
Abstract
Despite the development of antiretroviral therapy against HIV, eradication of the virus from the body, as a means to a cure, remains in progress. A “kick and kill” strategy proposes “kick” of the latent HIV to an active HIV to eventually be “killed”. Latency-reverting agents that can perform the “kick” function are under development and have shown promise. Management of the infected cells not to produce virions after the “kick” step is important to this strategy. Here we show that a newly synthesized compound, L-HIPPO, captures the HIV-1 protein Pr55Gag and intercepts its function to translocate the virus from the cytoplasm to the plasma membrane leading to virion budding. The infecting virus thus “locked-in” subsequently induces apoptosis of the host cells. This “lock-in and apoptosis” approach performed by our novel compound in HIV-infected cells provides a means to bridge the gap between the “kick” and “kill” steps of this eradication strategy. By building upon previous progress in latency reverting agents, our compound appears to provide a promising step toward the goal of HIV eradication from the body.
Similar content being viewed by others


Introduction
Although human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) has become amenable to control since 1997 by antiretroviral therapy (ART), HIV remains incurable to date1. In 2015, approximately 36.7 million people were living with HIV and 1.1 million people died of AIDS2. Most anti-HIV drugs work by blocking key steps of the viral replication cycle, making them effective against actively-replicating viruses, but have little effect on latent HIV-1 in that remains in cellular reservoirs throughout the body. The viral replication cycle is essentially shut down in the latent virus, allowing it to persist in the body even with the administration of comprehensive ART. A cure for HIV would mean the lack of viral activity after treatment which is not the case with current ART. Such strategies toward this end include the goal of eliminating the cellular reservoirs which continue to harbor HIV3. Previously, a “kick and kill” strategy was proposed to connect a “kick” process to a subsequent “kill” process for HIV-infected cells, wherein “kicked,” i.e. cells containing active HIV, can be placed in fatal crisis by such means as a heightened immune response or the induction of cell damage4. The discovery of latency reverting agents (LRA) that “kick” the latent virus-containing cells to activate the expression of HIV-1 protein has signified promising progress toward this approach4. However, efficient “kill” device remains to be developed4, 5. We report herein a novel “lock-in and apoptosis” compound that follows the “kick” process and facilitates eradication of HIV-infected cells.
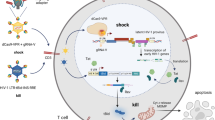
Generally, HIV recognizes and enters into CD4+ T lymphocytes. HIV proliferates inside the infected cells to release an enormous number of offspring virions that, in turn, propagates to other CD4+ T lymphocytes. Diminishing the release of new virions would help manage the virus population and reduce the burden of eradicating virus from the body. We sought to synthesize a small molecule that would hinder the budding of offspring virus, with the notion of turning an HIV-infected cell into a “prison cell” from which the invaded virus cannot escape (Fig. 1a and b). The host cell will eventually die without releasing the prisoner virus. Thus, if all reservoir viruses are kicked, locked in the host cell prison, and these cells apoptosed, the death of all such infected host cells would mark the eradication of HIV in the body.

Lock-in and apoptosis strategy using a cyclodextrin/dendrimer conjugate α-CDE and a man-made molecule L-HIPPO. (a) HIV-1 virion budding is mediated by viral protein Pr55Gag that binds to a membrane phospholipid PIP2. PIP2-bound Pr55Gag assembles in the membrane to form virions. (b) α-CDE delivers L-HIPPO into HIV-1-infected cell where L-HIPPO antagonizes the PIP2-Pr55Gag binding to interfere with virus budding. The infecting HIV-1 is enclosed and dies with the apoptosis of the host cell. (c) Structures of PIP2, D-HIPPO, L-HIPPO, cyclodextrin/dendrimer conjugates (α-CDE), and α-CDE conjugated with TRITC (α-CDE-TRITC).
It is known that the viral protein Pr55Gag is one of the products produced by hijacked host cells and mediates HIV-1 virion budding. Ordinarily, Pr55Gag migrates from the cytoplasm to the plasma membrane and binds to a specific inositol phospholipid, PIP2, in the membrane6. It was shown that the N-terminal MA (matrix antigen) domain of Pr55Gag interacts with PIP2 by prior NMR study7 and recent follow-up reports revising the role of acyl group of PIP28, 9. PIP2-bound Pr55Gag assembles each other in the membrane to form virions (Fig. 1a)6. As Pr55Gag-PIP2 binding triggers the virion budding, we considered that a small molecule that binds firmly to Pr55Gag could antagonize PIP2 in the Pr55Gag binding. Previously, we established a highly-sensitive in vitro assay to determine the dissociation constant (K d) for the binding of Pr55Gag to phosphoinositide derivatives using surface plasmon resonance (SPR) sensor analysis10, 11. We found that both negatively-charged inositol phosphates and the lipophilic acyl chains of phosphoinositide were essential for a strong interaction with Pr55Gag 10. As well, fully-phosphorylated inositol IP6 (phytic acid, “PA”) bound to the MA domain approximately 10 times stronger than IP3, the inositol head of PIP211. Accounting for each of these details, we had synthesized an artificial phosphoinositide, DL-HIPPO (DL-Heptanoylphosphatidyl Inositol Pentakisphosphate), as an isomeric mixture (a 1:1 mixture of D-HIPPO and L-HIPPO) (Fig. 1c). This compound was shown to have a MA-binding affinity 70-fold stronger than that of the less phosphorylated PIP2 derivative11. This activity was considered sufficient to antagonize PIP2 to suppress the membrane localization of Pr55Gag.
Here we examined the localization of HIV-1 Pr55Gag in HeLa cells using Venus fluorescent labeled Pr55Gag protein. HeLa cells were transfected with the plasmid vector pNL4-3/GagVenus12 having Venus yellow fluorescent protein at the C-terminus of Pr55Gag instead of a region called Pol of HIV-1 infectious clone pNL4-313, and incubated for 13 h. The cells were then fixed and observed by fluorescent confocal microscopy. In the absence of DL-HIPPO, Pr55Gag localized to “cytosol only” in 25.0% of cells and to “cellular membrane” in 75.0% of cells (Fig. 2a(A) and b(A)), well in accordance with previous findings12.

Effect of DL-HIPPO/D-HIPPO/L-HIPPO delivered into a cell by carrier on the cellular localization of Pr55Gag and HIV-1 release. (a) Effect of DL-HIPPO delivered into a cell by various carriers on the cellular localization of Pr55Gag. (A) Cellular localization of intact Pr55Gag. HeLa cells were transfected with a plasmid vector to express Gag-Venus (pNL4-3/GagVenus) and, after 13 h, Gag localization was observed by fluorescence microscopy. (B, C) Effect of DL-HIPPO-histone on Gag localization. HeLa cells were transfected with pNL4-3/GagVenus and, after 10 h, a complex prepared from DL-HIPPO (25 μM) and histone conjugated with TMR (25 μM) (DL-HIPPO-histone-TMR) was added. After a further 3 h incubation, localization of Gag and histone was observed by fluorescence microscope. (D, E) Effect of DL-HIPPO-dendrimer on Gag localization. The same experiment as that of (B, C) was performed using dendrimer instead of histone. (F, G) Effect of DL-HIPPO-α-CDE on Gag localization. The same experiment as that of (B, C) was performed using α-CDE instead of histone. (b) Effect of DL-HIPPO-carrier on the cellular localization of Pr55Gag. A total of approximate 100 cells in each experiment of (a) were observed and categorized into 4 types “dispersed in cytoplasm”, “punctate in cytoplasm”, “punctate in cytoplasm and membrane”, and “punctate in membrane” shown at the top of the table. The relative number of cells (%) in each category is shown. (c) Effect of DL-HIPPO-α-CDE on HIV-1 release. HeLa cells were transfected with pNL4-3 and, after 10 h, complex prepared from DL-HIPPO (25 or 50 μM) and α-CDE (25 μM) (DL-HIPPO-α-CDE) was added. After a further 3 h incubation, supernatant and the cells were harvested, lysed, and analyzed by immunoblotting using anti-p24 antibody. Wider images are shown in Supplementary Information F. (d) Quantification of effect of DL-HIPPO-α-CDE on HIV-1 release. Intensity of the bands in (c) were quantitated using ImageJ, and the amount of released virus, (amount of p24 in supernatant)/(amounts of p24 and Pr55Gag in cells), was calculated. Relative value to that of a control without additives is shown as percent. Data from three different experiments are shown as means ± standard deviations. P values were determined using Student’s t test. (e) Quantification of effect of D-HIPPO-α-CDE and L-HIPPO-α-CDE on HIV-1 release. The same experiment (3 h incubation after addition of HIPPO-α-CDE) as that in (c) and (d) was performed using enantiomerically pure D-HIPPO and L-HIPPO. Data from three different experiments are shown as means ± standard deviations. P values were determined using Student’s t test.
As DL-HIPPO, with its dense negative phosphate charges, was membrane non-permeable (data not shown), we sought to add an appropriate moiety possessing high positive charge in order to improve delivery of the compound across the cell membrane. We considered histone and dendrimer, known as PIP2 carriers14, and α-cyclodextrin/polyamidoamine dendrimer (G3) conjugate (α-CDE) (Fig. 1c)15,16,17 as potential carriers for the negatively charged molecule.
A histone complex, DL-HIPPO-histone-TMR, was prepared by mixing DL-HIPPO (25 μM) and histone conjugated with the fluorescent molecule tetramethylrhodamine [TMR, purchased from Echelon Biosciences (Salt Lake City, UT, USA)] (25 μM). A dendrimer complex (DL-HIPPO-dendrimer-TRITC) was prepared from DL-HIPPO (25 μM) and dendrimer conjugated with the fluorescent molecule tetramethylrhodamine isothiocyanate (TRITC)18 (25 μM). These complexes were added to the transfected cells. The cells were incubated for 3 h, fixed, and observed by fluorescence microscopy. When DL-HIPPO-histone-TMR was added, the ratio of Pr55Gag localized in “cytoplasm only” and “cellular membrane” were 61.6% and 38.4%, respectively, whereas Pr55Gag localized mainly in “cellular membrane” (73.1%) in the control experiment using histone-TMR alone (Fig. 2a(B)(C) and b(B)(C)). On the other hand, DL-HIPPO-dendrimer-TRITC resulted in less efficient effect compared with the histone complex (Fig. 2a(D)(E) and b(D)(E)).
The third candidate carrier, α-CDE17, 19, induced the most favorable ratio of Pr55Gag localization. α-CDE-TRITC was prepared from α-CDE according to a previous method20. α-CDE-TRITC and DL-HIPPO were mixed to form DL-HIPPO-α-CDE-TRITC, which was then added to HeLa cells. The ratio of Pr55Gag localized in “cytoplasm only” was 88.1% and that in “cellular membrane” was 11.9%, whereas the ratio of Pr55Gag localization in “cytoplasm only” and “cellular membrane” were 45.9% and 54.1%, respectively, in the experiment using α-CDE-TRITC alone (Fig. 2a(F)(G) and b(F)(G)). This weaker inhibitory effect would be traced back to that cyclodextrin abstracts cholesterol from cellular membrane and suppresses HIV-1 assembly21.
As DL-HIPPO-α-CDE-TRITC kept the most Pr55Gag away from the membrane of the tested candidates, its capability to lock the trigger for virion budding was examined more extensively. HeLa cells were transfected with the HIV-1 infectious clone pNL4-313 and incubated for 10 h. The DL-HIPPO-α-CDE-TRITC complex was added. After incubation for an additional 3 h, supernatant and cells were harvested and the amount of virus protein was analyzed by western blotting using anti-p24 antibody (Fig. 2c). This antibody reacts to both precursor protein Pr55Gag and its cleavage product, p24. Nearly half of the HIV-1 release was suppressed for both concentrations (25 and 50 μM) of DL-HIPPO complex tested, although the amount of membrane localized Pr55Gag was small (Fig. 2d).
Thus far we used DL-HIPPO that was synthesized as an equimolar mixture of D-HIPPO and L-HIPPO11. Usually, the biological activity of each isomer differs, i. e., in the case of D-HIPPO and L-HIPPO one isomer is potent and the other is less potent. Thus, the both isomers D-HIPPO and L-HIPPO were separately synthesized to identify the potent isomer (for the detail of synthesis, see Supplementary Information A).
Binding affinities of these two isomeric compounds to the MA domain of Pr55Gag were determined using the same SPR assay used previously10, 11. The K d for MA binding of D-HIPPO and L-HIPPO were 1.09 ± 0.47 and 0.18 ± 0.08 μM, respectively (Supplementary Information B). The K d determined for DL-HIPPO (0.25 ± 0.17 μM)11 was between the two values. The non-natural L-compound showed a higher binding affinity to MA compared with the natural D-isomer. We further assessed the HIV-1 release suppressing activity of the each isomer. The complex prepared from L-HIPPO (25 or 50 μM) and α-CDE (25 μM), L-HIPPO-α-CDE, inhibited more than 70% of virus release, while the other isomer D-HIPPO-α-CDE suppressed just 40% of the release (Fig. 2e).
The non-natural L-isomer was shown to be the potent isomer both in Pr55Gag binding and virus release suppression. To examine the possibility of apoptosis induction by L-HIPPO-α-CDE, we increased the incubation time beyond the three hours used in the virus-release experiment. Non-cytotoxicity of L-HIPPO-α-CDE to HeLa cells in the absence of HIV, even at high concentration (100 μM) and for prolonged incubation time (48 h), was shown by cell viability assay using MTT pigment. FACS analysis of cells treated with the complex using apoptosis indicator annexin V-Cy5 also confirmed the non-toxicity (for MTT assay and FACS, Supplementary Information C).
The effect of L-HIPPO-α-CDE on HIV-1 protein was then examined. HeLa cells were transfected with pNL4-3/Gag Venus and incubated for 10 h. Cells were treated with L-HIPPO-α-CDE, incubated for a further 12 h, stained with annexin V-Cy5, and analyzed by FACS (Fig. 3a). The number of cells stained with annexin V was very low (Q2: 5–6%) in control experiments using cells transfected with the vector and untreated with additives or treated with only carrier α-CDE (25 μM). More than half of the transfected cells treated with L-HIPPO-α-CDE (25 or 50 μM) were stained with annexin V. A dose of 50 μM of the complex showed the highest ratio (Q2: 81%) of cells stained, indicating the induction of apoptosis. Considerable amounts of cells were stained with annexin V in experiments with a shorter incubation time (4 h) after addition of the complex (25 or 50 μM) (Fig. 3b). However, cell population staining with necrosis indicator EthD-III (ethidium homodimer III) was almost the same as that seen in the control (Fig. 3c), excluding necrosis and showing induction of apoptosis. Worthy of note, two populations, high population and accompanying dim population, were seen in the FACS analysis of the cells transfected with pNL4-3/Gag Venus. Decrease of transfection efficiency showed increment of dim population, demonstrating the dim population is extracellular Gag-Venus particles that bind to the untransfected cells (Supplementary Information D). Apoptosis of the dim population (Fig. 3a,b and Supplementary Information D) is thought to be due to bystander effect of apoptosis of Gag-Venus expressing cells. We also confirmed cell death by microscopic analysis, after addition of the complex (25 μM) and subsequent incubation of the transfected cells for 12 h. The cells were responsive to both annexin V and EthD-III, demonstrating late stage of apoptosis in the longer incubation (Fig. 3d). Similarly, we observed by FACS that 68% of the same cells were stained with EthD-III (data not shown). These results, taken together, show that L-HIPPO-α-CDE induced apoptosis of cells expressing HIV-1 protein. Finally, we examined whether this effect of L-HIPPO-α-CDE is observed in T cells by experiments using Jurkat cell line. As results, L-HIPPO-α-CDE (100 μM) did not show toxicity (Supplementary Information E), and in the presence of this complex, larger amount of cells (67%) were shown to induce apoptosis than those without L-HIPPO (58%) in a cell population expressing Gag-Venus (Fig. 4).

Apoptotic effect of L-HIPPO-α-CDE with HIV-1 protein. (a) Apoptotic effect. HeLa cells were transfected with pNL4-3/GagVenus, and after 10 h, complex prepared from L-HIPPO (25 or 50 μM) and α-CDE (25 μM) (L-HIPPO-α-CDE) was added. After a further 12 h incubation, FACS analysis using annexin V-Cy5 was performed. (b) Apoptotic effect after a shorter incubation time. The same experiment as that in (a) was performed using 4 h incubation instead of 12 h incubation. (c) Necrotic effect after a shorter incubation time. The same experiment as that in (b) was performed using EthD-III instead of annexin V-Cy5. (d) Apoptotic effect observed by microscopy. The same experiment as (a) using L-HIPPO (25 μM) and α-CDE (25 μM) was performed by microscopic observation using annexin V-Cy5 and EthD-III instead of FACS analysis.

Apoptotic effect of L-HIPPO-α-CDE with HIV-1 protein in T cells. Jurkat cells were transfected with pNL4-3/GagVenus, and after 1 d, a complex prepared from L-HIPPO (100 μM) and α-CDE (25 μM) (L-HIPPO-α-CDE) was added. After a further 12 h incubation, FACS analysis using annexin V-Cy5 was performed.
To sum up, we have developed a novel analogue of phosphoinositide L-HIPPO that binds to HIV-1 protein Pr55Gag with strong affinity, and L-HIPPO-α-CDE complex suppresses membrane localization of Pr55Gag and subsequent virus release and induces apoptosis of the host cell. Thus L-HIPPO-α-CDE encloses the penetrated virus components, locks them in the host cell, and destroy virus by means of apopmechantosis of the host cell. L-HIPPO is “non-natural” because of (i) L-type stereochemistry and (ii) IP6 structure with lipophilic group(s). Thus this “non-natural” compound would not conflict the intracellular function of natural inositol phospholipid such as PIP2. Actually, L-HIPPO did not show toxicity on cells in this study (Supplementary Information C). As impact of apoptosis on surrounding cells, caused by direct contact, release of soluble factors such as cytokines, and so on, has been widely studied and revealed well22, 23, its regulation would be possible. While bystander effect was observed in the apoptosis, this method would be improved so as to induce apoptosis without affecting other normal cells.
The “kick and kill” strategy begins with the use of LRA to activate (“kick”) the viral replication cycle4, and satisfiable “kill” device has not been available yet4, 5. In this study we developed an alternative method to terminate the fate of infected cells that is not “kill” but “suicide (apoptosis)”. For the future development of orally available L-HIPPO, a prodrug approach may be conceivable. The “lock-in and apoptosis” approach presented in this work suppresses the virus release and induces apoptosis to become bases of the eradication of HIV. Furthermore, this “lock-in and apoptosis” would pave the way for new anti-virus strategy against various viruses.
Methods
Plasmids, cells and transfection
A plasmid vector pNL4-3/GagVenus12 was used to express HIV-1 Gag conjugated with Venus yellow fluorescent protein at its C-terminus using pNL4-313 as a full-length HIV-1 infectious clone. The human cervical cancer cell line HeLa and human leukemic T cell line Jurkat were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% heat-inactivated fetal bovine serum (FBS) and RPMI-1640 medium supplemented with 10% heat-inactivated FBS, respectively. Transfection of the HeLa cells with plasmids was performed using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA), unless otherwise stated. Transfection of Jurkat cells with plasmids was performed by Amaxa Cell Line Nucleofector Kit V (Lonza, Basel, Switzerland).
Synthesis and characterization of new compounds
See Supplementary Information.
Preparation and addition of complexes of phosphoinositide analogues with carriers to cells
TMR Labeled Shuttle PIP Carrier 2 (Histone H1) (Histone-TMR) was purchased from Echelon Biosciences (Salt Lake City, UT, USA). Dendrimer-TRITC was prepared according to a previous method18. A conjugate of α-cyclodextrin and polyamidoamine dendrimer (G3) (α-CDE) was synthesized from monotosyl-α-cyclodextrin and dendrimer (G3) as previously described17, 19. α-CDE-TRITC was prepared from α-CDE according to a previous method20. Each carrier and DL-HIPPO, D-HIPPO, or L-HIPPO were mixed to form complexes, and added to HeLa cells. The cells were incubated at 37 °C for 15 min, washed with PBS (×1), and fresh medium was added.
Fluorescence microscopy
Microscopic observations were performed using a Zeiss LSM 700 laser-scanning confocal microscopy (Carl Zeiss, Oberkochen, Germany), as previously described24.
Immunoblot analysis
Supernatant and cell lysate, prepared by the PBS-Laemmli buffer method, were analyzed by immunoblotting as previously described25. HIV-1 p24 Gag monoclonal (#24-4) (NIH AIDS Research and References Reagent Program)26, 27 (1:1000) and anti-β-actin clone AC-15 (Sigma-Aldrich, St Louis, MO, USA) were used as a detection antibody. Immunoreactivity was detected by chemiluminescence using ImmunoStar LD (Wako Pure Chemical Industries, Osaka, Japan). Intensity of the bands were quantitated using ImageJ software.
SPR studies
Dissociation constants (Kd) were determined by competition assay using a BIACORE 2000 (GE Healthcare, Uppsala, Sweden). In this analysis, biotinylated D-myo-inositol-1,3,4,5-tetrakisphosphate28 was immobilized to a streptavidin conjugated sensor chip. A solution of MA protein, prepared from pEF-Gag (p17) cFLAG10 vector-transfected 293 T cells, was applied to the chip using flow buffer [10 mM HEPES, 150 mM NaCl, 3.4 mM EDTA, 0.005% Tween 20, 2% (v/v) glycerol, 0.5 mg mL−1 BSA, and 5% DMSO (pH 7.8)]. The details of this protocol have been described previously11.
MTT assay
The MTT assay was performed as described previously29.
FACS analysis
The cells were detached from the plate by 0.05% trypsin, washed with PBS, and then incubated with annexin V-Cy5 (BioVision, Milpitas, CA, USA) or ethidium homodimer III (Takara Bio, Kusatsu, Japan) for 10 min. After washing the cells with annexin-binding buffer (BioVision), the cells were fixed with 2% paraformaldehyde, washed with PBS again, and analyzed using a BD FACSCalibur (Becton Dickinson, NJ, USA).
References
Marsden, M. D. & Zack, J. A. HIV/AIDA eradication. Bioorganic & medicinal chemistry letters 23, 4003–4010 (2013).
UNAIDS | 2016. AIDS by the numbers - AIDS is not over, but it can be. a pdf file http://www.unaids.org/sites/default/files/media_asset/AIDS-by-the-numbers-2016_en.pdf (2016).
Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F. & Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology 14, 55–60 (2016).
Cillo, A. R. & Mellors, J. W. Which therapeutic strategy will achieve a cure for HIV-1? Current opinion in virology 18, 14–19 (2016).
Kimata, J. T., Rice, A. P. & Wang, J. Challenges and strategies for the eradication of the HIV resernoir. Current Opinion in Imunology 42, 65–70 (2016).
Ono, A., Ablan, S. D., Lockett, S. J., Nagashima, K. & Freed, E. O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America 101, 14889–14894 (2004).
Saad, J. S. et al. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proceedings of the National Academy of Sciences of the United States of America 103, 11364–11369 (2006).
Charlier, L. et al. Coarse-grained simulations of the HIV-1 matrix protein anchoring: revisiting its assembly on membrane domains. Biophysical Journal 106, 577–585 (2014).
Mercredi, P. Y. et al. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. Journal of Molecular Biology 428, 1637–1655 (2016).
Anraku, K. et al. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 49, 5109–5116 (2010).
Tateishi, H. et al. Design and synthesis of lipid-coupled inositol 1,2,3,4,5,6-hexakisphosphate derivatives exhibiting high-affinity binding for the HIV-1 MA domain. Organic & biomolecular chemistry 12, 5006–5022 (2014).
Chukkapalli, V., Hogue, I. B., Boyko, V., Hu, W. S. & Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. Journal of virology 82, 2405–2417 (2008).
Adachi, A. et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of virology 59, 284–291 (1986).
Ozaki, S., DeWald, D. B., Shope, J. C., Chen, J. & Prestwich, G. D. Intracellular delivery of phosphoinositides and inositol phosphates using polyamine carriers. Proceeding of the National Academy of Sciences of the United States of America 97, 11286–11291 (2000).
Arima, H., Motoyama, K. & Higashi, T. Sugar-appended polyamidoamine dendrimer conjugates with cyclodextrins as cell-specific non-viral vectors. Advanced drug delivery reviews 65, 1204–1214 (2013).
Arima, H., Hayashi, Y., Higashi, T. & Motoyama, K. Recent advances in cyclodextrin delivery techniques. Expert opinion on drug delivery 12, 1425–1441 (2015).
Kihara, F., Arima, H., Tsutsumi, T., Hirayama, F. & Uekama, K. In vitro and in vivo gene transfer by an optimized alpha-cyclodextrin conjugate with polyamidoamine dendrimer. Bioconjugate chemistry 14, 342–350 (2003).
Arima, H. et al. Enhancement of gene transfer activity mediated by mannosylated dendrimer/alpha-cyclodextrin conjugate (generation 3, G3). Journal of controlled release: official journal of the Controlled Release Society 116, 64–74 (2006).
Arima, H., Kihara, F., Hirayama, F. & Uekama, K. Enhancement of gene expression by polyamidoamine dendrimer conjugates with alpha-, beta-, and gamma-cyclodextrins. Bioconjugate chemistry 12, 476–484 (2001).
Arima, H. et al. Inhibitory effect of siRNA complexes with polyamidoamine dendrimer/alpha-cyclodextrin conjugate (generation 3, G3) on endogenous gene expression. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences 44, 375–384 (2011).
Ono, A. & Freed, E. O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proceeding of the National Academy of Sciences of the United States of America 98, 13925–13930 (2001).
Duarte, S., Carle, G., Faneca, H., de Lima, M. C. P. & Pierrefite-Carle, V. Suicide gene therapy in cancer: Where do we stand now? Cancer Letters 324, 160–170 (2012).
Poon, I. K. H., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nature Reviews Immunology 14, 166–180 (2014).
Monde, K., Contreras-Galindo, R., Kaplan, M. H., Markovitz, D. M. & Ono, A. Human endogenous retrovirus K Gag coassembles with HIV-1 Gag and reduces the release efficiency and infectivity of HIV-1. Journal of virology 86, 11194–11208 (2012).
Fujita, M. et al. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes and infection/Institut Pasteur 6, 791–798 (2004).
Fouchier, R. A., Meyer, B. E., Simon, J. H., Fischer, U. & Malim, M. H. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. The EMBO journal 16, 4531–4539 (1997).
Simon, J. H. et al. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. Journal of virology 71, 5259–5267 (1997).
Anraku, K. et al. Design and synthesis of biotinylated inositol phosphates relevant to the biotin-avidin techniques. Organic & biomolecular chemistry 6, 1822–1830 (2008).
Karabacak, M. et al. Synthesis and Evaluation of New Pyrazoline Derivatives as Potential Anticancer Agents. Molecules (Basel, Switzerland) 20, 19066–19084 (2015).
Acknowledgements
HIV-1 p24 Gag Monoclonal (#24-4) was obtained from Dr. Michael H. Malim through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. The plasmid vector pNL4-3/GagVenus was kindly provided by Dr. Akira Ono, University of Michigan Medical School. This work was supported in part by a Grant-in-Aid for Scientific Research (B) from the Japanese Society for the Promotion of Science (23390028) (to M.O.).
Author information
Authors and Affiliations
Contributions
H.T. undertook the main experiments, i.e. the synthesis of compounds and biological experiments; K.M. performed specialty microscopic and FACS analysis; K.A. carried out SPR analysis; R.K. was responsible for the cellular studies; Y.H. synthesized carriers; T.H., K.M. and H.A. directed the study of the carriers and data elucidation; H.D., H.I.C., M.O. and M.F. directed the research and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tateishi, H., Monde, K., Anraku, K. et al. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci Rep 7, 8957 (2017). https://doi.org/10.1038/s41598-017-09129-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09129-w
This article is cited by
-
Evaluation of anti-glioma effects of benzothiazoles as efficient apoptosis inducers and DNA cleaving agents
Molecular and Cellular Biochemistry (2023)
-
Molecular mechanisms by which the HIV-1 latent reservoir is established and therapeutic strategies for its elimination
Archives of Virology (2023)
-
Selective cell death in HIV-1-infected cells by DDX3 inhibitors leads to depletion of the inducible reservoir
Nature Communications (2021)
-
Structural insight into host plasma membrane association and assembly of HIV-1 matrix protein
Scientific Reports (2021)
-
Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs
Cellular and Molecular Life Sciences (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
