. Predicting the deleterious effects of mutation load in fragmented populations. Conserv. Biol. 23, 207–218 (2009).
Sutherland, G. D., Harestad, A. S., Price, K. & Lertzman, K. P. Scaling of Natal Dispersal Distances in Terrestrial Birds and Mammals. 4, (2000).
Joshi, A., Vaidyanathan, S., Mondol, S., Edgaonkar, A. & Ramakrishnan, U. Connectivity of tiger (Panthera tigris) populations in the human-influenced forest mosaic of Central India. PLoS One 8, e77980 (2013).
Yumnam, B. et al. Prioritizing Tiger Conservation through Landscape Genetics and Habitat Linkages. PLoS One 9, e111207 (2014).
Thatte, P., Joshi, A., Vaidyanathan, S. & Ramakrishnan, U. Protected corridors preserve tiger genetic diversity and minimize extinction into the next century. bioRxiv 3323 (2016).
Pilot, M. et al. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity (Edinb). 112, 428–42 (2014).
Crandall, K. A., Bininda-Emonds, O. R. R., Mace, G. M. & Wayne, R. K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 15, 290–295 (2000).
Van Tassell, C. P. et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 5, 247–252 (2008).
Spurgin, L. G. et al. Museum DNA reveals the demographic history of the endangered Seychelles warbler. Evol. Appl. 7, 1134–1143 (2014).
Robinson, J. D., Coffman, A. J., Hickerson, M. J. & Gutenkunst, R. N. Sampling strategies for frequency spectrum-based population genomic inference. BMC Evol. Biol. 14, 254 (2014).
Schwartz, M. K. & McKelvey, K. S. Why sampling scheme matters: The effect of sampling scheme on landscape genetic results. Conserv. Genet. 10, 441–452 (2009).
Morin, P. A., Martien, K. K. & Taylor, B. L. Assessing statistical power of SNPs for population structure and conservation studies. Mol. Ecol. Resour. 9, 66–73 (2009).
Chattopadhyay, B. et al. Genome-wide data reveal cryptic diversity and genetic introgression in an Oriental cynopterine fruit bat radiation. BMC Evol. Biol. 16, 41 (2016).
Willing, E., Dreyer, C. & Oosterhout, C. V. Estimates of Genetic Differentiation Measured by F ST Do Not Necessarily Require Large Sample Sizes When Using Many SNP Markers. PLoS One 7, e42649 (2012).
Snyder-Mackler, N. et al. Efficient Genome-Wide Sequencing and Low Coverage Pedigree Analysis from Non-invasively Collected Samples. Genetics 203, 699–714 (2016).
Leonard, J. A., Vilà, C. & Wayne, R. K. Legacy lost: Genetic variability and population size of extirpated US grey wolves (Canis lupus). Mol. Ecol. 14, 9–17 (2005).
Milano, I. et al. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 23, 118–135 (2014).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7, e37135 (2012).
Cho, Y. S. et al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat. Commun. 4, 2433 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–303 (2010).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Alexander, D. H. & Novembre, J. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Research 19, 1655–1664, doi:10.1101/gr.094052.109 (2009).
Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Greenbaum, G., Templeton, A. R. & Bar-David, S. Inference and analysis of population structure using genetic data and network theory. Genetics 202, 1299–1312, doi:10.1534/genetics.115.182626 (2016).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50 (2005).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–75 (2007).
Szpiech, Z. A., Jakobsson, M. & Rosenberg, N. A. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 2498–2504 (2008).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180, 977–993 (2008).
Acknowledgements
We would like to acknowledge Noah Rosenberg, Fred Allendorf, Stephan Prost, Robin Vijayan and Krishnapriya Tamma for their comments, Srikrishna for help with Matlab, Swati Saini for help with the sampling map, Gili Greenbaum for help with NetStruct, Gideon Bradburd for Spacemix, Anuradha Reddy for samples and discussion, and the Forest Departments of Karnataka, Kerala, Rajasthan, Madhya Pradesh, Uttarakhand, Arunachal Pradesh, Assam, Orissa, West Bengal, Andhra Pradesh, Tamil Nadu, and Maharashtra for help in obtaining the samples. We acknowledge the Centre for Cellular And Molecular Platforms (C-CAMP) facility for the Illumina sequencing runs. We thank the National Tiger Conservation Authority and the Ministry of Environment, Forests and Climate Change for permissions and support. We would also like to thank Jeremy Hsu and Tanmoy Goswami for help with manuscript editing. Finally, we acknowledge logistical support and funding provided by Wildlife Conservation Society, India Program to MN. UR is currently supported as a Senior Fellow, Wellcome Trust/DBT India Alliance. This project was funded by the Department of Biotechnology under its environmental biotechnology taskforce, through project BT/PR13854/BCE/8/809/2010 to UR. MN was supported by BT/PR13854/BCE/8/809/2010. The research was also supported by internal NCBS funding to UR. GA was supported by the DBT funded program support on technological innovation and ecological research for sustainable use of bioresources in the Sikkim Himalaya, through the project BT/01/NE/PS/NCBS/09.
Author information
Authors and Affiliations
Contributions
U.R. and M.N. conceptualized the study. P.N., Y.V.J., A.Z., U.B. contributed to sample collection. P.N. and Y.V.J. helped in DNA extraction. M.N. contributed to library preparation. M.N., G.A. and U.R. participated in the data generation and analysis. M.N., G.A., and U.R. discussed and analyzed the results. M.N. and U.R. wrote the paper. Y.V.J. participated in manuscript editing.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Natesh, M., Atla, G., Nigam, P. et al. Conservation priorities for endangered Indian tigers through a genomic lens. Sci Rep 7, 9614 (2017). https://doi.org/10.1038/s41598-017-09748-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09748-3
This article is cited by
-
Recapitulating whole genome based population genetic structure for Indian wild tigers through an ancestry informative marker panel
Heredity (2022)
-
The tide of tiger poaching in India is rising! An investigation of the intertwined facts with a focus on conservation
GeoJournal (2022)
-
Exploiting genomic synteny in Felidae: cross-species genome alignments and SNV discovery can aid conservation management
BMC Genomics (2021)
-
Assembling mitogenome of Himalayan Black Bear (U. t. laniger) from low depth reads and its application in drawing phylogenetic inferences
Scientific Reports (2021)
-
Trends in Wildlife Connectivity Science from the Biodiverse and Human-Dominated South Asia
Journal of the Indian Institute of Science (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
