
Abstract
Despite considerable research, the location of an aliovalent dopant into SnO2 nanoparticles is far to be clarified. The aim of the present study on trivalent lanthanide doped SnO2 is to differentiate between substitutional versus interstitial and surface versus bulk doping, delineate the bulk and surface defects induced by doping and establish an intrinsic dopant distribution. We evidence for the first time a complex distribution of intrinsic nature composed of substitutional isolated, substitutional associates with defects as well as surface centers. Such multi-modal distribution is revealed for Eu and Sm, while Pr, Tb and Dy appear to be distributed mostly on the SnO2 surface. Like the previously reported case of Eu, Sm displays a long-lived luminescence decaying in the hundreds of ms scale which is likely related to a selective interaction between the traps and the substitutional isolated center. Analyzing the time-gated luminescence, we conclude that the local lattice environment of the lattice Sn is not affected by the particle size, being remarkably similar in the ~2 and 20 nm particles. The photocatalytic measurements employed as a probe tool confirm the conclusions from the luminescence measurements concerning the nature of defects and the temperature induced migration of lanthanide dopants.
Similar content being viewed by others

Introduction
There has been considerable research over the past decades on the n-type wide band gap metal oxide semiconductor, tin oxide (SnO2) due to its broad spectrum of applications. It is commonly used in transparent conducting electrodes and chemical sensors1, 2 production of batteries in conjunction with carbon based materials3, photocatalysts either in pure state, doped with non-lanthanide4, lanthanide ions (Ln)5 or in combination with another oxide (for example SnO2/TiO2 6, or SnO2/ZnO7) as well as photocatalysts with a post-illumination photocatalytic “memory”8. SnO2 has the rutile-type tetragonal structure belonging to the P42/mnm space group (lattice parameters a = b = 4.738 Å and c = 3.187 Å) with a band energy-gap situated between 3.5 and 3.8 eV according to both experimental results and theoretical calculations9, 10. Band- gap engineering has been used as an effective way to tune the band structure and optoelectronic properties of this oxide11. For this purpose, SnO2 has been synthetized by exploiting numerous approaches such as precipitation12, photochemical growth at the air–water interface9, 10, thermal decomposition13, sol-gel14, surfactant-assisted solvothermal10, hydrothermal synthesis15, 16 and sono-chemical method17.
Doping of SnO2 nanomaterials with metal cations proved to be a successful tool for tailoring their electrical, optical, and microstructural properties. The luminescence of pure SnO2, observed in the UV and/or visible region (350–550 nm) is generally correlated with the presence of crystalline defects resulting from the various synthesis processes18, 19. The literature agrees towards the oxygen vacancies as the most probable candidates for the recombination centers in the emission processes of SnO2 18, 19. Of the various metal dopants of SnO2, the aliovalent Ce3+ 18, 19, Mn2+ 18, 19 Co2+ 20, Ni2+ 21 or Cr3+ 22 revealed significant information on the relationships between doping, defects related luminescence, surface effects, changes in morphology and particle size.
Selection of a luminescence approach that is sensitive more to the local structure is expected to expose better the dopant location and distribution in SnO2. The trivalent Eu Ln metal with its characteristic orange/red emission is known to be highly sensitive to its local environment23. This explains why the Eu represents the most frequently investigated luminescent dopant for SnO2 with first reports dating back four decades ago24, 25. The information extracted from Eu luminescence can reveal whether the dopants segregate on the surface of nanosized SnO2 or/and they enter the oxide lattice via interstitial or substitutional way. In case of substitutional doping, substitution of Sn(4+) by bulkier Eu(3+) leads to both strain and electric effects due to the mismatch of both ionic radii (0.076 nm compared to 0.095 nm, in six-fold coordination26) and valence. The oxygen vacancies arising from charge-compensation can, in principle, locate either in the close vicinity or distant to Eu dopants leading to Eu- defects associates or substitutional isolated center, respectively. There are several reports that describe the emission shape of Eu as being dominated by relatively strong, three magnetic dipole (MD) emission lines around 590 nm, corresponding to 5D0–7F1 transition, in good agreement with the selection rules for the inversion low symmetry (C2h) at Sn sites. It is also established that SnO2 host acts as an efficient antenna that absorbs the UV excitation and subsequently transfers the excitation energy to the substitutional isolated Eu, compensating thus for the intrinsic low absorptions of the parity forbidden f – f transitions27, 28. Recently, the co-existence of uniform and exponential distributions in trap depths along with the thermal activation mechanism were suggested to explain a unique afterglow decay behavior in Eu - SnO2 27. It is established that, as the particle size decreases, the increasing number of surface and interface atoms generates stress/strain and concomitant structural perturbations28. By use of luminescence spectroscopy, a strongly distorted Eu environment has been suggested to occur in SnO2 nanoparticles with dimension close to the exciton Bohr radius (around 2 nm) compared to greater sized nanoparticles29. In addition, our literature survey evidences that a complex Eu related emission with both broad and narrow spectral features accompanies the characteristic emission of substitutional isolated Eu centers. The shape of this emission varies strongly with Eu concentration, synthesis approaches, thermal treatment and excitation conditions used in luminescence measurements28,29,30,31,32,33,34. The origin of this emission is undecided in the literature, being tentatively related to either Eu – defect associates, impurity phases (such as Eu2O3 or Eu2Sn2O7) or some unknown phase. In all, the literature data evidence a complex emission of still unknown origin for the Eu-SnO2 system. Elucidation of the specific location of the aliovalent dopant into the lattice is crucial since this influence significantly the functional properties of doped SnO2 nanomaterials. As an example, for a different significant semiconductor, anatase TiO2, it has been established that only Ln that enter substitutional in the TiO2 lattice contribute to the change of electronic structure and light absorption efficiency of the host35. For this host, substitutional doping mechanism was advanced at least for Nd, Sm, Eu, Er lanthanide ions36, whilst for Eu and Sm several lattice sites were identified already by site selective luminescence spectroscopy36,37,38.
To date, besides Eu, only Er Ln has been demonstrated by luminescence studies to substitute for the Sn lattice sites in the inversion C2h sites as an isolated substitutional dopant39. Indeed, Chen et al.38 measured an ultranarrow long-lived emission around 1500 nm which corresponds to the 4I13/2–4I15/2 transition having a strong magnetic dipole component40. For Nd, substitutional doping was suggested by luminescence and near edge X-ray absorption fine structure (NEXAFS) measurements41; however, the observance of a relatively strong 4F3/2 emission of Nd in the range of 800 to 1400 nm which is predominantly of electric dipolar nature sustains more a substitutional doping in association with a nearby defect. In such case, the association of the lanthanide with a defect in the nearest neighbour position remove the inversion symmetry around the dopant and thus the electric dipole transitions become allowed and their intensity dominate the spectra. For Sm42, 43, Tb44 or Dy45, 46 doped SnO2, the reported emission shapes deviate from the ones expected for an inversion local symmetry47,48,49, and thus, the isolated substitutional doping route remain uncertain.
It can be thus concluded that a critical question to be answered when studying doped SnO2 nanoparticles is How do we know if SnO 2 nanoparticles have been successfully doped? In this context, our study on the trivalent Ln doped SnO2 is aiming to differentiate between substitutional versus interstitial and surface versus bulk doping, delineate the bulk and surface defects induced by doping and establish an intrinsic dopant distribution. The experimental confirmation of successful doping of semiconductor nanoparticles with luminescent lanthanide activators is usually done in literature by a combination of the ex situ luminescence, X-ray diffraction (XRD), Diffuse Reflectance in the Ultraviolet/Visible (DR-UV/Vis) Raman, and Transmission Electron (TEM) or Scanning Electron (SEM) microscopy methods and techniques. Since for SnO2, reports show that the distribution of various metal dopants changes dramatically with temperature, from ordered, lattice sites to disordered, likely surface regions50, we use the in situ XRD, Raman and luminescence to provide additional insight into the dynamic evolution of dopant environment during thermal treatment51. In addition to conventional bulk doping by rapid hydrothermal or sol-gel methods, the impregnation of Ln on pre-calcined SnO2 nanoparticles was tested to differentiate between the bulk and surface defects. Photocatalytic oxidation of phenol was used as a reaction tool to further differentiate between the natures of Ln induced defects.
Results and Discussion
Overview of the texture, morphology and structure properties
As synthetized pure SnO2 consists of well-defined 2–4 nm sized nanoparticles with a near-spherical shape (Figure S1) aggregated into rather uniform structure with wormhole mesoporosity, originating from the diffusion controlled hydrolysis-polycondensation mechanism52. With thermal treatment at 400 °C, more strongly intergrown structure of 5–6 nm particles are evidenced along with sharp decrease of the surface area and increase of the pore size which continues with the increase of temperature at 700 and 1000 °C (Table 1). Calcination at 700 °C led to a bimodal pore size distribution combining mesopores and macropores whilst further increase at 1000 °C induced formation of a compact nonporous oxide.
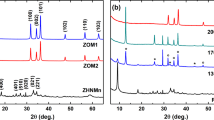
The purity of Ln (Eu, Sm) doped SnO2 samples was confirmed by X-ray fluorescence analysis (Figure S1). Doping SnO2 with 1% Ln did not cause any change in these textural properties and overall morphology. Generally, similar texture, morphology and structure properties were observed for Eu or Sm doped SnO2; therefore, we describe Eu- SnO2 in more detail. The ex situ XRD patterns of SnO2 and Eu-SnO2 calcined at 400, 700 and 1000 °C are presented in Fig. 1 (see also Figure S3 for the in situ with temperature XRD patterns of Eu-SnO2).

XRD patterns of SnO2 (a), Eu-SnO2 (b) and impregnated Eu-SnO2 (c, see text).
The diffraction angles at 26.3°, 33.8° and 51.8° from the diffractograms illustrated in Fig. 1, can be assigned to (110), (101) and (211) planes of the tetragonal SnO2 (PDF card 00-041-1445). In the limit of instrumental resolution, impurity phases, such as Eu2O3 or pyrochlore Eu2Sn2O7 were not detected. Broadening of XRD patterns with Eu doping together with the crystallite size and lattice parameters values listed in Table 2 demonstrate that Eu doping reduce the crystallite growth in large agreement with literature.
Similar effect was reported for other lanthanide ions and attributed to a solute drag and lattice distortion caused by the substitutional rather than surface doping53, 54. It is also worth noting that all crystallite sizes of the calcined samples exceed the quantum confinement region set by the exciton Bohr radius of SnO2 at 2.7 nm55.
Figure 2 shows the Raman spectra of SnO2 and Eu-SnO2 samples. The as - synthetized pure sample presents a broad and intense band at 570 cm−1 which is typically attributed to surface effects along with weak band around 760 cm−1 corresponding to B2g mode56. Samples calcined at 400, 700 and 1000 °C show in addition modes at 270–302, 474–510 (Eg), 626–641(A1g) and 680–700 and 768–782 cm−1 (B2g) in good agreement with literature57. Along with the increase of the temperature, a shift of the A1g and B2g lines towards higher wavenumbers and of the Eg line to lower wavenumbers is observed, associated to the increase of the particle size56. It should be remarked that doping with Eu is leading both to a stronger intensity of the broad bands around 500–570 cm−1 and decrease of the particle size as shown by the XRD patterns (Fig. 1, Table 2) which confirm the surface nature of this phonon mode.

Raman spectra of SnO2 (a), Eu-SnO2 (b) and impregnated Eu-SnO2 (c, see text).
DRIFTs spectra of SnO2 and Eu-SnO2 exhibit the typical profile for SnO2 58 (see Figure S4 and associated text). Pure samples show that the water content as well as hydroxyl absorption bands progressively disappear as the calcination temperature is increased whilst the presence of Eu appears to preserve more of the hydroxyl defects even at 1000 °C. The DR/UV-Vis diffuse reflectance spectra of SnO2 and Eu-SnO2 show a broad absorption band in the UV region (Figure S5). The shifts detected with the increase of the temperature may be attributed to the XRD observed changes in crystallinity and particle size (Fig. 1 and Table 2). The band-gaps (Eg) calculated by plotting (F(R)•hν)2 against hν18 (see also Experimental Section) are narrower than the value of 3.6 eV stated in literature9, 10 likely because of high concentration of oxygen vacancies/defects59. Further calcination at 1000 °C leads to the decrease of the band-gap up to 2.9 eV for pure SnO2 which narrowed to 2.7 eV for Eu-SnO2.
In situ short to long range investigations in the mild calcination regime
To get deeper insight into how the particle size, surface features, defects and order/disorder effects influence dynamically the local structure around the trivalent dopant, we further analyzed the Raman, luminescence and XRD data obtained by in situ measurements using Eu as a structural probe. In situ luminescence spectra were extracted from the in situ Raman spectra measured during heating and cooling cycles RT-500 °C-RT (RT stands for room-temperature, around 25 °C)60 while the in situ XRD patterns of Eu-SnO2 (Figure S3) were measured during both heating and cooling cycles (RT- 1000 °C- RT). As shown in Fig. 3, the position, width and intensity of the Raman modes of Eu- SnO2 change continuously with temperature during heating cycle. Up to 250 °C the Raman spectra of Eu-SnO2 are dominated by a broad band around 570 cm−1 which relates to the small particle size/surface effects56, 61 or to amorphous nature of the particles62. The average particle size is estimated to increase slightly from 2–4 nm before calcination to 6 nm at 500 °C (Figure S3).

Evolution with temperature of the in situ Raman and in situ luminescence of Eu-SnO2 during heating (Left) and cooling cycles (Right). Middle inset detail the 5D0–7F0 (normalized) emission. Deconvolution of the Raman spectra was done by use of Lorentzian functions. Excitation wavelength used in Raman measurements is 514 nm.
Besides this broad band (labelled as S1 in ref. 56) another broad phonon band is localized around 480 cm−1 (labelled as S2 in ref. 56). In the region of S2 band, the Eg mode around 470–480 cm−1 considered to be the most sensitive to oxygen vacancies, is expected to arise but its intensity is too low to be accounted for in the fitting57. Both S1 and S2 bands decrease in intensity with increase of particle size at the expense of A1g mode at 620–626 cm−1. The two Raman modes were both assigned to surface effects, more specifically to surface layer (around 1 nm as width) formation of a nonstoichiometric SnO2 with different symmetries than SnO2. However, the bands seem to differentiate in what concerns their evolution with temperature: while before calcination their relative intensities are rather similar, at 500 °C the S2 band exceed in intensity by a factor of four that of S1 band, equaling again after cooling down to RT. During cooling cycle, a gradual shift towards red, from 620 to 633 cm−1 is measured for A1g band parallels the slight increase of the particle size (Figure S3). As concern the in situ luminescence, this evidences broad shapes characteristic of surface Eu, dominated by strong emission around 612 nm which shifts and broadness continuously during heating and to less extent, during cooling cycles. To provide more insight into Eu distribution during the thermal treatment, the middle panel of Fig. 3 zoom the emission around 5D0–7F0 transition around 579 nm. This J = 0 − J = 0 transition is well known to be informative (in some conditions23) on the number of distinct Eu sites. Only single band assigned to average single Eu center could be detected with shape being progressively broadened and blue shifted from 578, 5 to 577, 4 nm during heating and then red shifted and slightly narrowed during cooling cycle. The luminescence evolution with temperature appears to be associated to either S1 or S2 or both phonon modes. The broadening of Eu emission during heating means an increased disorder around its nearest environment, likely induced by defects in the surface layer. Further series of experiments using pulsed laser excitation above 300 nm and long delay (time-gate mode) on the as-synthetized and calcined samples at 400 °C show a much weaker and narrower emission dominated by three 5D0–7F1 lines at 588 nm/593 nm/599 nm assigned to Eu substituting for Sn(4+) in C2h inversion sites24. In all, the in situ Raman and in situ luminescence results confirm that in the absence of calcination most Eu reside on the surface of ca 2 nm sized SnO2 nanoparticles with only few Sn inner lattice sites being substituted by Eu. Mild calcination up to 500 °C lead to a significant reduction of surface effects while the redistribution of dopants along with generation of defects induced by weak incorporation process remain limited to the surface layer of ~2–6 nm sized particles.
Substitutional doping, interaction with bulk defects and surface effects at high calcination temperatures
To date, literature generally describes a complex Eu related emission in SnO2 with both broad and narrow spectral features that changes strongly varies with Eu concentration, synthesis approaches, thermal treatment and excitation conditions used in luminescence measurements28,29,30,31,32, 34, 63. Here, we have extensively analyzed several tens of emission spectra of Eu-SnO2 calcined in air at 700 and 1000 °C at 80 and 300 K using various excitation wavelengths spanning the UV to Vis range and gate/time delays after the laser pulse (Figure S6). The specific aim of these investigations was to derive an intrinsic distribution of Eu described in terms of substitutional/interstitial doping, interaction with induced defects and surface effects.
By using a simple deconvolution procedure of the emission spectra64 backed also by the analysis of the excitation spectra and emission decays, at least five Sn centers, labelled as I to V were clearly differentiated (Fig. 5). The emission of center labelled as I is exclusively excited via SnO2 host as shown by the excitation spectrum measured around 588 nm (band at 290 nm). Its shape is characterized by relative strong three 5D0–7F1 lines around 590 nm being readily assigned to Eu substituting for the lattice Sn sites (with inversion C2h local symmetry) in line with previous reports24. In SnO2, the C2h inversion symmetry around Sn cation is shaped by the location of each of the two Sn in the unit cell of SnO2 amidst six oxygen atoms which approximately from the corners of a regular octahedron65 (Fig. 4). For Eu substituting for Sn with no defect nearby, the emission should be dominated by the allowed MD emission as the electric dipole (ED) transitions are forbidden in the inversion symmetry.

Crystal structure of SnO2 (a) Atomic arrangement in a rutile tetragonal unit cell with space group D4h 16 (P42/mnm). A unit cell contains two tin ions and four oxygen ions; (b) Perfect SnO6 tetrahedron with C2h local symmetry (with inversion center) around Sn.
In opposition to this center I, a much broader emission, excited exclusively by the f – f absorptions of Eu is observed and assigned to surface type Eu center, labelled as center V.
Besides the substitutional isolated C2h (I) and surface centers (V), at least three more Eu centers, labelled as II–IV (or non-C2h centers) could be differentiated for the first time (Fig. 5). The characteristic excitation spectra measured around their emissions around 606–612 nm, show a superposition of broad UV absorption bands around 300–320 nm along with narrow f-f absorptions of Eu. In the excitation spectra, the presence of O2−Eu3+ charge – transfer band (typically observed between 250–270 nm66) may be also masked on the high-energy side of broad UV absorption of SnO2 host. There is also a non - negligible interference from the 290 nm band characteristic of C2h center (I) due to incomplete spectral separation. The average lifetimes of non - C2h centers (1 ÷ 1.5 ms) are longer than that corresponding to surface center, V (0.7 ms), which is an expected result. The surface activators have unsaturated low symmetry coordinations being also subjected to efficient non-radiative energy transfer to surface defects, mainly OH groups still present at 700 and even 1000 °C (Figure S4b). Though the emission lifetime of C2h Eu is expected to be large due to its forbidden MD nature, it exceeds significantly the values of few ms measured for other hosts with inversion symmetry, such as Y2O3 (S6/C3i)47 or CeO2 (Oh)48.

Summary of the emission properties of Eu-SnO2 calcined at 700 °C/1000 °C and measured at 80 and 300 K. Characteristic excitation spectra (a), emission spectra (b) and emission decays (c) of Eu centers labelled as I–V centers (see text). Blue and orange spectra in (b) relate to cubic Eu2O3 and Eu2Sn2O7 pyrochlore, respectively. The guidelines in (b) evidence the distinctness of the emission spectra corresponding to II–IV centers.
Indeed, as shown in Fig. 5, the emission decay of C2h center displays an exceptional long decay time of 130 ms as estimated from the tail fitting at 80 K. Such a remarkable long-lived emission has recently been assimilated to an atypical persistent emission process that is thermally activated below (or above) 220 K via the co-existence of uniform and exponential distributions in trap depths27.
To check on the possibility that part of II–IV emissions relates to impurity phases such as Eu2O3 or Eu stannate oxide (Eu2Sn2O7), likely induced by the reportedly low solubility limit of Ln in tin oxide lattice, (below 1%), we compared the luminescence of calcined (700/1000 °C) Eu2O3 and Eu-SnO2 measured in identical conditions. There is obviously no resemblance between this emission (included as cyan and orange spectra in Fig. 5) and those of the II–IV centers. Next, the absence in the Raman spectra of the strongest characteristic phonon mode of stannate pyrochlore (around 510 cm−1 67) along with the strongly dissimilar emission exhibited by Eu2Sn2O7 68,69,70 grown by us using a sol-gel method71 (also included in Fig. 5 as orange spectrum) to Eu centers II–IV dismiss the occurrence of this phase in our samples.
Further, we asked whether these Eu II–IV centers were of intrinsic nature or resulted from the procedure of preparation or interaction with specific impurities. Comparison with an additional Eu-SnO2 sample grown by a simple sol-gel method give identical emission shapes to those obtained for samples grown by rapid hydrothermal approach. Besides, according to our extensive literature survey on the luminescence of Eu-SnO2 published over the last two decades, part of the emission related to centers II–IV can be observed as strongly non-separated, overlapped emission, though discernable in Eu-SnO2 grown by sol-gel30, 31, 34, 72 glass-ceramic waveguides32, hydrolysis73, impregnation and decomposition cycle method63, etc. Taken together, the emission shapes and narrowness (few cm−1, comparable to that of C2h center) suggest that centers II–IV are intrinsic to SnO2, resulting from the association of substitutional Eu with nearby bulk defects, most probably oxygen vacancy. Such defects arise from the charge-imbalance on substitution of tetravalent Sn by the trivalent Eu which in the Kroger-Vink74 notation is written as:
In a further set of experiments, we have surface doped SnO2 with Eu by impregnation of pre - calcined pure SnO2 at 400 and 1000 °C followed by calcination at 400 and 1000 °C. The XRD patterns, Raman spectra and DR-UV/Vis as well as particle size, lattice constants and estimated band-gap for these additional samples are summarized in Figs 1 and 2, Figures S5 and S6 and Table 2. Despite weak incorporation expected for the pre-calcination-impregnation-calcination procedures, the impregnated samples do show some emission of the substitutional isolated C2h center (I) (Figure not shown) embedded into a much broader emission like that of the surface center V. However, the notable result is that no emission can be associated to any of the defect centers (II–IV) giving thus a further confirmation that these centers are induced by Eu substituting for lattice Sn in close association with bulk defects.
Although similar emission properties were measured for Eu-SnO2 calcined at 700 and 1000 °C, there are some differences in the details. At higher calcination temperature, both weaker emission and appearance of a distinct distorted emission (Figure S7) suggest a migration of the substitutional Eu towards the nanoparticles surface where they may further associate with adsorbed OH defects75. Indeed, the DRIFTS spectra in Figure S4a,b confirm that the surface of the particles is terminated by adsorbed OH (bands centered at 3450 and 1640 cm−1) that persist even at 1000 °C and their contribution is exceeding that measured for pure SnO2.
The distribution found above for Eu is confirmed by a similar analysis applied to Sm. Compared to Eu, Sm was less frequently investigated as luminescent dopant for SnO2 48, 66, 67. Luminescence studies revealed an effective sensitization of Sm luminescence via host absorption67 similar to Eu case and also some dependency of the emission shape on the excitation wavelength. Figure 6 summarizes the analysis of luminescence properties of Sm–SnO2 calcined at 700/1000 °C that gives similar results for the employed methods (rapid hydrothermal and sol-gel). Under above bandgap excitation (280 nm) the emission of Sm (labelled as center I) displays narrow (~0.7 nm) and relative strong 4G5/2–6H5/2 emission around 568 nm which is mostly of MD nature 76. A weak intensity is observed for the 4G5/2–6H7/2 transition at 606 and 621 nm which is of mixed magnetic and electric dipole nature76 while no emission could be detected around 670 nm corresponding to the 4G5/2–6H7/2 transition which is mostly of electric dipolar nature (ED). In all, the emission shape is consistent with the substitution of Sn by Sm in the inversion C2h symmetry sites with no association with a nearby defect. Our results provide a first evidence of Sm emission assignable to the substitutional isolated C2h center (equivalent to center I of Eu). The emission shape is quite similar to that of Sm doped into other hosts with inversion symmetry sites, such as Y2O3 (S6/C3i)76 and CeO2 (Oh)48 recently reported by some of us. In addition, similar to Eu center I 27 (Fig. 5), the Sm center I displays also a remarkable long lived emission decaying on hundreds of ms scale with an estimated decay time from the tail fitting of 80 ms. It appears thus that that the long-lived emission is not Eu specific, given that it belongs to Ln substituting for Sn in the C2h sites (substitutional isolated center). It was previously suggested27 that the persistent luminescence of Eu in SnO2 may be associated with the co-existence of uniform and exponential distributions in trap depths. It should be noted that most of the persistent luminescence displayed by the lanthanide based phosphors is associated with ED and not to MD transitions27, 66.

Summary of the emission properties of Sm-SnO2 calcined at 700 °C/1000 °C and measured at 80 and 300 K. Characteristic excitation spectra (a), emission spectra (b) and decays (c) of Sm centers labelled as I–IV centers (see text). The guidelines in (b) evidence the distinctness of the emission spectra corresponding to II–IV centers.
In addition to the emission assignable to center I, we could separate by use of a similar procedure used for Eu- SnO2, at least three additional centers characterized by narrow (~0.7 nm) and shorter-lived emission (average lifetime around 1 ms) and labelled as centers II–IV in Fig. 6. The separation of the excitation spectra and decays of centers II–IV of Sm was however more difficult than in the case of Eu, therefore we have included only one excitation spectrum in Fig. 6. The average lifetimes of Sm centers II–IV is roughly estimated between 0.5 and 1ms. A broad and strongly distorted emission was observed and readily assigned to Sm surface center (corresponding center V of Eu). In addition to centers I and V, corresponding to isolated substitutional and surface centers of Eu and Sm, respectively, we can tentatively suggest a common nature also for centers IV of Eu and Sm. Both centers IV of Eu and Sm display a relative strong intensity of MD transition (5D0–7F1 for Eu and 4G5/2–6H5/2 for Sm) suggesting that the coordination around Eu and Sm centers is slightly deviated from the inversion C2h symmetry. Taken together, the luminescence properties of Eu and Sm in SnO2 are consistent with a common distribution, irrespective of the synthesis method, that includes substitutional isolated, associates with defects and surface centers.
We have also investigated the luminescence of Pr, Tb and Dy-SnO2 samples in the as-synthetized state and after calcination at 400 and 700 °C. Selected XRD patterns and DR-UV/Vis spectra as well as luminescence spectra of Pr, Tb and Dy-SnO2 are gathered in Figures S8 and S9, respectively. To the best of our knowledge, there is no literature report on Pr emission in SnO2, while for Tb44 or Dy45, 46 doped SnO2 the emissions appear to be related to surface location rather than substitutional doping. As a general observation, we note that the intensities of the Pr, Tb and Dy related emissions were poor, precluding advanced investigation. The emissions relate possibly to 1D2 level of Pr and definitely to 5D4 or4F9/2 levels of Tb or Dy. To decide whether the observed emission may relate to isolated substitutional doping, we included, in Figure S9 the emission spectra of Tb and Dy in the inversion sites of Y2O3 (S6/C3i) and CeO2 (Oh) reported recently by some of us47, 48. It can be observed that the emission shapes of Tb and Dy depart strongly from those expected for an inversion symmetry. Besides, the emissions are broad (~13 nm for Dy and ~9 nm for Tb) and short-lived (up to few hundreds of microsec). We therefore concluded that, that in contrast to Eu and Sm, Pr, Tb and Dy in SnO2 cannot substitute for Sn in the inversion lattice sites, preferring instead highly distorted environments, likely connected with the SnO2 surface.
Particle size effect on the local structure
It is well established that, as the particle size decreases, the increasing number of surface and interface atoms generates stress/strain and concomitant structural perturbations28. The comparison of luminescence properties of small (mean particle size comparable to the bulk exciton Bohr radius) and large sized Eu-SnO2 nanoparticles led to the conclusion that the Eu environment is strongly distorted in the small SnO2 while it is crystalline in the larger nanoparticles29. We re- asses here the particle size effect on the local structure selecting Eu-SnO2 particle sizes around 2 nm (as-synthetized) and 20 nm (calcined at 1000 °C). Figure 7 compares the steady-state emission spectra of the two Eu- SnO2 nanoparticles (excited above bandgap around 300 nm) that evidence a strongly distorted emission for the smaller sized nanoparticles (magenta spectrum). This may be used as a spectroscopic evidence that the local environment gets more distorted with decreasing particle size. However, using the time gated (delayed) mode, a narrow, long-lived type emission with orders of magnitude less intensity than the broad emission could be extracted with the 2 nm sized particles. Except for the slightly different widths, the delayed emission spectrum measured with the 2 nm particle is quite similar to that measured with the 20 nm particle. Both emissions correspond to the substitutional isolated Eu center (center I) with similar values of emission peaks (588.24, 593.04 and 599.08 nm) and intensity ratio (1/0.76/0.59) suggesting similar atomic scale environments of the inner lattice Sn, irrespective of the particle size. The reported distortions likely arises from the generic “surface effect” which is responsible for a broad and short-lived luminescence48 observed with wide range of doped nanoparticles, irrespective of host and luminescent activator.

Schematic representation of the similar local structures around Eu in SnO2 in the 2 and 20 nm sized SnO2 suggested by time-gated luminescence approach (δt notation means delay after the laser pulse).
We also highlight here that the advantage of using time-gated luminescence as a local technique over long-range XRD and medium range Raman techniques in detecting weak crystallinity features is obvious. In the Figs 1b and 2b only broad, non-crystalline patterns and surface phonon modes are observed for the as -synthetized Eu- SnO2 making difficult to assess the substitutional doping effect.
Surface versus bulk doping in the photocatalytic behavior
Recent literature shows that the Ln dopants increase the photocatalytic activity of SnO2 in the photo-oxidation of phenol under UV or solar simulated light5. Our photocatalytic measurements used here as an additional probe shows that pure, doped and impregnated Eu-SnO2 exhibit catalytic activity in the oxidation of phenol (Fig. 8a–c, Figure S11) irrespective of the calcination temperature and UV versus Vis irradiation range (see also Figure S1 for the spectral excitation profiles). The conversion decreased with the increase of the temperature in opposition to reports on non-structured catalysts15 being, however in line with textural analysis (surface and pore blockage by carbonaceous products and collapse of the structure by calcination (Table 1) and DR-UV-Vis measurements (Figure S5). Decrease of the conversion efficiency with calcination temperature (reflected in the decrease of the surface area, increase of the crystallite sizes (Table 1) and slight increase in the band gap (Table 2)) are due to modification of defects population in both pure and Eu doped/impregnated SnO2. The advantage of the materials structuration is demonstrated by the higher catalytic conversions of doped Eu-SnO2 (Figure S11b) and the smaller photocatalytic conversions on the impregnated Eu-SnO2 (Figure S11c) compared to pure SnO2. Although Eu presence does not affect strongly the band gap (Table 2), its dispersion in the bulk is generating a higher photoactivity compared to pure and impregnated samples. The luminescence investigations evidenced a migration of the lattice Eu towards the surface with increase of calcination temperature from 700 to 1000 °C while the impregnated samples exhibit mainly the surface defects (surface center V, Fig. 5) that along with a slightly wider band gap is reflected in a lower activity compared with the doped samples. Although photocatalysis is a surface process, the photons can penetrate at a certain depth in the nanoparticle where the presence of bulk defects may be beneficial. The difference of only 0.2 eV in the band-gap is not enough to explain the increased conversion with doped Eu-SnO2 compared to pure SnO2. Moreover, the impregnated Eu-SnO2 exhibits a higher band-gap than pure SnO2 or doped Eu-SnO2. On the other side, the activity of the impregnated samples increases with the increase in the temperature (pre-calcination and subsequent calcination) (Figure S11c), that is in agreement with previous reports15. The activity under Vis irradiation (allowed by the spectral profile illustrated in Figure S10) exceeds by almost 300 and 50% those of doped and pure SnO2 for the same (pre)calcination temperature of 400 °C. At 1000 °C, the Vis activity is comparatively weaker than at 400 °C which arises likely for the compromise of larger band gap (3.2 compared to 2.6 eV, Table 2) and reduction of bulk defects over surface ones (as Eu incorporation into the lattice is hindered by the higher pre-calcination temperature).

Conversion of phenol under 3 hours of UV and Vis irradiation using (a) SnO2, (b) Eu-SnO2 and (c) Eu-SnO2 (impregnated) and selectivity to Hydroquinone (HQ), 1,4 Benzoquinone (BQ) and Catechol (Cat) under UV irradiation using (d) SnO2, (e) Eu-SnO2 (conventional doping) and (f) Eu- SnO2 (impregnated) function of calcination temperature.
Figure 8d–f shows the evolution of the selectivity towards hydroquinone (HQ), catechol (Cat) and 1, 4-benzoquinone (BQ) intermediates77, 78 Under UV irradiation, the reaction with pure SnO2 advanced producing both BQ and Cat (Fig. 8d) with no Cat being produced after one hour of irradiation. In concordance to the above comments concerning the decrease in the population of the defects and changes in the band gap, the sample calcined at 400 °C afforded HQ and BQ in the first hour of irradiation, Cat being produced only after two hours, the sample calcined at 700 °C, after one hour afforded mainly HQ, and calcination at 1000 °C led to HQ as major product (100% selectivity after one hour), Cat not being produced. In all the cases, the plots suggest a consecutive/parallel oxidation process (Fig. 9). Doping SnO2 with Eu (Fig. 8e) changed the selectivity predominantly in the favor of HQ (see samples calcined at 700 and 1000 °C, respectively) following the same mechanism and preserving the effect of the calcination. However, the parallel reaction is slower in this case, with no Cat being produced. For the impregnated Eu-SnO2, a small amount of Cat is also detected (Fig. 8f) which is lower for the sample treated at 1000 °C than 400 °C following the same trend as pure SnO2.

Reaction pathways for the photo-oxidation of phenol with Eu-SnO2.
For both pure and Eu-SnO2 (doped or impregnated) samples, under Vis irradiation, the reaction stopped at hydroquinone (HQ) with complete selectivity (not shown). As the results showed, the presence of surface and bulk defects is beneficial for the photocatalytic performance of the samples. The neutral oxygen vacancies (Vo) in the structure of SnO2 that are produced by elimination of water during the Sn(OH)4 – SnO2 transition15. Although the insertion of Eu is supposed to enhance the population of the defects, except for the samples calcined at high temperatures, the band-gap remained basically unchanged. According to the equations included in Fig. 9, phenol can be oxidized by the O2 − species and OH radicals generated on the defected sites under the light irradiation. In all, the results described above suggest that the correlation of luminescence and photocatalytic properties, being both defects -related, can be used as an additional tool for improving the identification of bulk over surface defects in trivalent metal doped SnO2. Generally, the substitution of metal cations in oxide lattice by metal ions with different valence states disrupts the chemical bonds in the host oxide generating superficial active centers represented either by the oxygen atoms near the dopant or the dopant itself79. Low valence dopants such as the lanthanide ions, activate the near oxygen atoms, thus the doped host oxide is a better oxidant than the undoped one. The effect of higher valence dopants depends on the nature of the host oxide79. For irreducible oxides (e.g. Ta2O5), the host surface oxygen atoms are less active and the oxidation takes place merely by the surface adsorbed and activated O2. When the host oxide is reducible (e.g. TiO2, CeO2, SnO2), the dopant metal will donate electrons to the host cations (for the case of SnO2, reducing their valence as Sn4+ → Sn2+). For SnO2, in addition to the generation of active sites, the electronic control of energy levels in dopant clusters is involved in the activation of certain reactions by dopants80. On the other hand, the particle size of the doped metal oxide becomes smaller than the pure host oxide, as consequence of restriction of the motion of crystallites at the interaction on the boundaries between host and dopant crystallites81, as it can be seen in Table 2.
Conclusions
We have investigated pure and trivalent lanthanide doped SnO2 during and at various calcination temperatures up to 1000 °C, by use of a suite of in situ/ex situ techniques and methods. It is found that, before calcination most Eu resides on nanoparticle surface while mild calcination up to 500 °C lead to a significant reduction of surface effects. Besides, the redistribution of dopants along with induced defects remain limited to the surface layer of 2–6 nm sized particles. At higher calcination temperatures (≥700 °C) a complex distribution as substitutional isolated, associates with bulk defects and surface centers was evidenced for both Eu and Sm. The manifestation of a remarkable long-lived luminescence decaying on the hundreds of ms scale for both ions suggested that such phenomenon is not lanthanide specific, being tentatively associated to interaction between traps and the substitutional isolated Ln center. In contrast to Eu and Sm, Pr, Tb and Dy lanthanide dopants distributed mostly on the surface of SnO2 nanoparticles as judged by their characteristic emission. As both luminescence and photocatalytic properties are defects -related, their correlation proved effective in distinguishing the surface over bulk defects and confirming the temperature induced migration of dopants from the lattice sites towards the surface.
Materials and Methods
Synthesis
The synthesis of SnO2 nanoparticles was carried out by a rapid hydrothermal and sol-gel methods. In the first approach, a quick introduction of one gram of solid tin tetraisopropoxide isopropanol solvate (Multivalent Co UK) into 50 ml of boiling Milli-Q water with subsequent refluxing of the obtained milky dispersion for 30 min is used16. The mixture was then left for precipitation overnight, the supernatant was removed by decantation and the solid residue was dried at room temperature in air overnight. The Ln (Sm, Eu) doped SnO2 nanopowders were produced by the same procedure using instead of pure water a solution of stoichiometric amount of Ln(NO3)2 in 50 ml of water. Ln (1 wt%) was also deposed on pre-calcined SnO2 (at 400 and 1000 °C, respectively) via the wetness impregnation with an aqueous solution of Ln(NO3)3∙6H2O (Alfa Aesar) followed by drying overnight at room temperature and calcination at 400 and 1000 °C, respectively. In the sol-gel method, Ln(Eu, Sm, Pr, Tb and Dy, 1 wt%) doped SnO2 nanopowders were prepared by dissolving of 2 g stannous chloride dihydrate (SnCl2.2H2O) and the appropriate amount of dopant Ln(NO3)3∙6H2O in 100 ml distilled water. After complete dissolution, ammonia solution was added to the above solution by drop wise under stirring. The suspensions were left overnight for ageing, centrifuged and then dried at room temperature for 24 hours and 60 °C for 4 hours. The samples were calcined in air at the desired temperature (400, 700 and 1000 °C) with a rate of 10 °C/min.
Characterization
Microbeam X-ray fluorescence (micro-XRF) spectrometry was performed on a custom-made instrument with an X-ray tube: Oxford Instruments, Apogee 5011, Mo target, focus spot ∼40 µm, max. high voltage −50 kV, max current −1 mA, Amptek X-123 complete X-Ray spectrometer with Si-PIN detector. The key element of the micro-XRF instrument is an X-ray policapillary minilens (IfG-Institute for Scientific Instruments) which provides a focal spot size on the sample of 15–20 µm. X-Ray Diffraction (XRD) measurements were recorded on a Shimadzu XRD-7000 diffractometer using Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA) at a scanning speed of 2 degrees min−1 in the 10–90 degrees 2θ range. In-situ experiments were carried out in a cell accessory in the 50–1000 °C temperature range. The heating rate was 10 °C/min and the samples were kept for 10 min at the set temperature before collecting the diffractograms. The Raman spectra were acquired in the extended spectral region from 150 to 4000 cm−1. Raman analysis was carried out with a Horiba JobinYvon - Labram HR UV-Visible-NIR Raman Microscope Spectrometer, at 514 nm. In situ Raman spectra were recorded using the same apparatus equipped with an in-situ cell, until 600 °C. The heating rate was 10 °C/min and the sample was kept for 10 min at each temperature before collecting the spectra. Diffuse reflectance optical (DR-UV-Vis) spectra were recorded at room temperature on a Analytik Jena Specord 250 spectrophotometer with an integrating sphere for reflectance measurements and MgO as the reflectance standard. DR-UV-Vis spectra of the materials were recorded in reflectance units and were transformed in Kubelka–Munk remission function F(R). The estimation of the band-gap energy was made using the E = hc/λ (eV) formula, where h is the Planck constant (4.135 • 10−15 eVs), c is the speed of the light (3 • 108 ms−1), and λ the absorption threshold value for each sample determined from the spectra. The value of Eg (ie the band gap of the semiconductor) was derived from the spectra by plotting (F(R) • hν)2 against hν18. Diffuse Reflectance Fourier Transform Infrared (DRIFT) spectra were measured with a Thermo Electron Nicolet 4700 FTIR spectrometer with a Smart Accessory for diffuse reflectance measurements. The IR spectra were scanned in the region of 4000–400 cm−1 at the resolution of 4 cm−1. The final spectra are an accumulation of 400 scans. Transmission electron microscopy (TEM) investigations were made using a Tecnai G2 T20 microscope (FEITM, Holland) with an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) studies were carried out with Hitachi TM 1000 tabletop environmental microscope equipped with μ-DeX EDS detector system.
Luminescence measurements
The photoluminescence (PL) measurements were carried out using a Fluoromax 4 spectrofluorometer (Horiba) operated in both the fluorescence and the phosphorescence mode. The repetition rate of the xenon flash lamp was 25 Hz and 1 Hz for persistent luminescence decay, the integration window varied between 0.1 and 0.5 s, the delay after flash varied between 0.03 and 500 ms, and up to 30 flashes were accumulated per data point. For excitation spectra, the slits were varied from 5 to 29 nm while in emission the slits were varied from 1 nm to 3 nm. The PL decays were measured by using the “decay by delay” feature of the phosphorescence mode. The average decay time was calculated as integrated area of normalized decay. Time resolved emission spectra were recorded using a wavelength tunable (from 210 to 2300 nm) NT340 Series EKSPLA OPO (Optical Parametric Oscillator) operated at 10 Hz as excitation light source. The tunable wavelength laser has a narrow linewidth <4 cm−1 with scanning step varying from 0.05 to 0.1 nm. As detection system, an intensified CCD (iCCD) camera (Andor DH720) coupled to a spectrograph (Shamrock 303i, Andor) was used. The time-resolved emission spectra were collected in the spectral range of 450 nm < λem < 850 nm using the box car technique. The luminescence measurements were recorded at low-temperature (80 K) by use of FL-1013 Liquid Nitrogen (LN) dewar assembly (Horiba) and at 300 K.
Photocatalytic tests
Photocatalytic tests. In a typical phenol oxidation experiment, 15 mg of photocatalyst were added to a 5 mL aqueous solution of 50-ppm phenol. Two UV lamps (purchased from Vilber Lourmat) centered at 254 nm (VL-340.G 120 W) and 365 nm (VL-340.BL 120 W), respectively, were used as UV light sources for reactions under UV irradiation, and one 150 W Philips Master Colour CDM-T 150 W/830 visible lamp was used as visible light source for reactions under visible light irradiation. 30 μL of solution was collected at 60 min intervals for high performance liquid chromatography (HPCL, Agilent Technologies 1260 Infinity with DAD detector. Column Eurosphere C18, flow rate 1 mL/min, CAN: H2O = 40: 60, λ = 274.5 nm, Vinj = 10 µL).
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author (CT) on reasonable request.
References
Li, L. et al. Surface doping for photocatalytic purposes: relations between particle size, surface modifications, and photoactivity of SnO2:Zn2+ nanocrystals. Nanotechnology 20, 155706, doi:10.1088/0957-4484/20/15/155706 (2009).
Wang, L. L., Kang, L. P., Wang, H. Y., Chen, Z. P. & Li, X. J. Capacitive humidity sensitivity of SnO2:Sn thin film grown on silicon nanoporous pillar array. Sensors and Actuators B: Chemical 229, 513–519, doi:10.1016/j.snb.2016.02.025 (2016).
Wang, M. S. et al. Scalable preparation of porous micron-SnO2/C composites as high performance anode material for lithium ion battery. Journal of Power Sources 309, 238–244, doi:10.1016/j.jpowsour.2015.11.074 (2016).
Al-Hamdi, A. M., Sillanpää, M., Bora, T. & Dutta, J. Efficient photocatalytic degradation of phenol in aqueous solution by SnO2:Sb nanoparticles. Applied Surface Science 370, 229–236, doi:10.1016/j.apsusc.2016.02.123 (2016).
Al-Hamdi, A. M., Sillanpää, M. & Dutta, J. Photocatalytic degradation of phenol in aqueous solution by rare earth-doped SnO2 nanoparticles. Journal of Materials Science 49, 5151–5159, doi:10.1007/s10853-014-8223-2 (2014).
Hou, L. R., Yuan, C. Z. & Peng, Y. Synthesis and photocatalytic property of SnO2/TiO2 nanotubes composites. 139, 310–315, doi:10.1016/j.jhazmat.2006.06.035 (2007).
Zhang, M. et al. Novel preparation of nanosized ZnO–SnO2 with high photocatalytic activity by homogeneous co-precipitation method. 59, 3641–3644, doi:10.1016/j.matlet.2005.06.037 (2005).
Liu, L., Sun, W., Yang, W., Li, Q. & Shang, J. Post-illumination activity of SnO2 nanoparticle-decorated Cu2O nanocubes by H2O2 production in dark from photocatalytic “memory”. Scientific Reports 6, doi:10.1038/srep20878 (2016).
Wang, H. et al. Photochemical growth of nanoporous SnO2at the air–water interface and its high photocatalytic activityPhotochemical growth of nanoporous SnO2 at the air–water interface and its high photocatalytic activity. Journal of Materials ChemistryJournal of Materials Chemistry 20, 5641–5645, doi:10.1039/B926930D (2010).
Chu, D. et al. Enhanced Photocatalytic Properties of SnO2 Nanocrystals with Decreased Size for ppb‐level Acetaldehyde Decomposition. ChemCatChem 3, 371–377, doi:10.1002/cctc.201000334 (2011).
Mounkachi, O. et al. Band-gap engineering of SnO2. Solar Energy Materials and Solar Cells 148, 34–38, doi:10.1016/j.solmat.2015.09.062 (2016).
Esmaeili-Bafghi-Karimabad, A., Ghanbari, D., Salavati-Niasari, M., Nejati-Moghadam, L. & Gholamrezaei, S. Photo-catalyst tin dioxide: synthesis and characterization different morphologies of SnO2 nanostructures and nanocomposites. Journal of Materials Science: Materials in Electronics 26, 6970–6978, doi:10.1007/s10854-015-3316-5 (2015).
Ningthoujam, R. S. & Kulshreshtha, S. K. Nanocrystalline SnO2 from thermal decomposition of tin citrate crystal: Luminescence and Raman studies. Materials Research Bulletin 44, 57–62, doi:10.1016/j.materresbull.2008.04.004 (2009).
Zhang, G. & Liu, M. Preparation of nanostructured tin oxide using a sol-gel process based on tin tetrachloride and ethylene glycol. Journal of Materials Science 34, 3213–3219, doi:10.1023/A:1004685907751 (1999).
Wu, S., Cao, H., Yin, S., Liu, X. & Zhang, X. Amino acid-assisted hydrothermal synthesis and photocatalysis of SnO2 nanocrystals.Amino acid-assisted hydrothermal synthesis and photocatalysis of SnO2 nanocrystals. Journal of Physical Chemistry CJournal of Physical Chemistry C 113, 17893–17898, doi:10.1021/jp9068762 (2009).
Etacheri, V. et al. Ordered Network of Interconnected SnO2 Nanoparticles for Excellent Lithium‐Ion Storage. Advanced Energy Materials 5, 1401289, doi:10.1002/aenm.201401289 (2014).
Goswami, Y. C., Kumar, V. & Rajaram, P. Sonochemical growth of SnO2 hollow nanostructures on cellulose acetate flexible substrates. Materials Letters 128, 425–428, doi:10.1016/j.matlet.2014.04.174 (2014).
Gu, F. et al. Photoluminescence Properties of SnO2 Nanoparticles Synthesized by Sol−Gel Method. The Journal of Physical Chemistry B 108, 8119–8123, doi:S1089-5647(03)06741-5 (2004).
Zhang, C. & Lin, J. Defect-related luminescent materials: synthesis, emission properties and applications. Chemical Society Reviews 41, 7938–7961, doi:10.1039/C2CS35215J (2012).
Fang, L. et al. Microstructure and luminescence properties of Co-doped SnO(2) nanoparticles synthesized by hydrothermal method. Journal of Materials Science-Materials in Electronics 19, 868–874, doi:10.1007/s10854-007-9543-7 (2008).
Ahmed, A. et al. Band gap narrowing and fluorescence properties of nickel doped SnO2 nanoparticles. Journal of Luminescence 131, 1–6, doi:10.1016/j.jlumin.2010.07.017 (2011).
Garcia-Tecedor, M., Maestre, D., Cremades, A. & Piqueras, J. Influence of Cr Doping on the Morphology and Luminescence of SnO2 Nanostructures. Journal of Physical Chemistry C 120, 22028–22034, doi:10.1021/acs.jpcc.6b06682 (2016).
Tanner, P. A. Some misconceptions concerning the electronic spectra of tri-positive europium and cerium. Chemical Society Reviews 42, 5090–5101, doi:10.1039/C3CS60033E (2013).
Crabtree, D. F. The luminescence of SnO2-Eu3+. Journal of Physics D: Applied Physics 8, 107, doi:10.1088/0022-3727/8/1/019 (1975).
Matsuoka, T., Kasahara, Y., Tsuchiya, M., Nitta, T. & Hayakawa, S. The Preparation and Low Energy Electron (LEE) Excitation of SnO2: Eu Powder Phosphor. Journal of The Electrochemical Society 125, 102–106, doi:10.1149/1.2131371 (1978).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallographica Section A: Crystal Physics, Diffraction, Theoretical and General Crystallography 32, 751–767, doi:10.1107/S0567739476001551 (1976).
Kong, J. et al. Persistent luminescence from Eu3 + in SnO2 nanoparticles. Nanoscale 7, 11048–11054, doi:10.1039/C5NR01961C (2015).
Fernández‐García, M. & Rodriguez, J. A. Metal Oxide Nanoparticles. (John Wiley & Sons, Ltd, 2009).
Yanes, A. C. et al. Nanocrystal-size selective spectroscopy in SnO2:Eu3+ semiconductor quantum dots. Applied Physics Letters 85, 2343–2345 (2004). L03-5739.
Morais, E. A., Scalvi, L. V., Tabata, A., De Oliveira, J. B. & Ribeiro, S. J. Photoluminescence of Eu3+ ion in SnO2 obtained by sol–gel. Journal of Materials Science 43, 345–349, doi:10.1007/s10853-007-1610-1 (2008).
Brito, G. E. S. et al. Short Range Order Evolution in the Preparation of SnO2 Based Materials. Journal of Sol-Gel Science and Technology 8, 261–268, doi:10.1023/A:1026458212142 (1997).
Bhaktha, S. N. B. et al. Enhanced fluorescence from Eu3+ in low-loss silica glass-ceramic waveguides with high SnO2 content. Applied Physics Letters 93, 211904, doi:L08-09774R (2008).
Strauss, M., Destefani, T. A., Sigoli, F. A. & Mazali, I. O. Crystalline SnO2 Nanoparticles Size Probed by Eu3+ Luminescence. Crystal Growth & Design 11, 4511–4516, doi:10.1021/cg2007292 (2011).
Kiisk, V. et al. Structural and luminescence characteristics of SnO2:Eu and SnO2:Eu,Sb nanophosphors upon annealing at high temperatures. Materials Chemistry and Physics 130, 293–298, doi:10.1016/j.matchemphys.2011.06.042 (2011).
Li, W., Frenkel, A. I., Woicik, J. C., Ni, C. & Shah, S. I. Dopant location identification in Nd3+ -doped TiO2 nanoparticles. Physical Review B 72, 155315, doi:10.1103/PhysRevB.72.155315 (2005).
Chakraborty, A. et al. Identifying the Correct Host–Guest Combination To Sensitize Trivalent Lanthanide (Guest) Luminescence: Titanium Dioxide Nanoparticles as a Model Host System. The Journal of Physical Chemistry C 120, 23870–23882, doi:10.1021/acs.jpcc.6b08421 (2016).
Luo, W., Li, R., Liu, G., Antonio, M. R. & Chen, X. Evidence of Trivalent Europium Incorporated in Anatase TiO2 Nanocrystals with Multiple Sites. The Journal of Physical Chemistry C 112, 10370–10377, doi:10.1021/jp801563k (2008).
Yu, H. et al. Ultralong well-aligned TiO2:Ln3+ (Ln = Eu, Sm, or Er) fibres prepared by modified electrospinning and their temperature-dependent luminescence. Scientific Reports 7, 44099, doi:10.1038/srep44099 (2017).
Zhang, X. et al. Photoluminescence from Er3+ ion and SnO2 nanocrystal co-doped silica thin films. Chinese Optics Letters 10, 091603–091603, https://www.osapublishing.org/abstract.cfm?uri=col-10-9-091603 (2012).
Dodson, C. M. & Zia, R. Magnetic dipole and electric quadrupole transitions in the trivalent lanthanide series: Calculated emission rates and oscillator strengths. Physical Review B 86, 125102, doi:10.1103/PhysRevB.86.125102 (2012).
Bouras, K. et al. Insight into photon conversion of Nd3+ doped low temperature grown p and n type tin oxide thin films. Rsc Advances 6, 67157–67165, doi:10.1039/c6ra14460h (2016).
Faustino, B. M., Foot, P. J. S. & Kresinski, R. A. Synthesis and photoluminescent properties of Sm3+-doped SnO2 nanoparticles. Ceramics International 42, 18474–18478, doi:10.1016/j.ceramint.2016.08.183 (2016).
Yanes, A. C., Méndez-Ramos, J., del-Castillo, J., Velázquez, J. J. & Rodríguez, V. D. Size-dependent luminescence of Sm3+ doped SnO2 nano-particles dispersed in sol-gel silica glass. Applied Physics B 101, 849–854, doi:10.1007/s00340-010-4331-0 (2010).
Singh, L., Singh, N. & Srivastava, S. Terbium doped SnO2 nanoparticles as white emitters and SnO2:5Tb/Fe3O4 magnetic luminescent nanohybrids for hyperthermia application and biocompatibility with HeLa cancer cells. Dalton Transactions 44, 6457–6465, doi:10.1039/c4dt03000a (2015).
Pillai, S., Sikhwivhilu, L. & Hillie, T. Synthesis, characterization and photoluminescence properties of Dy3+ -doped nano-crystalline SnO2. Materials Chemistry and Physics 120, 619–624, doi:10.1016/j.matchemphys.2009.12.010 (2010).
Gu, F. et al. Preparation and luminescence characteristics of nanocrystalline SnO2 particles doped with Dy3+. Journal of Crystal Growth 255, 357–360, doi:10.1016/S0022-0248(03)01294-6 (2003).
Avram, D., Cojocaru, B., Florea, M. & Tiseanu, C. Advances in luminescence of lanthanide doped Y2O3: case of S6 sites. Optical Materials Express 6, 1635–1643, doi:10.1364/OME.6.001635 (2016).
Avram, D. et al. Toward a Unified Description of Luminescence–Local Structure Correlation in Ln Doped CeO2 Nanoparticles: Roles of Ln Ionic Radius, Ln Concentration, and Oxygen Vacancies. The Journal of Physical Chemistry C 119, 16303–16313, doi:10.1021/acs.jpcc.5b02240 (2015).
Lupei, A., Tiseanu, C., Gheorghe, C. and Voicu, F. Optical spectroscopy of Sm3+ in C2 and C3 isites of Y2O3 ceramics. Applied Physics B, doi:10.1007/s00340-012-5196-1 (2012).
Xu, C., Tamaki, J., Miura, N. & Yamazoe, N. Correlation between Gas Sensitivity and Crystallite Size in Porous SnO2-Based Sensors. Chemistry Letters 19, 441–444, doi:10.1246/cl.1990.441 (2006).
Tiseanu, C. et al. In situ Raman and Time-Resolved Luminescence Investigation of the Local Structure of ZrO2 in the Amorphous to Crystalline Phase Transition. The Journal of Physical Chemistry C 116, 16776–16783, doi:10.1021/jp3040538 (2012).
Seisenbaeva, G. A., Daniel, G., Kessler, V. G. & Nedelec, J. M. General Facile Approach to Transition‐Metal Oxides with Highly Uniform Mesoporosity and Their Application as Adsorbents for Heavy‐Metal‐Ion Sequestration. Chemistry - A European Journal 20, 10732–10736, doi:10.1002/chem.201402691 (2014).
Fu, C. et al. Effect of La doping on microstructure of SnO2 nanopowders prepared by co-precipitation method. Journal of Non-Crystalline Solids 357, 1172–1176, doi:10.1016/j.jnoncrysol.2010.10.019 (2011).
Bouras, K. et al. Optical and structural properties of Nd doped SnO2 powder fabricated by the sol–gel method. Journal of Materials Chemistry C 2, 8235–8243, doi:10.1039/C4TC01202J (2014).
Lee, E. J. et al. Photoluminescence in quantum-confined SnO2 nanocrystals: Evidence of free exciton decay. Applied Physics Letters 84, 1745–1747, doi:10.1063/1.1655693 (2004).
Dieguez, A., Romano-Rodrıguez, A., Vila, A. & Morante, J. R. The complete Raman spectrum of nanometric SnO2 particles. Journal of Applied Physics 90, 1550–1557, doi:10.1063/1.1385573 (2001).
Sun, S. H. et al. Raman scattering study of rutile SnO2 nanobelts synthesized by thermal evaporation of Sn powders. Chemical Physics Letters 376, 103–107, doi:10.1016/S0009-2614(03)00965-5 (2003).
Amalric-Popescu, D. & Bozon-Verduraz, F. Infrared studies on SnO2 and Pd/SnO2. Catalysis Today 70, 139–154, doi:10.1016/S0920-5861(01)00414-X (2001).
Li, N. et al. Effects of oxygen vacancies on the electrochemical performance of tin oxide. Journal of Materials Chemistry A 1, 1536–1539, doi:10.1039/C2TA01012G (2012).
Parvulescu, V. I. & Tiseanu, C. Local structure in CeO2 and CeO2–ZrO2 nanoparticles probed by Eu luminescence. Catalysis Today 253, 33–39, doi:10.1016/j.cattod.2015.02.013 (2015).
Lupan, O. et al. A rapid hydrothermal synthesis of rutile SnO2 nanowires. Materials Science and Engineering B-Advanced Functional Solid-State Materials 157, 101–104, doi:10.1016/j.mseb.2008.12.035 (2009).
Vijayarangamuthu, K. & Rath, S. Nanoparticle size, oxidation state, and sensing response of tin oxide nanopowders using Raman spectroscopy. Journal of Alloys and Compounds 610, 706–712, doi:10.1016/j.jallcom.2014.04.187 (2014).
Strauss, M., Destefani, T. A., Sigoli, F. A. & Mazali, I. O. Crystalline SnO2 Nanoparticles Size Probed by Eu3+ Luminescence. Crystal Growth & Design 11, 4511–4516, doi:10.1021/cg2007292 (2011).
Avram, D. et al. Lanthanide-lanthanide and lanthanide-defect interactions in co-doped ceria revealed by luminescence spectroscopy. Journal of Alloys and Compounds 616, 535–541, doi:10.1016/j.jallcom.2014.07.059 (2014).
Jarzebski, Z. M. & Marton, J. P. Physical Properties of SnO2 Materials. Journal of The Electrochemical Society 123, 199C–205C, doi:10.1149/1.2133010 (1976).
Zhang, H., Fu, X., Niu, S., Sun, G. & Xin, Q. Luminescence properties of Li+ doped nanosized SnO2:Eu. Journal of Luminescence 115, 7–12, doi:10.1016/j.jlumin.2005.02.008 (2005).
Zhang, T. T. et al. Synthesis and structural characterization of a series of lanthanide stannate pyrochlores. Journal of Physics and Chemistry of Solids 69, 2845–2851, doi:10.1016/j.jpcs.2008.07.014 (2008).
Fujihara, S. & Tokumo, K. Multiband Orange-Red Luminescence of Eu3+ Ions Based on the Pyrochlore-Structured Host Crystal. Chemistry of Materials 17, 5587–5593, doi:10.1021/cm0513785 (2005).
Blasse, G. & Van Keulen, J. Luminescence properties of Eu2Sn2O7. Chemical Physics Letters 124, 534–537, doi:10.1016/0009-2614(86)85071-0 (1986).
Pokhrel, M., Wahid, K. & Mao, Y. Systematic Studies on RE2Hf2O7:5%Eu3+ (RE = Y, La, Pr, Gd, Er, and Lu) Nanoparticles: Effects of the A-Site RE3+ Cation and Calcination on Structure and Photoluminescence. The Journal of Physical Chemistry C 120, 14828–14839, doi:10.1021/acs.jpcc.6b04798 (2016).
Mrázek, J., Surýnek, M., Bakardjieva, S., Buršík, J. & Kašík, I. Synthesis and crystallization mechanism of europium-titanate Eu2Ti2O7. Journal of Crystal Growth 391, 25–32, doi:10.1016/j.jcrysgro.2013.12.045 (2014).
Nogami, M., Enomoto, T. & Hayakawa, T. Enhanced fluorescence of Eu3+ induced by energy transfer from nanosized SnO2 crystals in glass. Journal of Luminescence 97, 147–152, doi:10.1016/S0022-2313(02)00217-X (2002).
Gonçalves, R. R., Messaddeq, Y., Aegerter, M. A. & Ribeiro, S. J. L. Rare Earth Doped SnO2 Nanoscaled Powders and Coatings: Enhanced Photoluminescence in Water and Waveguiding Properties. Journal of Nanoscience and Nanotechnology 11, 2433–2439, doi:10.1166/jnn.2011.3535 (2011).
Kröger, F. A. & Vink, H. J. Solid state physics. 307 (Academic, 1956).
Moon, T. et al. Hydroxyl-Quenching Effects on the Photoluminescence Properties of SnO2:Eu3+ Nanoparticles. The Journal of Physical Chemistry C 111, 4164–4167, doi:10.1021/jp067217l (2007).
Lupei, A., Tiseanu, C., Gheorghe, C. & Voicu, F. Optical spectroscopy of Sm3+ in C2 and C3i sites of Y2O3 ceramics. Applied Physics B 108, 909–918, doi:10.1007/s00340-012-5196-1 (2012).
Hudnall, P. M. Hydroquinone. Ullmann’s Encyclopedia of Industrial Chemistry, 10.1002/14356007.a13_499 (2017).
Cavallito, C. J., Soria, A. E. & Hoppe, J. O. Amino- and Ammonium-alkylaminobenzoquinones as Curarimimetic Agents. Journal of The American Chemical Society 72, 2661–2665, doi:10.1021/ja01162a088 (1950).
McFarland, E. & Metiu, H. Catalysis by Doped Oxides. Chemical Reviews 113, 4391–4427, doi:10.1021/cr300418s (2013).
Batzill, M. & Diebold, U. The surface and materials science of tin oxide. Progress in Surface Science 79, 47–154, doi:10.1016/j.progsurf.2005.09.002 (2005).
Sun, Y. et al. Metal Oxide Nanostructures and Their Gas Sensing Properties: A Review. Sensors 12, 2610–2631, doi:10.3390/s120302610 (2012).
Acknowledgements
DA and CT acknowledge the Romanian National Authority for Scientific Research and Innovation, the program NUCLEU, contract 4N/2016 and the Romanian National Authority for Scientific Research and Innovation (CNCS-UEFISCDI), project PN-III-P4-ID-PCE-2016-0305 for the partial financial support. GS and VK acknowledge Swedish Research Council (Vetenskapsrådet) for the grant 2014–3938.
Author information
Authors and Affiliations
Contributions
B.C. and D.A. performed structural, luminescence and photocatalytic experiments and processed the data, V.K. and G.S. synthesized the samples and performed SEM and TEM investigations, V.P. designed the photocatalytic study and CT designed the studies and wrote the manuscript. All authors contributed to the discussion of the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
41598_2017_9026_MOESM1_ESM.pdf
Supplementary Information Nanoscale insights into doping behavior, particle size and surface effects in trivalent metal doped SnO2
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cojocaru, B., Avram, D., Kessler, V. et al. Nanoscale insights into doping behavior, particle size and surface effects in trivalent metal doped SnO2 . Sci Rep 7, 9598 (2017). https://doi.org/10.1038/s41598-017-09026-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09026-2
This article is cited by
-
Structure-stability correlations in terms of microstructure during tin oxidation as examined by in situ high-temperature X-ray powder diffraction
Chemical Papers (2024)
-
Structure, Magnetic and Optical Characterization of Sn1−xLaxO2 Nanoparticles
Journal of Electronic Materials (2021)
-
Study of Copper Doped Zirconium Dioxide Nanoparticles Synthesized via Sol–Gel Technique for Photocatalytic Applications
Journal of Inorganic and Organometallic Polymers and Materials (2020)
-
Structural, Optical, and Magnetic Properties of Co-Doped SnO2 Nanoparticles
Journal of Electronic Materials (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
