
Abstract
The CD28 locus is associated with susceptibility to a variety of autoimmune and immune-mediated inflammatory diseases including primary sclerosing cholangitis (PSC). Previously, we linked the CD28 pathway in PSC disease pathology and found that vitamin D could maintain CD28 expression. Here, we assessed whether the PSC-associated CD28 risk variant A (rs7426056) affects CD28 expression and T cell function in healthy individuals (n = 14 AA, n = 14 AG, n = 14 GG). Homozygotes for the PSC disease risk allele (AA) showed significantly lower CD28 mRNA expression ex-vivo than either GG or AG (p < 0.001) in total peripheral blood mononuclear cells. However, the CD28 risk variant alone was not sufficient to explain CD28 protein loss on CD4+ T cells. All genotypes responded equally to vitamin D as indicated by induction of a regulatory phenotype and an increased anti-inflammatory/pro-inflammatory cytokine ratio. A genotypic effect on response to TNFα stimuli was detected, which was inhibited by vitamin D. Together our results show: (a) an altered gene expression in carriers of the susceptible CD28 variant, (b) no differences in protein levels on CD4+ T cells, and (c) a protective effect of the variant upon CD28 protein loss on CD4+ T cells under inflammatory conditions.
Similar content being viewed by others

Introduction
Primary sclerosing cholangitis (PSC) is a poorly understood chronic immune-mediated liver disease represented by widespread fibrotic strictures of the intra- and the extra-hepatic biliary tree. PSC is a devastating disease that lacks effective treatment and validated animal models. To date, several risk loci have been identified for PSC, with the large majority of them involving genes encoding molecules that serve essential functions in immune-related pathways1.
The CD28 locus is a newly recognized risk factor in PSC development2, 3; different genetic variants within the CD28/CTLA4 locus have been also associated with rheumatoid arthritis4, celiac disease5, alopecia areata6 and more recently with multiple sclerosis7 (an overview of the different SNPs and their location in relation to PSC risk variant is shown in Fig. 1). Because the CD28 protein is an important co-stimulatory molecule involved in the survival, clonal expansion, IL-2 production and metabolic activity of T cells8, it is predicted that such variants of CD28 will have functional impact on immune activation. From studies in several inflammatory diseases, including PSC, it is evident that the CD28 pathway has relevance to disease biology3 . However, thus far, the biological implications of such variants are not clear, limiting translation of genetic discoveries through to biologic impact.

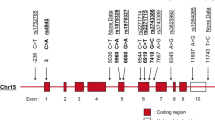
Location of rs7426056 single nucleotide polymorphism on CD28 locus. Rs7426056 SNP is located between CD28 and CTLA4 genes; approximately 3.5 kb downstream the CD28 3′UTR and approximately 120 kb upstream CTLA4 gene. Several risk variants in the CD28/CTLA4 locus have been associated with other autoimmune and immune-mediated diseases. Exons are indicated in black. (B) Table shows the linkage disequilibrium of rs7426056 with the other SNPs in CD28 and CTLA4 genes. PSC: primary sclerosing cholangitis, RA: rheumatoid arthritis, MS: multiple sclerosis, AA: alopecia areata, CEL: celiac disease.
The genetic variant “rs7426056” in the CD28 gene locus associated with PSC (minor allele A) is sufficiently common (0.229 in controls) to facilitate investigation in human lymphocytes1. Therefore, to probe our hypothesis that there are functional differences related to CD28 expression and function based on genetic background, we studied healthy subjects genotyped for this CD28 risk variant, evaluating: (a) basal CD28 expression and (b) phenotype and function of activated CD4+ T cells.
Results
CD28 mRNA expression is genotype dependent
The gender and age of all subjects was equal between groups [GG: 45 (range: 32–53 years), AA: 47.5 (range: 37–57 years), and AG: 45.5 (range: 33–53). CD28 mRNA expression was significantly lower in AA (2−ΔCt = 0.003) compared to GG (0.01, p < 0.001) and AG (0.009, p < 0.001) subjects when studying total peripheral blood mononuclear cells (see Methods) (Fig. 2A). However, we did not detect a statistically significant difference in peripheral blood CD4+CD28− T cell frequencies between the different genotypes (Fig. 2C,D). Studying CD28 cell surface MFI also revealed no statistically significant difference across genotypes [GG: median = 101 (range: 72–148), AA: 87 (63–154), AG: 99 (71–146)] (Fig. 2E). A positive correlation between frequency of CD4+CD28− T cells and CMV positivity was observed (Supplementary Figure 1A), as previously described9. After exclusion of CMV seropositive donors from the analysis, however, AA carriers still showed reduced CD28 mRNA expression (0.0029 median value) compared to GG (0.0098) and AG (0.0088) (Fig. 1B). Grubbs test analysis or Box plot revealed the presence of one extreme outlier in the AA group (value of 2−ΔCt of 0.03) and its removal from the statistical analysis showed that the CD28 mRNA expression levels in AA were significantly lower compared to GG (p = 0.02) and AG (p = 0.03). CD28 is expressed by both CD4 and CD8 T cells; mRNA expression was studied in total PBMCs thus the CD28 expression levels should be also attributed to CD8+ T cells. In CD8+ T cells no statistically significant differences in frequency of CD28− T cells was detected between the different genotypes (Supplementary Figure 1B). Exclusion of CMV seropositive donors when studying CD28 protein expression and specifically the frequency of CD28− T cells in the CD4 and CD8 population results in similar frequencies across genotypes [% of CD28− T cells in CD4 in GG: median = 0.279 (range 0.08–0.37), AA: 0.272 (0.15–1.13), AG: 0.294 (0.06–1.2)] [% of CD28− T cells in CD8 in GG: median = 21.1 (range 3.83–69.4), AA: 15.75 (8.63–54), AG: 18 (12.6–47.2)] (Supplementary Figure 1C,D). These data show that CMV seropositive donors have higher frequencies of CD28− T cells. However, it is noteworthy that in GG individuals only 3/13 (23%) were CMV seropositive, whereas in AA and AG 5/13 (38%) and 4/13 (31%), respectively, were CMV seropositive. Collectively, our data suggest that an interplay between viral infection and genotype might be important for CD28 expression.

AA subjects show reduced CD28 mRNA expression but similar frequencies of CD4+CD28− T cells with GG and AG. (A) RNA from freshly isolated peripheral blood mononuclear cells (PBMCs) was extracted, transcribed into cDNA and tested for CD28 mRNA expression in healthy individuals homozygous (AA) or heterozygous (AG) for the PSC risk variant and homozygous for the protective allele (GG) (n = 14 per group). Data are expressed as 2−ΔCt relative to beta actin. Lines indicate median values. **p = 0.002 and **p = 0.003 with Mann Whitney U test after exclusion of the outlier detected in the AA group by Grubbs test. (B) CD28 mRNA expression in total PBMCs in CMV seronegative donors. Data are expressed as 2−ΔCt relative to beta actin. AA and AG healthy individuals homozygotes and heterozygotes for the PSC risk variant and GG homozygotes for the protective allele. Lines indicate median values. *p = 0.02 and *p = 0.03 with Mann Whitney U test after exclusion of the outlier detected in the AA group by Grubbs test. (C) Representative flow cytometry plots showing the gating strategy to define CD4+CD28− T cells in PBMCs. CD3+CD4+ cells were selected after duplet exclusion and CD28 expression was studied in the CD4+ population. (D) Data show the proportion of CD4+ T cells that have lost CD28 expression. Line indicates median with interquartile range. (E) CD28 median fluorescence intensity (MFI) across genotypes. Line indicates median value.
The CD28 risk variant alone is not sufficient to explain CD28 loss
We have recently reported that CD4+CD28− T cells accumulate in livers of patients with PSC, where they localize close to the bile ducts and can induce the death and activation of the latter3. High levels of TNFα were detected in the liver of PSC patients and we showed that TNFα could downregulate the expression of CD28 on PSC T cells in vitro 3 . CD4+CD28− T cells serve as a surrogate marker of inflammation in several autoimmune and immune-mediated inflammatory diseases10,11,12, therefore we assessed whether there are biologic differences across the genotypes in the context of CD4+CD28− T cell expansion after TNFα stimulation.
After 21 days TNFα enhanced the expansion of CD4+CD28− T cells across all genotypes. This effect was not significantly different at day 14 for AA and AG genotypes, in keeping with a delayed response to TNFα-mediated loss of CD28 expression in at risk subjects (Fig. 3A). Even in untreated conditions where only two cycles of TCR stimulation were applied in the absence of TNFα, in cells with the GG genotype there was a greater expansion of CD4+CD28− T cells at day 14 compared to AA and AG (Fig. 3A).

Individuals with AA and AG genotypes are less susceptible to TNFα-mediated loss of CD28 expression. CD4+CD25− T cells were activated with aCD3/aCD28 beads (two cycles of activation) in the presence or absence of TNFα and in the presence or absence of 1,25(OH)2D3 for 14 and 21 days. (A and B) Data show the proportion of CD4+CD25− T cells that lost CD28 expression in the presence (TNFα) and absence of TNFα (Untreated) and in the presence or absence of 1,25(OH)2D3 after 14 and 21 days. (C) Data show the proportion of CD4+CD25− T cells that lost CD28 expression in the presence of TNFα and in the simultaneous presence of TNFα and 1,25(OH)2D3 after 14 and 21 days in culture. Healthy individuals homozygous (AA) (n = 14) or heterozygous (AG) (n = 14) for the PSC risk SNP “rs7426056” in the CD28 locus and n = 14 homozygous for the protective allele (GG). Data show mean with SEM. *p < 0.05, **p < 0.01, ***p < 0.001 with Wilcoxon matched-pairs signed rank test.
Carriers of the wild type allele (GG) are more susceptible to the pro-inflammatory effects of TNFα but also responsive to the protective effect of vitamin D
We have previously reported that active vitamin D (1,25(OH)2D3) can overcome the effect of TNFα upon loss of CD28 expression3, therefore we studied whether there are genotype-associated differences in the ability of T cells to respond to 1,25(OH)2D3 vitamin D. In GG, 1,25(OH)2D3 significantly reduced the frequency of CD4+CD28− T cells in the presence of TNFα at both day 14 and day 21 (5- and 4.3- fold, respectively, p < 0.001). In AA and AG, this effect was significant only at day 21 (2.5-fold mean suppression in both cases, p < 0.001, p < 0.01) (Fig. 3B and C), possibly because at day 14 there was not a significant induction of CD4+CD28− T cells by TNFα. Collectively, our data indicate that carriers of the wild type allele (GG) are more susceptible to the pro-inflammatory effects of TNFα but also remain responsive to the protective effect of vitamin D.
T cells from the different genotypes are equally responsive to 1,25(OH)2D3
We employed a system with fixed CHO-CD80 cells to stimulate CD4+CD25− T cells and study the ability of the latter to respond (by cytokine production, up-regulation of activation markers and proliferation) as this system has been well characterised in former published studies performed by our collaborators13, 14. These studies demonstrate the necessity of the CHO-CD80 co-stimulatory signal for T cell proliferation, even in the presence of anti-CD3, and its stronger ability to promote T cell proliferation compared to CD86, consistent with the greater affinity of CD80 for CD28 compared to CD86. Former studies have also shown the ability of the ligand binding domain of CTLA-4 (CTLA-4-Ig) to block the fixed CD80 signal provided by fixed CHO-CD80 cells, preventing T cell proliferation14.
T cell activation and CD28 signaling promote T cell proliferation, therefore we assessed whether the T cells from subjects with the risk variant show impaired proliferation capacity. Our data show that T cells from all genotypes were equally able to proliferate as indicated by the similar percentage of undivided cells [GG: median = 28.1% (range: 9.03–55.1), AA: median = 25.8% (range: 9.69–59.6), AG: median = 27.2% (range: 9.28–53.9)]. Treatment with vitamin D did not alter this frequency, which was again similar across genotypes [GG: median = 28.45% (range: 9.44–58.3), AA: median = 23.15% (range: 10.9–59.1), AG: median = 27.8% (range: 13.2–53.2)] (Fig. 4A). Evaluation of the proliferation index, which shows the average number of divisions for those cells that went into division, showed similar values across genotypes in the presence and absence of vitamin D (Fig. 4B). To account for cells that never divided we further calculated the division index, a measure of the average number of divisions that a cell in the original population has undergone. This was likewise similar across genotypes in the presence and absence of vitamin D (Fig. 4C).

GG, AA and AG individuals show similar levels of proliferation. CD4+CD25− T cells were activated in vitro with aCD3 antibody and CHO-CD80 cell line in the presence or absence of 1,25(OH)2D3 for 5 days. Data show (A) the percentage of undivided cells, (B) the proliferation index and (C) the division index across genotypes in the presence (VitD+) and absence (VitD−) of vitamin D.
T cell activation and CD28 signaling also promote the expression of Foxp3, CD25 and CTLA-4 molecules15, therefore we assessed whether the T cells from subjects with the risk variant are functionally defective in their CD28 signaling. CD4+CD25− T cells were stimulated for 5 days with anti-CD3 (aCD3) in the presence of CD80 expressed on CHO cells and the expression of the aforementioned markers was assessed by flow cytometry. No significant differences were detected between genotypes (Fig. 5). 1,25(OH)2D3 vitamin D has predominantly an immunosuppressive effect on the adaptive immune response promoting the development of regulatory T cells16, 17. We therefore further assessed the ability of 1,25(OH)2D3 vitamin D to induce a regulatory phenotype on CD4+CD25− T cells after 5 days in vitro activation by aCD3 and CHO-CD80. All groups (AA, AG, GG) upregulated CTLA-4 and Foxp3 expression (Fig. 5) upon vitamin D stimulation. In untreated conditions (without vitamin D) cells from GG, AA and AG individuals showed similar induction of CD25 as indicated by the % of CD25 expressing cells [GG: 66% ± 14, AA: 64% ± 14 and AG: 62% ± 19 (mean value ± SD)] and CD25 MFI [GG:151 ± 114, AA: 195 ± 152, AG: 157 ± 131 (mean value ± SD)]. Stimulation with vitamin D did not alter the % of CD25 expressing cells but resulted in significant induction of CD25 MFI in GG (from 151 to 229 mean value) (*p = 0.018) and AA (from 195 to 251 mean value) (*p = 0.016), thus causing a 1.5 and 1.3 fold induction, respectively. In AG, vitamin D induced a lower CD25 expression (from 157 to 191 mean value) (Fig. 5). It is known that CD25 expression is upregulated by TCR stimulation and CD28, as well as by vitamin D via CD28 signaling14. In our experiments vitamin D induced CD28 expression and IL-2 cytokine secretion at similar levels across genotypes (Supplementary Figures 2 and 3). It is also well known that high CD25 expression defines T regulatory cells, thus higher CD25 expression could possibly mean higher frequency of T regulatory cells. Our data however on Treg frequencies based on CD25 expression as well as Foxp3 and CTLA-4 expression revealed an induction of regulatory cells after vitamin D stimulation with no significant differences across genotypes (Fig. 6). CD25 is considered an activation marker, thus the higher expression on T cells from GG and AA may mean a more activated phenotype.

AA, AG and GG individuals express similar levels of CD25, Foxp3 and CTLA-4 molecules. CD4+CD25− T cells were activated in vitro with aCD3 antibody and CHO-CD80 cell line in the presence or absence of 1,25(OH)2D3 for 5 days. (A) Representative flow cytometry plots showing proliferating T cells expressing CD25, Foxp3 and CTLA-4. (B) Percentage of proliferating cells that express CD25, Foxp3 and CTLA-4 under no vitamin D treatment conditions (VitD−). (C–E) Data show the percentage of proliferating cells that express CD25, Foxp3 and CTLA-4 and their respective median fluorescence intensity values for GG, AA and AG genotypes of the PSC CD28 risk allele locus. Representative overlay histograms showing the expression of each marker in the presence and absence of 1,25(OH)2D3. *p < 0.05, **p < 0.01, ***p < 0.001 using Wilcoxon matched-pairs signed rank test.

GG, AA and AG individuals show similar frequencies of Tregs. CD4+CD25− T cells were activated in vitro with aCD3 antibody and CHO-CD80 cell line in the presence or absence of 1,25(OH)2D3 for 5 days. (A) Representative flow cytometry plots showing expression of Foxp3 and CTLA4 on CD25+ cells across genotypes in the presence (VitD+) and absence (VitD−) of 1,25(OH)2D3. Values in cross symbols represent the mean frequency (%) of Foxp3hi CTLA4+ and Foxp3int CTLA4+ plus/minus standard deviation across genotypes in all samples (n = 14 per group). (B) Frequency of Foxp3hi CTLA4+ in CD25+ cells in the presence and absence of vitamin D. (C,D) Foxp3 and CTLA4 median fluorescence intensity (MFI) on Foxp3hi CTLA4+ cells.
Treg cell induction was further assessed based on the co-expression of CD25, Foxp3 and CTLA-4. Tregs were therefore defined as CD25+ Foxp3hi CTLA4+. Our data show a similar frequency of Tregs across genotypes in basal conditions (VitD−) [GG: median = 11% range (4.2–21.7), AA: median = 11% range (3.7–29.0), AG: median = 9% (range: 5.6–23.6); values show the proportion of CD25+ cells expressing Foxp3 and CTLA4]. The presence of vitamin D (VitD+) significantly increased the proportion of Foxp3hi CTLA4+ cells in the CD25+ population in each group [GG: median = 25% (range: 5.0–43.8), AA: median = 25% (range: 7.7–43.7), AG: median = 20% (range: 6.7–46.2)] (Fig. 6A,B). No significant differences were detected in the proportion of induced Tregs in the presence of vitamin D across genotypes (Fig. 6A,B). The median fluorescence intensity of Foxp3 and CTLA-4 molecules was similar across genotypes in the presence and absence of vitamin D (Fig. 6C,D).
All groups (AA, AG, GG) downregulated OX40 and PD-1, with no significant differences across the groups being detected. The expression of ICOS activation marker on CD4+CD25− T cells after in vitro stimulation with anti-CD3 antibody and CHO-CD80 cell line was similar across genotypes albeit slightly lower in GG and AG compared to AA individuals [MFI in GG: 78 ± 35, AA: 94 ± 33 and AG: 76 ± 24 (mean value ± SD)]. In GG donors stimulation with 1,25(OH)2D3 induced ICOS MFI [from 78 ± 35 to 96 ± 48 (mean value ± SD) by 1.2 fold (p = 0.042)]. In AA there was no change in ICOS MFI after stimulation with vitamin D and in AG there was only minimal ICOS induction (Fig. 7). The significant upregulation of ICOS MFI on T cells from GG donors after vitamin D stimulation is possibly because there was lower ICOS expression in untreated conditions as a starting point thus the difference in its expression was seen more robust after vitamin D stimulation. It is known that ICOS expression requires TCR activation and CD28 co-stimulation. We and others have shown that vitamin D can increase the median fluorescence of CD28 on CD4+ T cells3, 18, and our current data show an induction of CD28 MFI after vitamin D stimulation at similar levels across genotypes (Supplementary Figure 3)], thus the differential level of ICOS MFI on CD4+ T cells across genotypes seems not to be attributed to effects of vitamin D and CD28 expression. Notably, vitamin D receptor (VDR) binding regions have been detected closer to CD28 and CTLA-4 genes (one close to the promoter region of CD28, two in between CD28 and CTLA-4, and one in the CTLA-4 gene itself). In either side of ICOS gene there have been detected some VDR binding sites but they are more distant, therefore CD28 and CTLA-4 are more likely targets of direct regulation by vitamin D than is ICOS19. ICOS plays an important role in the development and effector functions of Th1, Th2, Th17 and Treg cells20, thus the higher expression of ICOS on GG donors may suggest differential effects on immune cell activation after its stimulation. Therefore, these data further verify the immunoregulatory role of vitamin D and the equal responsiveness of T cells across different genotypes to 1,25(OH)2D3.

AA, AG and GG individuals express similar levels of ICOS, OX40 and PD-1 molecules after T cell activation. CD4+CD25− T cells were activated in vitro with aCD3 antibody and CHO-CD80 cell line in the presence or absence of 1,25(OH)2D3 for 5 days. (A) Representative flow cytometry plots showing proliferating T cells expressing ICOS, OX40 and PD-1. (B) Percentage of proliferating cells that express ICOS, OX40 and PD-1 under no vitamin D treatment conditions (VitD−). (C–E) Data show the percentage of proliferating cells that express ICOS, OX40 and PD-1 and their respective median fluorescence intensity values for GG, AA and AG genotypes of the PSC CD28 risk allele locus. Representative overlay histograms showing the expression of each marker in the presence and absence of 1,25(OH)2D3. *p < 0.05, ***p < 0.001, ****p < 0.0001 using Wilcoxon matched-pairs signed rank test.
The level and type of co-stimulation that T cells receive plays an important role in determining their differentiation and cytokine production21. Therefore, we also assessed the effect of CD28 genotype upon CD4+ T cell cytokine production. Genotype had no effect upon cytokine production in the absence of 1,25(OH)2D3 and T cells from all three genotypes were able to respond to 1,25(OH)2D3 as indicated by the increased ratio of anti-inflammatory IL-10 versus the pro-inflammatory cytokines IFNγ, IL-17, IL-9, IL-21 and IL-22 (Fig. 8, Supplementary Figure 2). Therefore, these data also indicate an equivalent ability of each CD28 genotype to transmit the co-stimulation signal to respond to 1,25(OH)2D3.

AA, AG and GG individuals respond to vitamin D and produce regulatory cytokines. CD4+CD25− T cells were activated in vitro with aCD3 antibody and CHO-CD80 cell line in the presence or absence of 1,25(OH)2D3. At day 3, cell-free supernatant was collected and tested for the presence of secreted cytokines. Data show the ratio of IL-10 versus pro-inflammatory cytokines IFNγ, IL-17, IL-2, IL-9, IL-21 and IL-22. **p < 0.01, ***p < 0.001 using Wilcoxon matched-pairs signed rank test.
Discussion
Studies on the biological significance of identified genetic susceptible loci are limited. Many polymorphisms linked to human autoimmune and immune-mediated diseases have been identified through genome-wide association studies (GWAS), however the functional relevance of most of these genetic variations remain undefined. Here we explored the association of the confirmed PSC disease risk allele rs7426056 in the CD28 locus with expression of CD28 and T cell function and responsiveness to variable stimuli in healthy individuals. We confirm that: (i) basally there is genotype dependent effect on gene expression in carriers of the susceptible CD28 variant; (ii) an interplay between viral infection and genotype might be also important for CD28 expression; (iii) signaling of CD28 is not different across the genotypes and neither is their response to vitamin D; and (iv) there is a genotype effect on response to TNFα inflammatory stimuli, which can be overcome by vitamin D.
The rs7426056 CD28 genetic variant is located 3.5 kb downstream of the CD28 3′UTR and approximately 120 kb upstream of CTLA4. It is in almost complete linkage disequilibrium (r2 = 0.99) (https://caprica.genetics.kcl.ac.uk/~ilori/ld_calculator.php) with the genetic variant rs1980422, also within the CD28 locus, that has been associated with rheumatoid arthritis locus4. The large majority of disease-associated SNPs are located in between genes and such non-coding SNPs may lead to decreased or enhanced production of the transcript and/or its translation into protein by affecting enhancers, microRNAs, or through long-range transcription regulation22. Our data show an altered gene expression in carriers of the susceptible CD28 variant, however no differences in protein levels were detected, at least on CD4+ T cells that were studied. A significant correlation between another CD28 variant (rs6435203), which is in close linkage disequilibrium with the studied rs7426056 and CD28 mRNA expression has been also reported7. The triggers of expansion of CD28− T cells are unclear; continuous antigenic stimulation23 and in vitro replicative senescence24 have been reported to influence CD28 expression. An association between HLA-DRA and loss of CD28 expression on memory CD8+ T cells has been reported in healthy subjects25 therefore a combination of genetic and environmental factors may further contribute to loss of CD28 expression. In addition, our data show that the signaling of CD28 and specifically the ability of CD4+ T cells to respond to vitamin D is not different across genotypes as indicated by the induction of a regulatory phenotype and an increased anti-inflammatory/pro-inflammatory cytokine ratio in vitamin D treated samples.
Identifying the functional significance of GWAS-identified disease-associated variants is now a major but important challenge as it will allow enhanced understanding of disease pathologies from which novel preventative and therapeutic strategies can be developed. Overall, our study into the functional significance of the PSC-associated CD28 variant, rs7426056, has identified a possible inhibitory effect of the SNP upon CD28 mRNA expression, although CD28 protein ex vivo remained unchanged in CD4+ T cells and we observed a protective effect of the variant upon CD28 loss under inflammatory conditions. To note, our system used a mixed CD4 population consisting of both naïve and memory cells thus unable to distinguish between polarization of naïve cells and reactivation of memory cells. However, a mixture of CD4 populations is expected to be present in vivo, thus we believe our approach was a more realistic approach. The mixture of naïve and memory cells was similar across genotypes, thus this should not interfere with the validity of findings.
The functional effect of this CD28 SNP in PSC pathology therefore remains slightly unclear. It is likely that rs7426056, is not an independent regulator of CD28-mediated T cell responses but becomes significant in PSC when inherited with other PSC-associated gene variants. Our study does not suggest an interaction of rs7426056 with the environmental variant vitamin D but other PSC-associated environmental risk factors could affect its biological effect. One interesting regulator of immune responses that is gaining much attention as a regulator of immune responses, is metabolism. Notably, CD28 co-stimulation has been shown to enhance the expression of glucose transporters, glucose uptake, and glycolysis in human T cells in a PI3K dependent manner26. Whether rs7426056 affects T cell metabolism is therefore of future interest.
An alternative explanation for the association of rs7426056 with PSC is its co-inheritance with a functional disease variant by linkage disequilibrium. Indeed, it is very likely that many GWAS-identified disease variants are essentially tags for a limited number of functional disease-causing variants. Studies to identify the roles of GWAS variants should therefore focus initially on those in or surrounding genes for which a functional involvement of the encoded factor has already been demonstrated. However, as our study suggests, multiple genetic and environmental factors could affect the functional manifestation of the mutation and its involvement in disease. Understanding the potential role of genetic association signals in disease biology will therefore require complex and carefully controlled studies. To date, genome wide association studies in PSC have identified 23 susceptibility loci and this is mirrored by even more risk loci for other autoimmune diseases. Therefore, in this context, whilst of course ultimately one wishes to study gene effects in patients (and more specifically in target tissues and target cell types, controlled for risk, stage and disease treatment), it is scientifically logical to start evaluating a gene risk locus in healthy controls: this allows investigators to attempt to model the impact of the genetic variation more closely, without all the confounding effects of multiple other disease risk gene effects, the consequences of disease and its progression, as well as the impact of therapies.
A methodological constraint of our study and one that makes the task of analyzing healthy individuals in which a single variant of interest is being studied, is the power calculation analysis. Initial power calculation analysis based on our previous findings3 revealed that to detect a difference of 50% between the control and vitamin D treated groups a sample size of 14 patients per group would be sufficient to detect a difference of this size with alpha = 0.05 and power of 80%. Based on the sample size we have used (n = 14 per group of healthy individuals) one-way ANOVA comparing the logged values of CD4+CD28− T cell frequencies showed a 6.7-fold minimal detectable difference between the groups at 80% power and 5% alpha. Our sample size was limited however by substantial ethical and logistic practicalities (i.e. need for same day shipping of blood in working hours, day of the week the bleeding should take place on, availability and willingness of subjects to take part, and season of the year [acquisition and processing of 42 samples in total lasted 5 months]) and therefore it is acknowledged that our study power could in the ideal world have been augmented, and future much larger studies overcoming the logistics of working with fresh samples from large numbers of healthy volunteers should account for this. Our main genotype difference is between GG vs AG/AA, at around 2.5-fold (geometric mean: 0.35 vs 0.88/0.81), and for this to be statistically significant confirmed finding, a much larger number of healthy individuals (approaching 200 in total across genotypes) would be needed, something we could not overcome in this study.
To conclude, our study is one of the very few that attempts to address the biological significance of CD28 genetic variant in the pathology of an immune-mediated liver disease of unknown etiology. Understanding the effects of the different genotypes on gene expression, susceptibility to inflammatory stimuli and environmental factors has started to be the focus of several research attempts. Therefore, hopefully extension of such studies as ours will lead to better understanding of how genetic risk loci associated with disease impact meaningful biologic pathways.
Methods
Study population
Peripheral blood from healthy individuals with known risk (AA and AG) and control genotypes (GG) (n = 14 per group) was collected via the Cambridge NIHR BioResource. All samples were obtained with Local Research and Ethics Committee approval and informed patient consent. All experimental protocols were approved by NHS Health Research Authority and NRES Committee West Midlands - South Birmingham (REC 2003/242) and all methods were carried out in accordance with relevant guidelines and regulations.
Table 1 illustrates the characteristics (sex and age) of all individuals included in the study. All data were assessed blinded to genotype.
Cell Isolation
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lympholyte (Cedarlane Laboratories, Burlington, Canada) for 20 min at 800 g. CD4+CD25− T cells were purified from PBMCs using a custom made negative selection antibody cocktail (StemCell Technologies), according to manufacturer’s instructions.
Cell Culture
Chinese hamster ovary (CHO) cells expressing CD80, were cultured in DMEM (Life Technologies, Paisley, UK) supplemented with 10% v/v FBS (Life Technologies), 1% penicillin, streptomycin (Life Technologies) and incubated at 37 °C in a humidified atmosphere of 5% CO2.
CD4+ CD25− T cell in vitro stimulation
CD4+CD25− T cells, enriched (on average) to >94% were cultured in RPMI1640 media supplemented with 1% penicillin-streptomycin-glutamine (PSG) and 10% foetal calf serum (FCS) (Life Technologies). For some studies as described, T cells were stimulated with aCD3/aCD28 Dynabeads (1 μl/well (~1:10 bead:cell); Life Technologies), IL-2 (50U/ml; Peprotech) in the presence/absence of TNFα (10ng/ml; Peprotech) with or without vitamin D (1,25(OH)2D3 10 nM); ENZO Life Sciences). At 4-days, beads were removed using a magnet and cells were split to 0.5 × 106 cells/ml. At day 6 cells were re-activated with aCD3/aCD28 beads for another cycle of activation for 4 days. At day 10 beads were removed, cultures were assessed and maintained at 0.5–1 × 106 cells/ml. After that, cultures were assessed every 4 days and maintained at 0.5–1 × 106 cells/ml. Cytokines and 1,25(OH)2D3 were also re-supplemented at the time of cell culture assessment. The concentration of IL-2 was increased to 100U/ml after 1 week. Cells were cultured for up to 21 days. CD28 expression was assessed at days 14 and 21 using flow cytometry.
For separate experiments, CD4+CD25− T cells were labelled with cell trace violet (Life Technologies) per manufacturer’s instructions. CHO-CD80 cells were trypsinized and fixed with 0.025% glutaraldehyde for 3 mins at room temperature followed by quenching with media containing FCS. Fixed cells were washed and finally resuspended in RPMI 1640 + 10% FCS + 1% PSG. Cell trace violet-labelled T cells (0.5 × 106 cells/ml) were co-cultured with CHO-CD80 (at 5:1 ratio) with anti-CD3 (OKT3, at 0.5 μg/ml), with or without 10 nM 1,25(OH)2D3 (ENZO Life Sciences) in 96-U plates for 5 days. Cell free supernatant was collected at day 3 for assessment of cytokine expression.
Luminex cytokine analysis
Cell free supernatant from T cell-CHO-CD80 day 3 co-cultures was collected and assessed for cytokine expression using the human Th1/Th2/Th17/Th22/Treg Procartaplex Multiplex Immunoassay (eBiosciences catalog number EPX180-12165-901), according to manufacturer’s instructions. The cytokines measured were: GM-CSF, IFNγ, IL-1, IL-12, IL-13, IL-18, IL-2, IL-5, IL-6, TNFα, IL-4, IL-10, IL-17, IL-21, IL-22, IL-23, IL-27, IL-9.
Flow Cytometry
Flow cytometric analysis was performed on peripheral blood T cells using a Cyan flow cytometer (Beckman Coulter, Bucks, UK), and analysed using FlowJo (version 9). CD28 expression on unstimulated PBMCs (at day of isolation) was studied by surface staining. Cultured cells were collected (at days 14 and 21) and stained with live/dead marker (Zombie, Biolegend) for 20 minutes at room temperature, followed by washes and surface staining with CD3-FITC (clone HIT3a), CD4-APC (clone RPA-T4) and CD28-PE (clone 28.2) (all from BD Biosciences). After 5-days T cell – CHO-CD80 co-culture, cells were stained with: CD4-PerCP-Cy5.5 (clone RPA-T4, BD Biosciences), CD28-PeCY7 (clone 28.2, Biolegend), ICOS-FITC (clone REA192, Miltenyi Biotec), OX40-PE (clone ACT35, BD Biosciences) and PD-1-APC (clone MIH4, BD Biosciences).
For intracellular and nuclear protein detection cells were stained for live/dead marker prior to fixation and permeabilization using the Foxp3/transcription factor staining buffer set according to manufacturer’s instructions. After fixation and permeabilization cells were stained with: CD4-PerCP-Cy5.5 (clone RPA-T4, BD Biosciences), CD28-PeCY7 (clone 28.2, Biolegend), CD25-BB515 (clone 2A3, BD Biosciences), CTLA-4 (clone BNI3, BD Pharmingen) and Foxp3-APC (clone PCH101, eBioscience). Analysis of activation markers (CD25, PD-1, ICOS, CTLA-4, OX40) and transcription factors (Foxp3) was focused on activated/proliferating cells as inclusion of non-activated cells would have skewed the results.
Analysis of CMV positivity in plasma samples
Plasma samples were used to determine CMV IgG titres. A CMV IgG ELISA assay developed at the University of Birmingham by Professor Paul Moss’s group was used. A sample from a known CMV seropositive donor (positive control) and a known CMV seronegative donor (negative control) for which multiple aliquots were frozen was run with every single plate in order to document assay precision and reproducibility. All samples were run in duplicate in CMV lysate coated wells, as well as mock lysate coated wells, and the average absorbance value seen in the mock lysate coated wells (background non-specific binding) was subtracted from the average absorbance value in the CMV lysate coated plate.
Quantification of mRNA expression levels by RT-PCR
Total RNA was extracted from uncultured peripheral blood mononuclear cells using the RNeasy mini kit (Qiagen, UK) according to manufacturer’s instructions. The eluted RNA concentration was measured using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). 50 μg of extracted RNA was transcribed into cDNA using iScript cDNA synthesis kit (BioRad, Hercules, CA). Quantitative analysis of CD28 mRNA expression was performed using Taqman Fluorogenic 5′ nuclease assays using gene-specific 5′FAM labelled probes (Hs01007422_m1, Life Technologies) run on ABI 7900 sequencer with beta-actin used as endogenous control (Hs01060665_g1, Life Technologies). Values are represented as the difference in Ct values normalized to β-actin for each sample as per the following formula: Relative RNA expression = 2−ΔCt where ΔCt = Ct of CD28 – Ct of β-actin27.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism. Data not normally distributed were evaluated using Wilcoxon matched-pairs signed rank test and Kruskal-Wallis test. Values of P < 0.05 were considered significant.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Liu, J. Z. et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature genetics 45, 670–675, doi:10.1038/ng.2616 (2013).
Liu, J. Z. et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nature genetics 44, 1137–1141, doi:10.1038/ng.2395 (2012).
Liaskou, E. et al. Loss of CD28 expression by liver-infiltrating T cells contributes to pathogenesis of primary sclerosing cholangitis. Gastroenterology 147, 221–232, e227, doi:10.1053/j.gastro.2014.04.003 (2014).
Raychaudhuri, S. et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nature genetics 41, 1313–1318, doi:10.1038/ng.479 (2009).
Dubois, P. C. et al. Multiple common variants for celiac disease influencing immune gene expression. Nature genetics 42, 295–302, doi:10.1038/ng.543 (2010).
Betz, R. C. et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun 6, 5966, doi:10.1038/ncomms6966 (2015).
Lill, C. M. et al. Genome-wide significant association with seven novel multiple sclerosis risk loci. J Med Genet 52, 848–855, doi:10.1136/jmedgenet-2015-103442 (2015).
Noel, P. J., Boise, L. H. & Thompson, C. B. Regulation of T cell activation by CD28 and CTLA4. Advances in experimental medicine and biology 406, 209–217 (1996).
Chanouzas, D. et al. CD4+ CD28- T-cell expansions in ANCA-associated vasculitis and association with arterial stiffness: baseline data from a randomised controlled trial. Lancet 385(Suppl 1), S30, doi:10.1016/S0140-6736(15)60345-2 (2015).
Thewissen, M. et al. CD4+ CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol 179, 6514–6523 (2007).
Broux, B., Markovic-Plese, S., Stinissen, P. & Hellings, N. Pathogenic features of CD4+ CD28- T cells in immune disorders. Trends in molecular medicine 18, 446–453, doi:10.1016/j.molmed.2012.06.003 (2012).
Seyda, M., Elkhal, A., Quante, M., Falk, C. S. & Tullius, S. G. T Cells Going Innate. Trends in immunology 37, 546–556, doi:10.1016/j.it.2016.06.004 (2016).
Manzotti, C. N. et al. Integration of CD28 and CTLA-4 function results in differential responses of T cells to CD80 and CD86. European journal of immunology 36, 1413–1422, doi:10.1002/eji.200535170 (2006).
Gardner, D. H. et al. 1,25(OH)2D3 Promotes the Efficacy of CD28 Costimulation Blockade by Abatacept. J Immunol 195, 2657–2665, doi:10.4049/jimmunol.1500306 (2015).
Scotta, C., Soligo, M., Camperio, C. & Piccolella, E. FOXP3 induced by CD28/B7 interaction regulates CD25 and anergic phenotype in human CD4+ CD25- T lymphocytes. J Immunol 181, 1025–1033 (2008).
Jeffery, L. E. et al. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol 183, 5458–5467, doi:10.4049/jimmunol.0803217 (2009).
Jeffery, L. E. et al. Availability of 25-hydroxyvitamin D(3) to APCs controls the balance between regulatory and inflammatory T cell responses. J Immunol 189, 5155–5164, doi:10.4049/jimmunol.1200786 (2012).
Kickler, K., Ni Choileain, S., Williams, A., Richards, A. & Astier, A. L. Calcitriol modulates the CD46 pathway in T cells. PloS one 7, e48486, doi:10.1371/journal.pone.0048486 (2012).
Ramagopalan, S. V. et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome research 20, 1352–1360, doi:10.1101/gr.107920.110 (2010).
Simpson, T. R., Quezada, S. A. & Allison, J. P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Current opinion in immunology 22, 326–332, doi:10.1016/j.coi.2010.01.001 (2010).
Zhang, R., Borges, C. M., Fan, M. Y., Harris, J. E. & Turka, L. A. Requirement for CD28 in Effector Regulatory T Cell Differentiation, CCR6 Induction, and Skin Homing. J Immunol 195, 4154–4161, doi:10.4049/jimmunol.1500945 (2015).
Manolio, T. A. Genomewide association studies and assessment of the risk of disease. The New England journal of medicine 363, 166–176, doi:10.1056/NEJMra0905980 (2010).
Vallejo, A. N., Brandes, J. C., Weyand, C. M. & Goronzy, J. J. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J Immunol 162, 6572–6579 (1999).
Effros, R. B. et al. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: a possible cause for both in vivo and in vitro immunosenescence. Exp Gerontol 29, 601–609 (1994).
Orru, V. et al. Genetic variants regulating immune cell levels in health and disease. Cell 155, 242–256, doi:10.1016/j.cell.2013.08.041 (2013).
Frauwirth, K. A. et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002).
Yuan, J. S., Reed, A., Chen, F. & Stewart, C. N. Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics 7, 85, doi:10.1186/1471-2105-7-85 (2006).
Acknowledgements
We are grateful to PSC Partners Seeking a Cure who funded this project and to Cambridge NIHR BioResource for accessing blood samples from healthy individuals with/without the risk allele. We are thankful to all the individuals who donated their blood for this research. Finally, we are thankful to James Hodson for his support on power calculation and statistical analysis. EL and GMH have received support from the Birmingham Liver Biomedical Research Unit and FP7 Career Integration Grant Award entitled: “Primary Biliary Cirrhosis: Translating Genetic Discovery into Patient Benefit”. This paper presents independent research supported by the NIHR Birmingham Liver Biomedical Research Unit based at the University of Hospital Birmingham NHS Foundation Trust and the University of Birmingham. The views expected are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
E.L., L.J., B.S., D.S., G.M.H. contributed to study design; E.L., L.J., D.C. performed experiments; E.L., L.J., D.C., M.F.S. performed data analysis; E.L., L.J., D.C., M.F.S., G.M.H. interpreted the data, E.L. drafted the manuscript. All authors critically revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liaskou, E., Jeffery, L., Chanouzas, D. et al. Genetic variation at the CD28 locus and its impact on expansion of pro-inflammatory CD28 negative T cells in healthy individuals. Sci Rep 7, 7652 (2017). https://doi.org/10.1038/s41598-017-07967-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07967-2
This article is cited by
-
CD28 co-stimulation: novel insights and applications in cancer immunotherapy
Nature Reviews Immunology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
