
Abstract
With increasingly stringent environmental regulations, the removal of nitrogen-containing compounds (NCCs) from gasoline fuel has become a more and more important research subject. In this work, we have successfully synthesized TiO2/α-Fe2O3 heterogeneous photocatalysts with different mass ratios of TiO2 vs. α-Fe2O3. Taking photocatalytic denitrification of typical alkali NCCs, pyridine, in gasoline fuel under visible light irradiation (λ ≥ 420 nm) as the model reaction, the TiO2/α-Fe2O3 hybrids have exhibited enhanced photocatalytic activity compared with pure TiO2 and α-Fe2O3, giving a pyridine removal ratio of ∼100% after irradiation for 240 min. The improved photocatalytic performance can be attributed to the integrative effect of the enhanced light absorption intensity and more efficient separation of photogenerated electron-hole pairs. Importantly, this type of heterogeneous photocatalysts can be easily separate in the reaction medium by an external magnetic field that is very important for industrial purpose. In addition, major reaction intermediates have been identified by the liquid chromatograph-mass spectrometer (HPLC-MS) and a tentative photocatalytic denitrification mechanism has been proposed.
Similar content being viewed by others

Introduction
The nitrogen-containing compounds (NCCs) in gasoline fuel are one of the most alarming environmental concerns to date. Crude gasoline fuel naturally contains a high concentration of NCCs, such as pyridine, indoles, nitrides and their derivatives1. Once released into the atmospheric environment, their combustion products (e.g., NO2, NO and unburned hydrocarbon particles) will cause photochemical smog and resulting in serious hazardous effects on ecosystems and human health1, 2. The removal of nitrogen containing compounds from gasoline is currently achieved by catalytic hydrodenitrification, adsorptive denitrogenation, oxidative denitrogenation, and photocatalytic denitrogenation3,4,5,6,7,8. Among the above methods, photocatalytic is a promising technique since it achieves the one-pot removal of NCCs by utilizing sunlight7, 8.
Semiconductor titanium oxide (TiO2) has always been regarded as one of the most common photocatalyst for the treatment of NCCs, because of its physical and chemical stability, simple preparation, nontoxicity, low cost, and unique electronic and optical properties9, 10. Nevertheless, two main drawbacks should be tackled before TiO2 can meet the actual application in large-scale NCCs denitrogenation. First, due to its large band gap (3.2 eV), TiO2 does not absorb photons in the visible region of the electromagnetic spectrum, which significantly reduces its solar energy conversion efficiency. Even worse, as frequently reported, the low charge mobility in TiO2 contributed to higher recombination rate of photogenerated electrons and holes, thereby limiting the catalytic activity11, 12. Many attempts have been made to realize the actual applications of TiO2 photocatalysts, such as nanostructuring (nanofibers, hollow sphere)13,14,15 and coupling with other materials (CuS, graphene, noble metal)16,17,18,19,20. Actually, combining TiO2 with other semiconductors to construct heterostructures is considered as one of the best approaches to effectively improve its solar energy conversion and effectively accelerate the separation of photoexcited charge carriers21. Therefore, the exploration of efficient semiconductor-coupled TiO2 nanocomposites with highly visible-light photocatalytic denitrogenation performance has become an attractive area of investigation.
Based on its abundance, stability, nontoxic nature, and much smaller band gap (2.3 eV), iron oxide (α-Fe2O3) in particular is a promising candidate for the development of efficient solar photocatalysts22,23,24,25. However, α-Fe2O3 has one significant drawback: its photocatalytic performance is limited by the high recombination rate of the photogenerated charge carriers. The introduction of magnetic component α-Fe2O3 might not only offer some synergetic enhancement of the catalytic activity by forming the hybrid structure, but also prevent the agglomeration of the catalyst nanoparticles during recovery26. TiO2 could be one of the best surface catalysts due to its matched band position with α-Fe2O3. Thus, it is reasonable to believe that this heterostructure has the enormous potential to increase the separation and transfer efficiency of photongenerated charge carriers and meanwhile conquer the drawbacks of pure TiO2 and α-Fe2O3 27, 28. Although there have been some reports on TiO2 or α-Fe2O3 photocatalysts. It should be noted that they always focus on the elimination of NOx from the flue gas10, 29, 30. Especially, the utilization of TiO2/α-Fe2O3 composites in photocatalytic denitrification of NCCs from the original gasoline fuel has remained unavailable so far.
Herein, we report for the first time that the TiO2/α-Fe2O3 composites can be utilized as photoactive and durable photocatalysts toward the denitrification of one kind of typical NCCs, pyridine, in original gasoline fuel under ambient conditions. Alkali nitride, pyridine, has been chosen as the target of the research owing to its widely existing and greatly influence the quality of fuel oil. The results have demonstrated that, under the irradiation of visible light, the TiO2/α-Fe2O3 exhibit enhanced photocatalytic performance, compared to individual α-Fe2O3 at the same conditions. The origin accounting for the improved photoactivities and the underlying reaction mechanisms have been studied in terms of a series of characterization and trapping experiments. Moreover, the possible photocatalytic reaction mechanism has also been investigated in detail.
Results
Characterizations
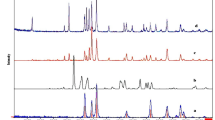
Figure 1 shows the XRD patterns of the as-prepared TiO2, Fe2O3 and TiO2/α-Fe2O3 composites (with 30, 50, and 70 wt.% of Fe2O3, designated as TiO2/Fe2O3-3, TiO2/Fe2O3-5, and TiO2/Fe2O3-7, respectively). It is obvious that the all the diffraction peaks of the TiO2 belong to pure anatase structure of TiO2 (JCPDS 21–1272). Meanwhile, six crystal peaks at 2θ = 24.1°, 33.2°, 35.6°, 40.8°, 49.5°, and 54.1°can be indexed as (012), (104), (110), (113), (024) and (116) reflection of Fe2O3 (α-Fe2O3, JCPDS 89–8103)28. For the TiO2/Fe2O3 composites with different mass ratios, all the reflection peaks could be indexed to hematite phases of Fe2O3 and anatase phases of TiO2, indicating the successfully combined anatase TiO2 with α-Fe2O3 in the composites. No peak from impurity phase was found, indicating the high purity of the as-prepared composites. It is easy to observe that the main characteristic diffraction peaks of the TiO2/Fe2O3 composites did not noticeably change after TiO2 hybridized with α-Fe2O3, suggesting that the calcination process could not destroy the crystal of TiO2.

XRD patterns of pure TiO2, pure α-Fe2O3 and TiO2/Fe2O3 composites.
The surface morphology and microstructure information of the as-synthesized samples have been characterized by SEM and TEM. As seen in Fig. 2(a), the as-prepared TiO2 exhibit the 1D morphology with an average length of ca. 5–7 μm and an average diameter of ca. 50–100 nm, which is consistent with the previous report31. After the deposition of α-Fe2O3 onto the TiO2 microrods, Fe2O3 particles are densely coated onto the surface of the TiO2 microrods to form a hetero-interface between TiO2 microrods and Fe2O3 particles (Fig. 2(b–f)). It can be also found that, the quantity of α-Fe2O3 coated on the surface of the TiO2 increases gradually along with the enhancement of the loading amount of α-Fe2O3. Simultaneously, it could be observed from the TEM images (Fig. 2(g and h)), α-Fe2O3 particles are tightly combined with TiO2 microrods via a simple calcination process. This is further confirmed from a representative HRTEM image of the TiO2/Fe2O3-5 composite. From the Fig. 2(i), we could clearly observe two different crystal lattices corresponding to anatase-TiO2 (d101 = 0.35 nm) and α-Fe2O3 (d110 = 0.25 nm). The EDS elemental mapping also confirms that Fe, Ti and O elements are uniformly distributed in the TiO2/Fe2O3-5 (Fig. S1). All these results gave solid evidence that TiO2 microrods and α-Fe2O3 were successfully coupled together to form TiO2/Fe2O3 photocatalysts.

SEM images of (a) TiO2, (b) α-Fe2O3, (c) TiO2/Fe2O3-3; (d,e) TiO2/Fe2O3-5 (f) TiO2/Fe2O3-7, and (g,h) TEM images of as-prepared TiO2/Fe2O3-5; (i) HRTEM images of the TiO2/Fe2O3-5.
X-ray photoelectron spectroscopy (XPS) is performed to study the structural and chemical state of the elements present in TiO2/Fe2O3 nanocomposites. As shown in Fig. 3(a), the survey spectrum of the TiO2/Fe2O3-5 shows the pronounced featured signal of O 1 s, Fe 2p and Ti 2p, indicating that the Fe2O3 particles are successfully immobilized on the surface of the TiO2. The weak signal of Ti is maybe due to the covering effect of Fe2O3 in the TiO2/Fe2O3-5. Figure 3(b) shows the high-resolution XPS spectrum of the O1s. Beside the component of O1s lattice centered at 529.4 eV, two bands at 530.8 eV and 532.1 eV are detected and attributed to the presence of coordinatively unsaturated oxygen species (surface defects)32, which belong to the lattice oxygen combined with Fe3+ and Ti4+. Figure 3(c) shows the high-resolution XPS spectra of Fe 2p. The binding energies of 710.3 and 724.2 eV with a satellite signal at 718.9 eV are characteristic of Fe(III) in Fe2O3, which is due to spin-orbit splitting. The peak separation, namely, Δ = 2p1/2–2p3/2 = 13.9 eV, which is very similar to those reported for α-Fe2O3 33. Figure 3(d) shows peaks at 458.5 and 464.1 eV and are assigned to Ti 2p3/2 and Ti 2p1/2 core levels. The difference between the Ti 2p core levels is 5.6 eV, indicating the normal state of Ti4+ in the anatase TiO2 34, 35. The shoulder of the Ti 2p3/2 peak corresponds to a band at 458.5 eV. This band is assigned to formation of a Ti-O-Fe bond in the interface of TiO2/Fe2O3, which indicated the formation of TiO2/Fe2O3. Similar observation was also reported for TiO2/Fe2O3 coatings by Zhang and Lei36. The amount of Fe2O3 deposited on TiO2/Fe2O3 has been determined by ICP. It is found that the loading percentage of Fe2O3 in samples of TiO2/Fe2O3-3, TiO2/Fe2O3-5 and TiO2/Fe2O3-7 are 27.1%, 45.6%, 66.0%, respectively. It is also demonstrated that the calcination approach is an effective technique of immobilizing Fe2O3 onto the TiO2, because the content of Fe2O3 only slightly less than the theoretical content. For further information of the surface acidity of TiO2/Fe2O3 composites, the temperature-programmed desorption (TPD) of ammonia has been carried out. As shown in Fig. S2, the peaks at 560 °C can be assigned to desorption of ammonia from Lewis acid sites, which is due to the presence of unsaturated surface Ti4+ ions37. Moreover, The appearance of peak at 275 °C in NH3-TPD curve of TiO2/Fe2O3-5 can be associated with ammonia desorption from surface Fe3+, which play role of medium Lewis acid centers38. The NH3-TPD measurements results have confirmed the strong surface acidity of TiO2/Fe2O3 composites. Thus, the Lewis acid surface of TiO2/Fe2O3 is benefit to absorbing Lewis base, pyridine to obtain a better photocatalytic activity.

XPS patterns of TiO2/Fe2O3-5.
The optical properties of the as-synthesized pure TiO2, α-Fe2O3 and TiO2/Fe2O3 composites have been characterized by UV-vis diffuse reflectance spectroscopy (DRS) and the results are shown in Fig. 4. TiO2 shows a typical absorption band edge at 390 nm, which originated from its band gap of 3.18 eV and in accordance with the reported value in the literatures39. As for the α-Fe2O3, it exhibits strong light absorption over the visible range, even extending to the infrared region, which is caused by 2(6A1) → (4T1) ligand field transition of Fe3+. With the integration of Fe2O3, the optical absorption of the composites in the visible light region is greatly improved, which is in accordance with the color change of the samples from white to reddish brown. Therefore, the enhanced absorbance of light is expected to improve the visible-light-driven photocatalytic activity for a target reaction. This inference is well verified by the photocatalytic testing of TiO2/Fe2O3 composites toward denitrogenation of NCCs under visible light irradiation.

UV-vis absorption spectra of TiO2, pure α-Fe2O3 and TiO2/Fe2O3 composites.
Photocatalytic properties
The photocatalytic activities of TiO2/α-Fe2O3 composites have been evaluated by the photocatalytic denitrogenation of pyridine under visible light irradiation (λ ≥ 420 nm). Blank experiments have been first carried out to demonstrate the photocatalytic nature of the reaction (Fig. S3). Apparently, the denitrogenation of pyridine hardly occurs in the absence of photocatalyst or light. Instead, the denitrogenation of pyridine proceeds smoothly in the presence of photocatalyst. Importantly, TiO2/Fe2O3-5 composites exhibit much higher photocatalytic activity than that of TiO2 (~0%) and α-Fe2O3 (44%) under identical experimental conditions, respectively. The reduction ratio is rapidly increased to ~100% after visible light irradiation (λ ≥ 420 nm) for 240 min. Moreover, such photoactivity is higher than that of TiO2 + α-Fe2O3, which is prepared by simply mixing TiO2 and α-Fe2O3 in proper proportions under identical conditions (Fig. 5(a)). Fig. 5(b) is the photocatalytic reaction kinetics of pyridine photocatalytic denitrogenation in octane based on the data plotted in Fig. 5(a). As can be observed, photocatalytic denitrogenation of pyridine approximately follows pseudo-first-order kinetics, as evidences by the linear plot of ln(C0/Ct) vs. reaction time t. As displayed in Fig. 5(b), the TiO2/Fe2O3-5 composite has the highest rate constant (0.0097 min−1) among all of the samples. The kinetic rate constants follow the order TiO2/Fe2O3-5 (0.0097 min−1) > TiO2/Fe2O3-7 (0.0050 min−1) > TiO2/Fe2O3-3 (0.0047 min−1) > α-Fe2O3 (0.0024 min−1) > TiO2 (0.0001 min−1). Therefore, we can draw the conclusion that coating TiO2 microrods with an ultrathin α-Fe2O3 layer could lead to the obvious photoactivity enhancement toward denitrogenation reactions. The results indicate that the effective interfacial hybridization between TiO2 and α-Fe2O3 contributes to the remarkably enhanced photoactivity, thus making TiO2/Fe2O3 composites be an efficient photocatalyst for denitrogenation of NCCs.

(a) Photocatalytic denitrogenation of pyridine over TiO2, α-Fe2O3 and TiO2/Fe2O3 composites with different mass ratios of Fe2O3; (b) The pseudo-first order rate constants of pyridine photocatalytic denitrogenation over TiO2, α-Fe2O3 and TiO2/Fe2O3 with different mass ratios of Fe2O3. Reaction conditions: 40 mg of photocatalyst, 40 mL of 100 μg/g pyridine.
Besides the excellent photo-denitrogenation efficiency, the stability and recyclability of photocatalysts is another significant factor in their practical application. To confirm the photostability of the as-prepared photocatalysts, the recycling tests for pyridine denitrogenation have been conducted with sample TiO2/Fe2O3-5. After each cycling experiment, the photocatalyst was separated from the aqueous suspension by filtration and washed with ethanol several times. And then, the photocatalyst was centrifuged at 4500 rpm for 5 min and dried in vacuum at 100 °C for 2 h. As shown in Fig. S4(a), the results of recycling tests indicate no significant loss of photocatalytic activities after four cycles, suggesting that the TiO2/Fe2O3-5 photocatalyst is stable during the photocatalytic reaction. XRD results reveal that no significant changes are observed in the crystal structure of TiO2/Fe2O3-5 before and after the catalytic reaction (Fig. S4(b)). Additionally, the separability of the TiO2/Fe2O3-5 magnetic composites has also been tested by placing a magnet near the glass bottle after dispersing the TiO2/Fe2O3-5 in octane (Fig. S4(c)). It is observed that the magnetic particles are attracted towards the magnet within 1 min. The magnetic properties of the resultant TiO2/Fe2O3 have been investigated at room temperature by vibrating sample magnetometry (VSM) in the field range from - 4 to + 4 KOe (Fig. S5). The samples at room temperature have been measured to be 8.3 emu/g−1, revealing strong magnetic properties. The curve presents a magnetic hysteresis loop, which also depicts the strong magnetic response to a varying magnetic field. The above results directly demonstrate the convenient separation of the TiO2/Fe2O3-5 from liquids using an external magnetic field.
Discussion
In order to investigate the reasons for obvious photoactivity enhancement toward denitrogenation of pyridine over the photocatalysts, the surface area measurement and photoelectrochemical experiments have been performed. The Brunauer-Emmett-Teller (BET) surface areas of the as-prepared composites have been investigated using nitrogen adsorption-desorption experiments. As shown in Fig. S6, the isotherm for three samples exhibited a type IV with a H3 hysteresis loop according to the IUPAC classification. The BET surface areas of the pristine TiO2, and Fe2O3 have been calculated to be 19.5 and 33.8 m2/g, respectively. To clearly see the variations of original-TiO2 after α-Fe2O3 decoration, we summarize the BET surface areas of the samples in Table 1. The results show that after coupling with α-Fe2O3, the surface area of the composites revealed a slight increase compared with TiO2. However, for all of these samples, there are no line-relationship between the surface areas and the observed photoactivity order, that is, TiO2/Fe2O3-5 > TiO2/Fe2O3-7 > TiO2/Fe2O3-3 > α-Fe2O3 > TiO2, cannot be attributed to the difference of surface area.
It is well established that TiO2 and Fe2O3 are provided with matchable energy band position, together with intimate interfacial contact confirmed by our TEM results, which is able to result in the efficient charge carriers transfer. This reference is verified by the electrochemical impedance spectra (EIS). It can be obviously seen from Nyquist impedance plots (Fig. 6(a)) that the TiO2/Fe2O3-5 shows depressed semicircles at high frequencies as compared to blank TiO2 and α-Fe2O3, suggesting that the charge-transfer resistance decreases. Therefore, a consensus is reached that the integration of TiO2 with α-Fe2O3 could improve the transfer of charge carriers, thereby efficiently hampering the recombination of electron-hole pairs40. The improved charge carrier separation and the prolonged lifetime of photogenerated electron-hole pairs can be confirmed by the photo-electrochemical experiments. As displayed in Fig. 6(b), the introduction of α-Fe2O3 enhances the photocurrent significantly, indicating a more efficient separation of the photoexcited electron-hole pairs.

(a) Transient photocurrent response of TiO2, α-Fe2O3 and TiO2/Fe2O3 composites in 0.2 M Na2SO4 aqueous solution under irradiation of visible light (λ ≥ 420 nm); (b) Nyquist impedance plots of TiO2, α-Fe2O3 and TiO2/Fe2O3-5.
A sequence of controlled experiments using different radical scavengers has been carried out in order to deeply understand the role of photogenerated radical species in the photocatalytic denitrogenation of pyridine over the photocatalysts. The results of adding different radical scavengers (ethanol for holes, CCl4 for electrons and isopropyl alcohol for OH·) over TiO2/α-Fe2O3 systems are shown in Fig. S7. When the holes scavenger, ethanol is added into the reaction system, the removal ratio of pyridine remarkably decrease, manifesting that the denitrogenation of pyridine is mainly related to photoinduced holes. Thus, the adsorbed pyridine in solution interacts with holes to form the corresponding radical cations, which further reacts with the trace H2O molecular and dioxygen, leading to the formation of CO2, H2O, NH3 and HCOOH (Fig. S8). For further information of the denitrogenation pathway of pyridine, the liquid chromatograph-mass spectrometer (HPLC-MS) has been carried out. As displayed in Fig. S9, the peak intensity m/z 81.5 is reduced after the photocatalytic fuel denitrification, meaning that the pyridine has been degraded successfully. Two new peaks appeared at m/z 85.0 and m/z 46.1 after the photocatalytic fuel denitrification, meaning that the pyridine has been translated into the protonated of intermediate products C4H4O2 and CH3NH2.
Based on the discussion presented above and the experimental results, a synergistic photocatalytic mechanism of the TiO2/Fe2O3 catalyst was proposed, as illustrated in Fig. 7. It is clearly shown that the photogenerated electrons of α-Fe2O3 will be excited from valence band (VB) to its different energy-level conduction band (CB) position under the excitation of visible light, including high-energy region (−0.05 eV ~ −1.0 eV vs. SHE) and low-energy region (0.1 eV ~ −0.05 eV vs. SHE) in this system. Thus, the photogenerated electrons at low-energy level would quickly relax to the VB bottom of α-Fe2O3, then to recombine with holes. Meanwhile, partial high-energy electrons would thermodynamically transfer to the CB of TiO2 due to their matchable energy band position and intimate interfacial contact, thus resulting in the improved fate of photogenerated electron-hole pairs. Taking pyridine denitrogenation as the model reaction, the TiO2/α-Fe2O3 photocatalysts have exhibited enhanced photocatalytic activity compared with pure TiO2 and α-Fe2O3. The higher photoactivity of TiO2/Fe2O3 can be attributed to the enhanced visible light absorption, efficient charge-carrier separation as well as the synergistic effect between TiO2 and α-Fe2O3. To the best of our knowledge, this work represents the first example to use the TiO2/α-Fe2O3 semiconductor composite photocatalyst for photocatalytic denitrification of NCCs. It is expected that our work could offer new inroads into explore heterojunction photocatalysts for photocatalytic denitrification of gasoline fuel.

Possible mechanism of photocatalytic denitrogenation of pyridine over TiO2/Fe2O3-5.
Methods
Reagents and chemicals
All reagents and solvents were used as received from commercial suppliers without further purification. Tetrabutoxytitanium was supplied by Aladdin Reagent Co., Ltd. (Shanghai, China). Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), ethylene glycol, pyridine, octane were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
Synthesis of TiO2 sample
TiO2 were prepared using the method previously reported by Peng et.28. 3 mL of tetrabutoxytitanium was added to 30 mL ethylene glycol in a rockered flask. The solution was treated at 180 °C for 2 h under continuous magnetic stirring, the white slurry was formed. Then the solution was cooled to room temperature naturally. The final white solid products were centrifuged and washed with ethanol several times to ensure total removal of the excess ethanol and the dried at room temperature. The assynthesized white solid products were titanium glycolate. Finally, the titanium glycolate precursor was calcined at 450 °C for 2 h to form TiO2.
Synthesis of TiO2/Fe2O3 samples
The TiO2/Fe2O3 composites were synthesized by wet impregnation, drying, ethanol washing, and calcination process. Typically, 30 mL of 0.3 M (0.6 M or 0.9 M) Fe(NO3)3·9H2O in ethanol (EtOH) was added to 1.5 g of TiO2 powder, stirring for 30 min at room temperature and then sonicating for 30 min. After that, the suspension was evaporated at 50 °C to obtain solid sample. Then the sample was calcined at 300 °C for 10 min. Next, the sample was washed by ethanol thoroughly. Finally, the sample was once again calcined at 300 °C for 6 h. The loading of α-Fe2O3 in the composites was about 30, 50, and 70 wt% for 0.3, 0.6, and 0.9 M Fe(NO3)3·9H2O, designated as sample TiO2/Fe2O3-3, TiO2/Fe2O3-5, and TiO2/Fe2O3-7, respectively.
Characterization of materials
XRD patterns were carried on a Bruker D8 Advance X-ray diffrractometer operated at 40 kV and 40 mA with Ni-filtered Cu Kα irradiation (λ = 0.15406 nm). The data were recorded in the 2θ range of 10–80°. The Brunauer-Emmett-Teller (BET) surface area was measured with an ASAP2020M apparatus (Micromeritics Instrument Corp., USA). Before the test, the samples were degassed in vacuum at 240 °C for 6 hours. The nitrogen adsorption and desorption isotherms were measured at 77 K. UV-vis diffuse reflectance spectra (UV-vis DRS) were obtained by a UV-vis spectrophotometer (Shimadzu UV-2700) with BaSO4 as a reflectance standard. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a PHI Quantum 2000 XPS system equipped with a monochromatic Al Kα X-ray source to obtain the surface elemental composition of the sample. The concentration of Fe2O3 in the sample was detected by the Ultima2 ICP optical emission spectrometer. The magnetization curves were measured at room temperature under a varying magnetic field from −4 to + 4 kOe on a BHV-55 vibrating sample magnetometer (VSM). Temperature-programmed desorption (TPD) of ammonia was conducted in a flow apparatus on a Micrometrics 2910 Autochem analyzer. In a typical NH3-TPD experiment, about 0.2 g of the sample was loaded in U-shaped quartz cell above a small amount of quartz wool. Before the experiments, the samples were pretreated for 2 h at 400 °C in a flow of helium. NH3-TPD was carried out in helium flow after purging the sample at 50 °C during 60 min to decrease the amount of physisorbed ammonia. The temperature was increased with a rate of 10 °C/min up to 700 °C. The electrochemical measurements were performed in a conventional three electrode cell, Ag/AgCl electrode was used as the reference electrode and a Pt plate was used as the counter electrode. The photocurrent measurements were conducted with a BAS Epsilon workstation. The liquid chromatograph-mass spectrometer (HPLC-MS) methods for analyzing pyridine was performed using an Agilent 1200 series (Palo Alto, CA, USA) equipped with an Agilent Zorbax Eclipse XDB-C18 column (2.1 mm × 100 mm, 3.5 m). The column was maintained at 30 °C during the sample analysis. The measurement for pyridine was performed in an isocratic elution program with methanol/acetone = 70:30 (v/v) as mobile phase. Flow rate was kept at 0.2 mL/min, and the injection volume was10 μL.
Evaluation of photocatalytic activity
Simulated NCCs-containing gasoline fuel of 100 μg/g was prepared by dissolving 70 mg of pyridine in 1.0 L of octane. The octane interaction with hydroxylated surfaces of TiO2/Fe2O3 has been shown to be very weak and nonspecific, while the interaction of polar compounds such as pyridine is expected to be stronger. Furthermore, considering that octane is the main ingredient of gasoline, which is low-cost and low toxicity. Therefore, we have chosen octane as the optimum reaction solvent in our reaction system. The photocatalytic denitrification of pyridine was carried out at 30 °C in a 100 mL quartz reactor containing 40 mg of TiO2/Fe2O3 and 40 mL of pyridine/octane solution (100 μg/g). The suspension was stirred in the dark for 1 h to ensure the establishment of adsorption-desorption equilibrium, the suspensions were irradiated by a 300 W Xe lamp (PLS-SXE 300, Beijing Perfectlight Co. Ltd) with a UV-CUT filter to cut off light of wavelength < 420 nm. During illumination, 2 mL of suspension was taken from the reactor at a scheduled interval and centrifuged to separate the photocatalyst. The pyridine content in the supernatant solution was determined colorimetrically at 251 nm using a Cary 50 UV-vis spectrophotometer (Varian Co.). In order to quantitatively understand the reaction kinetics of the pyridine photocatalytic denitrification in our experiments, we applied the pseudo-first order model as expressed by Eq. (1), which is generally used for photocatalytic degradation process if the initial concentration of pollutant is low:
where C0 and Ct are the concentrations of the pollutants in solution at time 0 and t, respectively, and k is the pseudo-first order rate constant.
References
Nguyen, T. B., Laskin, J., Laskin, A. & Nizkorodov, S. A. Nitrogen-containing organic compounds and oligomers in secondary organic aerosol formed by photooxidation of isoprene. Environ. Sci. Technol. 45, 6908–6918 (2011).
Lobert, J. M., Scharffe, D. H., Hao, W. M. & Crutzen, P. J. Importance of biomass burning in the atmospheric budgets of nitrogen-containing gases. Nature 346, 552–554 (1990).
Yao, H. et al. Deep hydrodenitrification of pyridine by solid catalyst coupling with ionic liquids under mild conditions. Green Chem. 19, 1692–1700 (2017).
Ahmed, I., Khan, N. A. & Jhung, S. H. Adsorptive denitrogenation of model fuel by functionalized UiO-66 with acidic and basic moieties. Chem. Eng. J. 321, 40–47 (2017).
Zhang, H., Li, G., Jia, Y. & Liu, H. Adsorptive removal of nitrogen-containing compounds from fuel. J. Chem. Eng. Data 55, 173–177 (2010).
Qu, D. et al. Adsorption of heterocyclic sulfur and nitrogen compounds in liquid hydrocarbons on activated carbons modified by oxidation: capacity, selectivity and mechanism. RSC Adv. 6, 41982–41990 (2016).
Zheng, L. et al. Visible-light photocatalytic denitrogenation of nitrogen containing compound in petroleum by metastable Bi20TiO32. Int. J. Hydrogen Energy 39, 13401–13407 (2014).
Teders, M., Gómez-Suárez, A., Pitzer, L., Hopkinson, M. N. & Glorius, F. Diverse Visible-light-promoted functionalizations of benzotriazoles inspired by mechanism-based luminescence screening. Angew. Chem. Int. Edit. 56, 902–906 (2017).
Wang, W. et al. Single-step one-pot synthesis of TiO2 nanosheets doped with sulfur on reduced graphene oxide with enhanced photocatalytic activity. Sci. Rep. 7, 46610–46619 (2017).
Su, C., Ran, X., Hu, J. & Shao, C. Photocatalytic process of simultaneous desulfurization and denitrification of flue gas by TiO2-polyacrylonitrile nanofibers. Environ. Sci. Technol. 47, 11562–11568 (2013).
Luttrell, T. et al. Why is anatase a better photocatalyst than rutile? - Model studies on epitaxial TiO2 films. Sci. Rep. 4, 4043–4051 (2014).
Tang, X., Huang, L., Zhang, W., Jiang, R. & Zhong, H. Photocatalytic activities of plant hormones on semiconductor nanoparticles by laser-activated electron tunneling and emitting. Sci. Rep. 5, 8893–8902 (2015).
Qin, N. et al. Highly efficient photocatalytic H2 evolution over MoS2/CdS-TiO2 nanofibers prepared by an electrospinning mediated photodeposition method. Applied Catalysis B 202, 374–380 (2017).
Cai, J. et al. Influence of TiO2 hollow sphere size on its photo-reduction activity for toxic Cr(VI) removal. J. Colloid Interf. Sci. 490, 37–45 (2017).
Hou, H. et al. Efficient photocatalytic activities of TiO2 hollow fibers with mixed phases and mesoporous walls. Sci. Rep. 5, 15228–15237 (2015).
Lu, Y. Y. et al. In situ loading of CuS nanoflowers on rutile TiO2 surface and their improved photocatalytic performance. Appl. Surf. Sci. 370, 312–319 (2016).
Kumar, S. G. & Rao, K. S. R. K. Comparison of modification strategies towards enhanced charge carrier separation and photocatalytic degradation activity of metal oxide semiconductors (TiO2, WO3 and ZnO). Appl. Surf. Sci. 391, 124–148 (2017).
Liu, H., Liu, S., Zhang, Z., Dong, X. & Liu, T. Hydrothermal etching fabrication of TiO2@graphene hollow structures: mutually independent exposed {001} and {101} facets nanocrystals and its synergistic photocaltalytic effects. Sci. Rep. 6, 33839–33851 (2016).
Demirci, S. et al. Synthesis and characterization of Ag doped TiO2 heterojunction films and their photocatalytic performances. Appl. Surf. Sci. 390, 591–601 (2016).
Gomathi Devi, L. & Kavitha, R. A review on plasmonic metal/TiO2 composite for generation, trapping, storing and dynamic vectorial transfer of photogenerated electrons across the Schottky junction in a photocatalytic system. Appl. Surf. Sci. 360, 601–622 (2016).
Low, J., Yu, J., Jaroniec, M., Wageh, S. & Al-Ghamdi, A. A. Heterojunction photocatalysts. Adv. Mater. 29, 1601694–1601714 (2017).
Zhu, T., Li Ong, W., Zhu, L. & Wei Ho, G. TiO2 Fibers Supported β-FeOOH Nanostructures as Efficient Visible Light Photocatalyst and Room Temperature Sensor. Sci. Rep. 5, 10601–10616 (2015).
Zhu, M. & Wachs, I. E. Iron-based catalysts for the high-temperature water-gas shift (HT-WGS) reaction: a review. ACS Catal. 6, 722–732 (2016).
Mishra, M. & Chun, D.-M. α-Fe2O3 as a photocatalytic material: A review. Appl. Catal. A: Gen. 498, 126–141 (2015).
Sivula, K., Le Formal, F. & Grätzel, M. Solar water splitting: progress using hematite (α-Fe2O3) photoelectrodes. ChemSusChem 4, 432–449 (2011).
Liu, Y. et al. A magnetically separable photocatalyst based on nest-like γ-Fe2O3/ZnO double-shelled hollow structures with enhanced photocatalytic activity. Nanoscale 4, 183–187 (2012).
Luan, P., Xie, M., Liu, D., Fu, X. & Jing, L. Effective charge separation in the rutile TiO2 nanorod-coupled α-Fe2O3 with exceptionally high visible activities. Sci. Rep. 4, 6180–6187 (2014).
Peng, L., Xie, T., Lu, Y., Fan, H. & Wang, D. Synthesis, photoelectric properties and photocatalytic activity of the Fe2O3/TiO2 heterogeneous photocatalysts. Phys. Chem. Chem.Phys. 12, 8033–8041 (2010).
Yuan, Y. et al. Simultaneous removal of SO2, NO and mercury using TiO2-aluminum silicate fiber by photocatalysis. Chem. Eng. J. 192, 21–28 (2012).
Maggos, T., Bartzis, J. G., Leva, P. & Kotzias, D. Application of photocatalytic technology for NOx removal. Appl. Phys. A 89, 81–84 (2007).
Zhou, W. et al. Mesoporous TiO2/α-Fe2O3: Bifunctional composites for effective elimination of arsenite contamination through simultaneous photocatalytic oxidation and adsorption. J. Phys. Chem. C 112, 19584–19589 (2008).
Barreca, D. et al. Controlled vapor-phase synthesis of cobalt oxide nanomaterials with tuned composition and spatial organization. CrystEngComm 12, 2185–2197 (2010).
Jiao, J., Qiu, W., Tang, J., Chen, L. & Jing, L. Synthesis of well-defined Fe3O4 nanorods/N-doped graphene for lithium-ion batteries. Nano Res. 9, 1256–1266 (2016).
Akhavan, O., Azimirad, R., Safa, S. & Larijani, M. M. Visible light photo-induced antibacterial activity of CNT-doped TiO2 thin films with various CNT contents. J. Mater. Chem. 20, 7386–7392 (2010).
Akhavan, O. Thickness dependent activity of nanostructured TiO2/α-Fe2O3 photocatalyst thin films. Appl. Surf. Sci. 257, 1724–1728 (2010).
Zhang, X. & Lei, L. Preparation of photocatalytic Fe2O3-TiO2 coatings in one step by metal organic chemical vapor deposition. Appl. Surf. Sci. 254, 2406–2412 (2008).
Watanabe, M., Aizawa, Y., Iida, T., Nishimura, R. & Inomata, H. Catalytic glucose and fructose conversions with TiO2 and ZrO2 in water at 473K: Relationship between reactivity and acid-base property determined by TPD measurement. Appl. Catal. A: Gen. 295, 150–156 (2005).
Ramis, G., Larrubia, M. A. & Busca, G. On the chemistry of ammonia over oxide catalysts: Fourier transform infrared study of ammonia, hydrazine and hydroxylamine adsorption over iron-titania catalyst. Top. Catal. 11, 161–166 (2000).
Hansen, J. et al. Unravelling site-specific photo-reactions of ethanol on rutile TiO2 (110). Sci. Rep. 6, 21990–22001 (2016).
Moniz, S. J. A., Shevlin, S. A., An, X., Guo, Z.-X. & Tang, J. Fe2O3-TiO2 nanocomposites for enhanced charge separation and photocatalytic activity. Chem. Eur. J. 20, 15571–15579 (2014).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21473096, 21603112 and 51672048), the Natural Science Foundation of Fujian Province (2014J01052 and 2015J05029), the Fujian Provincial Key Laboratory of Featured Materials in Biochemical Industry (FJKL_FMBI201606) and the Scientific Research and Development Foundation of Ningde Normal University (2016FZ16 and 2016Q40).
Author information
Authors and Affiliations
Contributions
X.W., L.W. and G.Y. designed project and carried out data analyses. R.L. performed the materials characterization. H.F. and S.Y. participated in analyzing the results. R.H. wrote the manuscript. All authors reviewed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, R., Liang, R., Fan, H. et al. Enhanced Photocatalytic Fuel Denitrification over TiO2/α-Fe2O3 Nanocomposites under Visible Light Irradiation. Sci Rep 7, 7858 (2017). https://doi.org/10.1038/s41598-017-08439-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08439-3
This article is cited by
-
Impact of Cu, Fe2O3, and Cu/Fe2O3 on the magnetic and structural characteristics of FeTiO2 nanocomposite synthesized through mechanical alloying processes
Applied Physics A (2024)
-
Sn0.94Bi0.03X0.03O2 (X = Cu, Zn) semiconductor compositions: enhanced physical, optical, electrical, dielectric, and photocatalytic properties for the elimination of organic contaminants
Journal of Sol-Gel Science and Technology (2024)
-
S-Scheme α-Fe2O3/TiO2 Photocatalyst with Pd Cocatalyst for Enhanced Photocatalytic H2 Production Activity and Stability
Catalysis Letters (2022)
-
Improvement of water resistance by Fe2O3/TiO2 photoelectrocatalysts for formaldehyde removal: experimental and theoretical investigation
Environmental Science and Pollution Research (2022)
-
In situ reduction of TiO2 nanoparticles on cotton fabrics through polydopamine templates for photocatalysis and UV protection
Cellulose (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
