
Abstract
Phages infecting Aeromonas salmonicida subsp. salmonicida, the causative agent of the fish disease furunculosis, have been isolated for decades but very few of them have been characterized. Here, the host range of 12 virulent phages, including three isolated in the present study, was evaluated against a panel of 65 A. salmonicida isolates, including representatives of the psychrophilic subspecies salmonicida, smithia, masoucida, and the mesophilic subspecies pectinolytica. This bacterial set also included three isolates from India suspected of being members of a new subspecies. Our results allowed to elucidate a lytic dichotomy based on the lifestyle of A. salmonicida (mesophilic or psychrophilic) and more generally, on phage types (lysotypes) for the subspecies salmonicida. The genomic analyses of the 12 phages from this study with those available in GenBank led us to propose an A. salmonicida phage pan-virome. Our comparative genomic analyses also suggest that some phage genes were under positive selection and A. salmonicida phage genomes having a discrepancy in GC% compared to the host genome encode tRNA genes to likely overpass the bias in codon usage. Finally, we propose a new classification scheme for A. salmonicida phages.
Similar content being viewed by others


Introduction
The Gram-negative bacterium Aeromonas salmonicida subsp. salmonicida is a major fish pathogen that is responsible for significant economic losses in the aquaculture industry worldwide1. More recently, genomic studies on A. salmonicida have led to a better understanding of its evolutionary history and global diversity, including its large number of mobile genetic elements2,3,4 as well as its lifestyle (psychrophilic or mesophilic)4, 5.
As with other bacteria, A. salmonicida subsp. salmonicida can be infected by viruses, namely bacteriophages or phages. Moreover, it is generally recognized that phages play significant roles in microbial ecology through bacterial lysis, reprograming of host metabolism and horizontal gene transfer6,7,8. One of the first studies on phages infecting A. salmonicida was published in 1933 and reported the presence of these biological entities in English rivers9. Another study on A. salmonicida phages published in 197010 identified various lysotypes (i.e., groups of bacterial strains based on their sensitivity to specific phages), suggesting that A. salmonicida is subject to infection by dynamic and diverse groups of phages. It was later shown that the A-layer, a bacterial surface structure implicated in virulence11, may also be a phage receptor in A. salmonicida 12. Both virulent phages, which produce virions after lysis of the host cell, and temperate phages, which can integrate their genome into the host bacterial chromosome, have been isolated for A. salmonicida 10, 13,14,15,16,17,18,19.
With the rise of bacterial strains resistant to antibiotics, phages are now being revisited as a potential complement or alternative to antibiotics to treat bacterial infections20. For example, phages were explored to treat or prevent furunculosis, a disease caused by A. salmonicida subsp. salmonicida 21. Although phage therapy remains a promising strategy, there are potential drawbacks associated with the use of phages as antibacterial agents. For example, lysogenic conversion, the process whereby the host capitalizes on the genes encoded by prophage to enhance its own fitness, has conferred new capabilities in a wide range of bacterial species. It has now been established that some genes related to drug resistance22, metabolism23 and virulence24 in several bacteria are a result of lysogenic conversion. Thus, before using phages as therapeutics, it is important to characterize them at the genomic and phenotypic levels.
Although viruses are the most abundant biological entities on earth and are implicated in various important ecological processes25, their genomic sequences represent only a small fraction in public databases. For example, only 10 complete genome sequences of phages infecting A. salmonicida are currently available (phages 25, 56, 31, 65, 44RR2.8t, Aes508, AS4, AS5, AS7, and PX29). These phages are classified in the Myoviridae family (dsDNA genome, long contractile tail), except phage AS7, which is a member of the Podoviridae family (dsDNA genome, short tail). The genomes of five Aeromonas phages (Aeh1, Aes012, CC2, pAh6-C, ΦO18P) infecting other species (hydrophila, media) are also available in GenBank.
Based on comparative genomic analyses with other phages17, 26, A. salmonicida phages have been classified by the International Committee on Taxonomy of Viruses (ICTV) into several different taxa. For example, the Secunda5virus genus of the Myoviridae family includes the viral species Aeromonas virus 25, Aeromonas virus 31, Aeromonas virus Aes508, and Aeromonas virus AS4. The Biquartavirus genus of the Myoviridae family comprises the species Aeromonas virus 44RR2.8t. In addition, there are several species of Aeromonas myophages that have yet to be assigned a genus, such as Aeromonas virus 65.
Here, we have significantly increased the genomic information of Aeromonas phages. The complete genomes of 12 phages infecting A. salmonicida subsp. salmonicida were obtained and analyzed. The antimicrobial potential of these phages was also evaluated against a panel of 65 strains of A. salmonicida. Additionally, we propose a revised classification scheme for phages infecting A. salmonicida subsp. salmonicida.
Results and Discussion
A. salmonicida phages
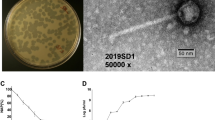
One of the goals of this study was to isolate and characterize new virulent phages able to infect A. salmonicida subsp. salmonicida and to assess the diversity of phages infecting this species. Water samples were collected from three rivers, including two passing through fish farms within the Province of Quebec (Canada). All samples tested were found to contain A. salmonicida subsp. salmonicida phages. Three phages were purified and named SW69-9, L9-6 and Riv-10. As shown in Fig. 1, the three phages belong to the Myoviridae family with an elongated capsid and a contractile tail. As determined by one-step growth curves (see Supplementary Fig. S1), the burst sizes were very low at 3, 2 and 2 new virions per infected cell for phages SW69-9, L9-6 and Riv-10, respectively while the latent period was long at 145, 150 and 142 minutes. Such low burst sizes were not unexpected since Aeromonas virus 31, which is genetically close (see Pan-genome analysis section), is known to have a burst size of 7 new virions per infected cell27, 28.

Micrographs of phages SW69-9, L9-6 and Riv-10. The average head/tail length and diameter are indicated below each phage. The bars represent 50 nm.
Nine other virulent phages infecting A. salmonicida were obtained from the Félix d’Hérelle Reference Center for Bacterial Viruses (phage 3 (HER84), 31 (HER105), 32 (HER106), 51 (HER108), 56 (HER109), 59.1 (HER100), 65 (HER110), 44RR2.8t (HER98), and Asp37 (HER99)). The electron micrographs of these phages are available online on the Félix d’Hérelle Reference Center website (http://www.phage.ulaval.ca) and are all classified in the Myoviridae family.
Genomic characterization
The complete genomes of the 12 phages (9 from the Félix d’Hérelle Reference Center for Bacterial Viruses and the 3 phages isolated from the environmental samples in this study) were sequenced using Illumina technology and de novo assembled, resulting in a final database of 18 A. salmonicida subsp. salmonicida phage genomes, which includes the six genomes already available through GenBank, namely the podophage AS7 and the myophages 25, Aes508, AS4, AS5, and PX29 (Table 1). It should be noted that we resequenced the genomes of phages 31, 56 [deposited as vB_AsaM-56], 65, and 44RR2.8t and subsequently renamed them 31.2, 56, 65.2 and 44RR2.8t.2. Non-synonymous mutations were present between the two versions of the four genomes (see Supplementary Table S1), suggesting that either these phages have evolved or that these discrepancies are due to the use of a different sequencing technology and genome assembly software.
A striking feature was the pronounced divergence in GC% between phages with small (56% ± 1.29) or medium/large (42.27% ± 2.19) genome (Table 1). These results were surprising because in general the GC% of phage genomes tend to correspond with the GC content of their bacterial hosts29, which in the case of A. salmonicida subsp. salmonicida, is 58.5%. A previous study on codon co-evolution showed that A. salmonicida phage genomes with small GC% encode tRNA genes that permit the phages to presumably bypass codons overused in the phage genes, since the repertoire of tRNAs from the host is likely inadequate to translate efficiently phage mRNAs30. Our larger dataset of phages allowed us to investigate whether phages with low or high GC% can be statistically differentiated based on (1) the relative synonymous codon usage (RSCU) and (2) the amino acids composition. Principal component analysis (PCA) showed in both cases a clear separation between genomes having low or high GC% (see Supplementary Fig. S2). Interestingly, although the genome with the lowest GC% (phage 65.2 with 37%) generally clustered with other low-GC% genomes (see Pan-genome analysis section), it was a distinct outlier, which is concordant with its extreme GC%. Also, an analysis of our dataset showed a perfect correlation between the presence or absence of tRNA genes and the GC% of the genomes (Table 1).
Pan-genome analysis
The pan-genome (i.e., whole gene repertoire of a study group) of our current phage dataset was determined. An analysis that included all the ORFs from all 18 phage genomes (3,599 sequences) grouped them into 1,222 clusters. As expected considering phage diversity, no cluster contained ORFs from all phage genomes. Consequently, there is no core-genome when one takes all 18 genomes into consideration (see Supplementary Fig. S3). Although via manual curation we were able to identify a large terminase subunit that was present in all of the genomes, the percent sequence similarity was so low between some sequences (see Supplementary Fig. S4) that they were not considered as homologous by GET_HOMOLOGUES (see Methods section). However, even with a high degree of sequence divergence, phylogenetic clusters based on large terminase subunit sequences are well known to correlate with DNA packaging strategies31. We assembled a database of the large terminase subunit sequences of the 18 A. salmonicida phages from this study and 78 other terminase sequences from phages known to have different DNA packaging strategies, many of which were experimentally validated, to generate a large-scale molecular phylogeny (see Supplementary Fig. S5). The results of this analysis suggest that most of the 18 phages from our study have a headful packaging strategy, although one phage (AS7) has short direct terminal repeats (DTRs) and four (3, Asp37, 32 and 59.1) use 5′ protuberant cos ends. These findings support the view that a diverse phage population infects strains of A. salmonicida.
However, it also complicates their phylogenetic analysis because the sequences do not share a sufficient number of valuable sites to infer robust relative phylogenetic positions among the phages without introducing a clear functional bias. Even by using a Bayesian phylogeny approach with the site-heterogeneous model CAT32, an approach known to reduce artefacts due to long branch attraction33, we recovered a tree topology with poor statistical support where the podophage AS7 was basal to the clade comprised of phages with medium and large genomes (see Supplementary Fig. S6A). We assessed the potential saturation of the phylogenetic matrix by plotting the uncorrected p-distance with the pairwise distances in branch lengths from the tree34. Since the resulting plot clearly showed a non-linearity, we concluded that any true evolutionary signals were obfuscated by saturation and homoplasy (see Supplementary Fig. S6B). In order to bypass this issue, we encoded the pan-genome matrix generated by the previous analysis in binary (i.e. presence/absence of gene clusters) and then performed hierarchical clustering (Fig. 2). The nodes within this tree were well supported (Approximately Unbiased (AU) p-value greater than 95), with some exceptions. With this approach, the phage genomes clustered based on their size (small: 45,329 bp ± 2,502; medium: 169,191 bp ± 6,036; large: 227,947 bp ± 7,641). By coupling this tree with a sequence alignment (Fig. 2), we were able to classify the phages into six groups (I to VI), with the fourth group further divided into subgroups A and B. We supported this grouping by combining the tree with a resulting identity matrix based on BLASTP (see Supplementary Fig. S7). Interestingly, phages also clustered together based on their predicted mode of DNA packaging (Fig. 2). These results show that even if phages infect the same host, they may exhibit considerable genomic diversity.

Clustering based on the gene repertoire. Phages having a small, medium and large size genome are in red, green and blue, respectively. The AU (Approximately Unbiased) p-value is indicated at each node when inferior to 100. Alignment of genomes and the proposed grouping are coupled to the tree. The inferred mode of DNA packaging is indicated for each cluster.
The group III contained the only Aeromonas phage of the Podoviridae family. Our phylogeny clustered, Aeromonas viruses 25, Aes508, and AS4 into the same subgroup (IV-B), indicating that they are closely related, and likely in the same species. In contrast the ICTV has classified these three phages as different species albeit in the same genus (Secunda5virus). Moreover, Aeromonas viruses 31.2 and 44RR2.8t.2, which are also considered different species by the ICTV, clustered in subgroup IV-A along with the three new phages isolated in this study (Riv-10, SW69-9, and L9-6). As shown in Fig. 2 and with a focus on the group IV-A (see Supplementary Fig. S8), the new isolated phages share genomic features, even if they were isolated from various sites within the Province of Quebec. It is also interesting to note that the three new phages from the Province of Quebec are as genetically distant between them (~97.8% of whole genome identity) as Aeromonas virus 31.2 is from 44RR2.8t.2 (97.2% of whole genome identity). The latter were isolated in France and Ontario, respectively (see Supplementary Fig. S9) and are the most similar phage genome sequences available in GenBank. Phages Asp37, 3, 32 and 59.1 were clustered in group I, while phages 51 and 56 were combined in group II. Phages PX29 and AS5 were found in group V and finally, phage 65.2 was placed in group VI. Clearly, ICTV uses a very stringent approach to speciate phages, which otherwise seem highly similar at the genomic level. A 95% DNA sequence identity (BLASTN algorithm) was apparently chosen by the ICTV as the criterion for demarcation of species in the Secunda5virus genus.
Because no gene clusters were shared by all the genomes, we identified the core-genome for each group and verified the number of shared core-clusters among them. We were unable to identify a core cluster among the small genome phages (I, II and III) or shared between the small and the medium size phage genomes (IV-A and IV-B), underlining the extreme sequence heterogeneity among these viruses (see Supplementary Fig. S10). Additionally, we could not identify a core gene cluster among the small genomes and group V, a group that contains phages with larger genomes. There is a small core-cluster, however, shared between the genome of phages AS7 (III) and 65.2 (VI). This is surprising given that AS7 belongs to the Podoviridae family while 65.2 is a myophage. The shared gene encodes a hypothetical protein without any known function. A BLASTP analysis uncovered an orthologous gene in the genome of CC2, a phage that infects Aeromonas hydrophila 35. But again, this putative phage protein has yet to be assigned a function.
The medium and large size genomes are less heterogeneous in their gene repertoire and thus allowed us to identify a larger core genome (Fig. 3A). Our analysis identified 37 clusters that were shared amongst medium and large genomes. These genes were grouped by functional categories accordingly to another study (Fig. 3B)36. More than half (~66%) of the coding sequences (CDSs) can be grouped into two categories: non-structural (i.e. DNA replication, recombination, repair, packaging), and structural (capsid, tail etc.) proteins. Interestingly, 16% of the CDSs could not be assigned to a functional category. These CDSs included four hypothetical proteins, a putative nicotinamide phosphoribosyl transferase and a lysozyme.

Shared core-clusters analysis. The shared core-clusters between medium (IV-A and IV-B) and large (V and VI) genomes are represented as a Venn diagram (A). The 37 core-clusters shared by the medium- and large-size genomes were grouped by functional categories (B). The categories are: (A) Transcription, (B) Translation, (C) Nucleotide metabolism, (D) DNA replication, recombination, repair, packaging, and processing, (E) Virion proteins, (F) Chaperonins/assembly catalysts and (L) Others.
Genes under positive selection
Our next objective was to investigate whether the orthologous genes that we had identified in the previous analysis were under positive selection (also known as diversifying or Darwinian selection). A standard procedure to quantify the selection of a gene is by calculating the dN/dS ratio, dS being the synonymous substitution rate (assumed to be neutral) and dN being the non-synonymous substitution rate (an indicator of positive selection since the amino acid composition of the encoded protein was modified)37. When two phages co-infect a bacterial cell, their genomes may exchange genetic segments through recombination38. Recombination can drive genome evolution but also bias in the detection of selection by increasing the number of false positively selected sites38. We evaluated the recombination events and partitioned each gene based on where these events may have taken place. We then used an algorithm optimized to infer positive selection from the recombining coding sequences identified in the previous analysis.
Our results identified five phage genes that were significantly (p < 0.05) under positive selection (Table 2). The gene under positive selection with the smallest p-value was ndd, a gene that encodes a protein implicated in the disruption of the bacterial nucleoid39. The second most significant gene under positive selection was gene 6, encoding the baseplate protein gp6. This protein is likely present in multiple copies and forms a continuous ring around the central hub while playing a critical role in the assembly and function of the baseplate40, 41. During the infection of an Escherichia coli cell by the myophage T4, six long-tail fibers interact reversibly to the cell-surface of the host, followed by the attachment of short-tail fibers that bind to additional receptors on the cell surface in an irreversible fashion. The attachment of the short-tail fibers triggers a conformation change of the baseplate from a dome-shaped to a star-shaped42. Recent studies have shown that gp6 is one of the key proteins in the signal transmission from the short-tail fibers to the central region of the baseplate during this conformational shift40, 41. Gene 6 was found in medium and large size genomes, with the exception of phage AS4. Deeper investigation led to the identification of a 6-like gene within the genome of AS4, but with multiple frameshifts. It is tempting to speculate that these frameshifts may be the result of sequencing artefacts given the vital role of gp6. It is not clear why gene 6 is under positive selection, but if gp6 is implicated in host infection as it appears, mutations to this gene may alter attachment kinetics of the phage to the cell surface, leading to changes in host specificity.
The last three genes under positive selection code for a hypothetical protein, the clamp-loader subunit gp44 and a topoisomerase II large subunit. It is worth noting that the gene encoding the hypothetical protein is the only gene from phages with small genomes found to be under positive selection. Additionally, the topoisomerase II large subunit is the product of two genes (39 and 60) in phage T443 and thus may be a genomic region under disproportionate positive selection.
Host range of the phages
Temperate phages are able to integrate their genomes into the chromosome of their host. In some instances, these phage-encoded genes are used by the host to its own advantage. We investigated each of the 18 genomes for the presence of an integrase gene, the hallmark of temperate phages. A gene coding for an integrase was found in all the genomes of phages in group I (3, Asp37, 59.1, and 32). This suggests that these phages are likely capable of lysogeny and are therefore not good candidates for phage therapy. The genomes of phages 51 and 56, both from the group II, appeared to harbour a gene encoding for a truncated integrase, however we have low confidence in the gene’s identity. The podophage AS7 (group III) is the only phage having a small genome that does not harbour a gene encoding for an integrase. No genes coding for antibiotic resistance and/or virulence factors were identified in any genome.
We assessed the host range of the 12 phages with a panel of 65 A. salmonicida isolates (Fig. 4). To investigate the specificity of these phages, isolates from subspecies other than salmonicida were added: smithia, masoucida, pectinolytica as well as three Indian strains suspected of being part of a new A. salmonicida subspecies4. The resulting host range patterns coupled to a clustering (heatmap) allowed us to classify the phages into three groups.

Clustering and heatmap based on a panel of 65 A. salmonicida isolates challenged with 12 phages. The genomic cluster of each phage is indicated in parentheses. The mesophilic isolates are shown in red while the psychrophilic ones are in black. High and low lytic activities are represented in purple and yellow, respectively. In addition, the dilution ranges obtained by spot tests used to encode the matrix are indicated below the legend.
The first group contained phages (phages 59.1 (genomic cluster I) and 56 (II)) with the narrowest host range, where only few bacterial isolates (<5 out of 65) were sensitive to the phages based on spot tests (Fig. 4). As indicated previously, the genome of phage 59.1 possesses an integrase gene while it is not clear if phage 56 has a functional integrase gene. Neither of these phages was reported as temperate by the original studies that characterized them10,14. However, the bioinformatics tool PHACTS45, which predict the lifestyle of a phage, indicated that these two phages are likely temperate.
The second group, composed of phages 32 (group I), 3 (group I), 65.2 (group VI) and Asp37 (group I), displayed an intermediate host range where 10 to 30 bacterial isolates were sensitive to the phages (Fig. 4). Phages 32, 3 and Asp37 have a gene encoding for an integrase, consequently with the potential to be temperate under some conditions. Even though we were unable to identify a gene coding for an integrase in the phage 65.2 genome, it has the highest GC% discrepancy (37%) with the genome GC% of its host (58.5%), raising the possibility that this phage is less efficient at lysing its host.
The third group included phages within the genomic cluster group IV-A and phage 51 (cluster II). Members of this group had the broadest host range (>44 out of 65) (Fig. 4). Consequently, phages from this group have the most potential from a phage therapy perspective. For example, phage 44RR2.8t.2 infected 57 out of the 65A. salmonicida strains. Similarly, the newly isolated phage SW69-9 infected 56 out of the 65 strains. The host range of phages 44RR2.8t.2 and SW69-9 was overlapping and as such, all A. salmonicida subsp. salmonicida strains could be infected by one of the two phages or both (Fig. 4).
Surprisingly, phage 51 was able to lyse 41 strains, even if the original study reported it to be temperate10. The tool PHACTS also predicted the lifestyle of this phage as temperate, but with an uncertain probability (0.515 ± 0.042). This was even more surprising because the other phage within the cluster II, phage 56, had a very limited host range (lysed 4 strains). A global alignment of both phage genomes (51 and 56) resulted in only four non-synonymous SNPs. The first two were located in genes coding for hypothetical proteins, each having a predicted conserved unknown domain (DUF2213 and DUF2184). While the last two SNPs are in a gene encoding for a tail or truncated integrase protein. One SNP led to change from a methionine to a valine and the other one from a proline to leucine. Further work is required to determine which mutation(s) is responsible for the expanded host range.
Strains belonging to other psychrophilic subspecies (smithia and masoucida) were also sensitive to phages infecting the subspecies salmonicida. However, the three mesophilic strains from India4 (Y577, Y567 and Y47) and the strain of the subspecies pectinolytica (also having a mesophilic lifestyle5) were insensitive to all 12 phages, at the exception of pectinolytica, which is sensitive to phage 3. The A-layer, which is a structure composed of a protein and lipopolysaccharides implicated in virulence11, could be one of the phage receptors in A. salmonicida 12. Analysis of the various bacterial genomes showed that the gene vapA, encoding the protein forming the A-layer, is present in psychrophilic isolates but absent in mesophilic isolates. In order to better understand host phage dynamics in mesophilic A. salmonicida, it could be interesting to isolate phages able to infect them and to identify the phage receptor.
It is well documented that prophages can provide resistance to infection from other phages, by superinfection exclusion (Sie) systems found in both Gram negative and positive bacteria46. At the exception of the mesophilic/psychrophilic separation, the heatmap in Fig. 4 shows no clear bacterial clustering suggesting that such resistance could be provided (see Supplementary Fig. S11). This is of interest because A. salmonicida subsp. salmonicida isolates usually harbour two prophages sharing structural similarities with the temperate phage ΦO18P 47, found in Aeromonas media. Moreover some A. salmonicida subsp. salmonicida strains also have a new recently discovered prophage (prophage 3) and variants of a genomic island named AsaGEI 2, 3, 48. None of these mobile elements actually have a known function and we can reasonably here rule out their implication in protection against phages.
In conclusion, we investigated the genomes of 18 phages infecting A. salmonicida and characterized their diversity, which led to a robust classification scheme based on their genomic composition. We argue that our approach to classifying these phages will result in a more accurate characterization and classification of new A. salmonicida phages in the future. We also evaluated the infectivity of 12 phages, including three newly isolated phages, on a panel of 65 isolates of A. salmonicida. Overall, these phages showed a heterogeneous host range. Phages with overlapping and large host range were identified and hold potential to contribute to a phage cocktail to control this fish pathogen in the aquaculture industry.
Methods
Environmental sampling, phage isolation, and characterization
Skin mucus samples from furunculosis-infected fishes from four fish farms in the province of Quebec (Canada) as well as water samples (50 ml) from the same fish farms and rivers in the same region were collected and kept at 4 °C. The mucus was swabbed and diluted in 5 ml of phage buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 8 mM MgSO4). Each environmental sample was centrifuged for 10 min at 3,200 × g and filtered (0.45 μm, Sarstedt, Canada). The filtrate was then mixed with the same volume of 2X TSB (EMD Millipore, Canada) and inoculated with 1% (v/v) of A. salmonicida subsp. salmonicida in exponential growth phase. The bacterial strains used were those recommended by the Felix d’Hérelle Reference Center for Bacterial Viruses (http://www.phage.ulaval.ca). All cultures were incubated at 18 °C overnight and agitated at 200 RPM before being filtered again. The above amplification procedure was repeated four times or until the appearance of cell lysis (replacing 2X TSB with 1X TSB after each round). In parallel, controls without environmental sample were inoculated with the bacterial host to compare the growth and whether lysis had occurred in challenged host cultures.
The new phages were isolated from single plaques as follows: in 3 ml of TSB soft agar (0.75%) kept at 55 °C, 100 μl of filtrate and 100 μl of bacterial host (A. salmonicida subsp. salmonicida 08-2783 host of L9-6 and A. salmonicida subsp. salmonicida M15879-11 host of SW69-9 and Riv-10) were mixed and poured onto TSA plates (EMD Millipore, Canada) before being incubated at 18 °C overnight. Plaques (up to 10 per sample) were picked up with sterile tips and suspended in 500 μl of phage buffer. Phages were allowed to diffuse for 30 min at room temperature before serial dilutions in phage buffer. The above plaque isolation procedure was repeated two more times. Subsequently, phages were amplified in 10 ml of TSB incubated with bacterial host at 18 °C overnight and agitated at 200 RPM. Aliquots of the resulting filtrates were either stored at −80 °C in 15% glycerol or stored at 4 °C for up to six months until subsequent analysis.
Transmission electron microscopy was used to observe the three new phages as described elsewhere49. Briefly, 1.5 mL of phage lysate was centrifuged at 23,500 × g for 1 h at 4 °C and the pellet washed twice with ammonium acetate (0.1 M, pH 7.0). The resulting phage preparation was then used to prepare observation grids, which were stained with uranyl-acetate (2%) and observed with a JEOL 1230 at the microscopy platform of the Institut de Biologie Intégrative et des Systèmes (U. Laval). Capsid size and tail length were determined by measuring at least 15 different specimens.
Phage one-step growth curve assays of phages were performed in triplicate, as previously reported elsewhere50. Approximately 109 CFU/ml of precultured cells that just reached stationary phase were harvested by centrifugation and resuspended in 900 µl of TSB. Each phage was respectively added at a multiplicity of infection (MOI) of 0.05 and allowed to adsorb for 20 minutes at 18 °C. Then, infected cells were harvested by centrifugation and the pellet washed twice with 1 ml of fresh TSB. Finally, the pellet containing the infected cells was resuspended in a final dilution of 0.001 in a glass tube containing 10 ml of TSB and incubated at 18 °C at 200 RPM. Every 30 min, an aliquot of 100 µl was taken to determine the phage titer, up to 210 minutes. The burst size was calculated by dividing the average phage titer after the exponential phase by the average titer before the infected cells began to release virions51. The latent period was evaluated according to the median of the exponential curve.
The DNA of the amplified phages was extracted using a standard phenol/chloroform protocol and then analyzed by restriction profiles using the enzymes DraI, SspI and MseI (NEB, Canada) according to the manufacturer recommendations at 37 °C for one hour. Restricted phage DNA was run on a 1% agarose gel for 30 min at 90 V. Three different restriction profiles were observed and the distinct phages were named SW69-9, L9-6 and Riv-10.
Sequencing and de novo assembly
Sequencing libraries were prepared from purified phage DNA using the Nextera XT DNA Library Preparation Kit and sequenced on an Illumina MiSeq apparatus. The resulting sequencing reads were de novo assembled by the pipeline A5-miseq version 2015052252 to obtain an initial sequencing depth. Given their small size and the high-throughput capabilities of the Illumina platform, viral genomes are usually sequenced with very high depth, causing an unnecessary complexity for subsequent de novo assembly53. Consequently, the reads of each sequenced genome were randomly down-sampled by seqtk (https://github.com/lh3/seqtk) to obtain assemblies (also with A5-miseq version 20150522) having around 100X of sequencing depth.
We compared the resequenced phage genomes 44RR2.8t, 31, 56 and 65 by generating kmers with a length of 300 nt and then mapping them with BWA version 0.7.12-103954 to reference sequences. Mutations were called by using samtools version 0.1.19-44428cd55 and VarScan version 2.4.256. Finally, the effect of mutations was evaluated based on SnpEff version 4.257 (with the reference sequences added beforehand in the database).
Annotation, pan-genome and other bioinformatics analyses
Each genome, including those already available in GenBank, was annotated through the webserver RAST58 by choosing the “virus” parameter. A list of all predicted genes is available for each new phage: SW69-9, L9-6 and Riv-10 (see Supplementary Table S2). Annotated CDSs were downloaded and evaluated by GET_HOMOLOGUES version 2.0.1644. The sequences were clustered by using the COG and OMCL algorithms, both included in GET_HOMOLOGUES. Only the clusters found by both algorithms were used for downstream analyses (see Supplementary Fig. S12). The presence/absence binary matrix was evaluated by Pvclust59 under the binary distance method with 10,000 bootstrap replicates through the statistical framework R (https://www.r-project.org) resulting in a clustering of the phages based on their gene repertoire. An identity matrix was calculated with the BLASTP scores among protein sequences using also GET_HOMOLOGUES.
Genes under positive selection and other analysis
We codon aligned the nucleotide sequences corresponding to each cluster found by GET_HOMOLOGUES and having at least four sequences by PRANK version 15080360. Alignments were then evaluated by GARD through HyPhy version 2.2.461 to find potential recombination events. Finally, positive selection was evaluated with the PARRIS algorithm, also through HyPhy version 2.2.4.
Each genome was screened for antibiotic resistance genes by the RGI tool of the webserver CARD62. Homology searches were performed locally with BLASTP between all the CDSs annotated by RAST and the curated PATRIC_VF database63 which contained 1,570 sequences of CDSs known to be implicated in virulence (downloaded on July 8th 2016) and also a database containing all integrase sequences available in the Protein database of the NCBI (1, 347, 621 sequences on July 8th, 2016).
Host range
The host range of 12 phages was determined using a set of 65 isolates of A. salmonicida (see Supplementary Table S3). Cells stocks were thawed, streaked on TSA plates and grown for three days at 18 °C. Isolated colonies were inoculated in 3 ml TSB in sterile snap cap tubes and incubated overnight at 18 °C and agitated at 200 RPM. For each strain, 100 μl of overnight bacterial culture was added to 3 ml of soft agar and poured onto a TSA plate. Ten-fold serial dilutions of each phage (up to 10−7) were done in phage buffer and 5 μl of each dilution was spotted onto the inoculated TSA plates. The plates were incubated at 18 °C overnight and the sensitivity or insensitivity was recorded.
References
Dallaire-Dufresne, S., Tanaka, K. H., Trudel, M. V., Lafaille, A. & Charette, S. J. Virulence, genomic features, and plasticity of Aeromonas salmonicida subsp. salmonicida, the causative agent of fish furunculosis. Vet. Microbiol. 169, 1–7 (2014).
Emond-Rheault, J.-G. et al. Variants of a genomic island in Aeromonas salmonicida subsp. salmonicida link isolates with their geographical origins. Vet. Microbiol. 175, 68–76 (2015).
Emond-Rheault, J.-G. et al. AsaGEI2b: a new variant of a genomic island identified in the Aeromonas salmonicida subsp. salmonicida JF3224 strain isolated from a wild fish in Switzerland. FEMS Microbiol. Lett. 362, fnv093 (2015).
Vincent, A. T. et al. Increasing genomic diversity and evidence of constrained lifestyle evolution due to insertion sequences in Aeromonas salmonicida. BMC Genomics 17, 1–12 (2016).
Pavan, M. E., Abbott, S. L., Zorzópulos, J. & Janda, J. M. Aeromonas salmonicida subsp. pectinolytica subsp. nov., a new pectinase- positive subspecies isolated from a heavily polluted river. Int. J. Syst. Evol. Microbiol. 50, 1119–1124 (2000).
Koskella, B. & Brockhurst, M. A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931 (2014).
Klimenko, A. I., Matushkin, Y. G., Kolchanov, N. A. & Lashin, S. A. Bacteriophages affect evolution of bacterial communities in spatially distributed habitats: a simulation study. BMC Microbiol. 16, 31–41 (2016).
Stern, A. & Sorek, R. The phage-host arms race: Shaping the evolution of microbes. BioEssays 33, 43–51 (2011).
Todd, C. The Presence of a Bacteriophage for B. salmonicida in River Waters. Nature 131, 360 (1933).
Popoff, M. & Vieu, J.-F. Bactériophages et lysotypie d’Aeromonas salmonicida. Comptes rendus l’Académie des Sci. 270, 2219–2222 (1970).
Kay, W. W. et al. Purification and disposition of a surface protein associated with virulence of Aeromonas salmonicida. J. Bacteriol. 147, 1077–1084 (1981).
Ishiguro, E. E., Ainsworth, T., Shaw, D. H., Kay, W. W. & Trust, T. J. A lipopolysaccharide-specific bacteriophage for Aeromonas salmonicida. Can. J. Microbiol. 29, 1458–1461 (1983).
Kim, J. H. et al. Complete Genome Sequence of Bacteriophage phiAS7, a T7-Like Virus That Infects Aeromonas salmonicida subsp. salmonicida. J. Virol. 86, 2894–2895 (2012).
Paterson, W. D., Douglas, R. J., Grinyer, I. & McDermott, L. A. Isolation and Preliminary Characterization of Some Aeromonas salmonicida Bacteriophages. J. Fish. Res. Board Canada 26, 629–632 (1969).
Kim, J. H. et al. Complete genomic sequence of a T4-like bacteriophage, phiAS4, infecting Aeromonas salmonicida subsp. salmonicida. Arch. Virol. 157, 391–395 (2012).
Kim, J. H. et al. Complete genome sequence and characterization of a broad-host range T4-like bacteriophage phiAS5 infecting Aeromonas salmonicida subsp. salmonicida. Vet. Microbiol. 157, 164–171 (2012).
Petrov, V. M., Ratnayaka, S., Nolan, J. M., Miller, E. S. & Karam, J. D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 7, 292 (2010).
Ishiguro, E. E., Kay, W. W. & Trust, T. J. Temperate bacteriophages for Aeromonas salmonicida. FEMS Microbiol. Lett. 8, 247–250 (1980).
Ishiguro, E. E., Ainsworth, T., Harkness, R. E., Kay, W. W. & Trust, T. J. A temperate bacteriophage specific for strains of Aeromonas salmonicida possessing A-layer, a cell surface virulence factor. Curr. Microbiol. 10, 199–202 (1984).
Reardon, S. Phage therapy gets revitalized. Nature 510, 15–16 (2014).
Richards, G. P. Bacteriophage remediation of bacterial pathogens in aquaculture: a review of the technology. Bacteriophage 4, e975540 (2014).
Modi, S. R., Lee, H. H., Spina, C. S. & Collins, J. J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499, 219–22 (2013).
Paul, J. H. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2, 579–589 (2008).
Waldor, M. K. & Mekalanos, J. J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914 (1996).
Sime-Ngando, T. Environmental bacteriophages: viruses of microbes in aquatic ecosystems. Front. Microbiol. 5, 1–14 (2014).
Comeau, A. M. et al. Phage morphology recapitulates phylogeny: the comparative genomics of a new group of myoviruses. PLoS One 7, e40102 (2012).
Popoff, M. Etude sur les Aeromonas salmonicida. II. Characterisation des bacteriophages actifs sur les Aeromonas salmonicida et lysotypie. Ann. Rech. Vet 2, 35–45 (1971).
Popoff, M. Study of Aeromonas salmonicida II Characterization of the Phages Acting on Aeromonas salmonicida and Phage-typing, at http://www.dfo-mpo.gc.ca/Library/26450.pdf (1973).
Xia, X. & Yuen, K. Y. Differential selection and mutation between dsDNA and ssDNA phages shape the evolution of their genomic AT percentage. BMC Genet. 6, 20 (2005).
Prabhakaran, R., Chithambaram, S. & Xuhua, X. Aeromonas phages encode tRNAs for their overused codons. Int. J. Comput. Biol. Drug Des. 7, 168–182 (2014).
Merrill, B. D., Ward, A. T., Grose, J. H. & Hope, S. Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genomics 17, 679 (2016).
Lartillot, N. & Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109 (2004).
Lartillot, N., Brinkmann, H. & Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 7(Suppl 1), S4 (2007).
Borowiec, M. L., Lee, E. K., Chiu, J. C. & Plachetzki, D. C. Extracting phylogenetic signal and accounting for bias in whole-genome data sets supports the Ctenophora as sister to remaining Metazoa. BMC Genomics 16, 987 (2015).
Shen, C., Liu, Y. & Lu, C. Complete Genome Sequence of Aeromonas hydrophila Phage CC2. J. Virol. 86, 10900 (2012).
Miller, E. S. et al. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156 (2003).
Mugal, C. F., Wolf, J. B. W. & Kaj, I. Why time matters: Codon evolution and the temporal dynamics of dN/dS. Mol. Biol. Evol. 31, 212–231 (2014).
Pérez-Losada, M., Arenas, M., Galán, J. C., Palero, F. & González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30, 296–307 (2015).
Snustad, D. P. et al. Mutants of bacteriophage T4 deficient in the ability to induce nuclear disruption. J. Mol. Biol. 89, 675–687 (1974).
Yap, M. L. et al. Role of bacteriophage T4 baseplate in regulating assembly and infection. Proc. Natl. Acad. Sci. 113, 2654–2659 (2016).
Taylor, N. M. I. et al. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 533, 346–352 (2016).
Kostyuchenko, V. A. et al. Three-dimensional structure of bacteriophage T4 baseplate. Nat. Struct. Biol. 10, 688–693 (2003).
Huang, W. M., Wei, L. S. & Casjens, S. Relationship between bacteriophage T4 and T6 DNA topoisomerases. T6 39-protein subunit is equivalent to the combined T4 39- and 60-protein subunits. J. Biol. Chem. 260, 8973–8977 (1985).
Contreras-Moreira, B. & Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701 (2013).
McNair, K., Bailey, B. A. & Edwards, R. A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 28, 614–618 (2012).
Labrie, S. J., Samson, J. E. & Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–27 (2010).
Beilstein, F. & Dreiseikelmann, B. Temperate bacteriophage PhiO18P from an Aeromonas media isolate: characterization and complete genome sequence. Virology 373, 25–9 (2008).
Long, M. et al. Aeromonas salmonicida subsp. salmonicida strains isolated from Chinese freshwater fish contain a novel genomic island and possible regional-specific mobile genetic elements profiles. FEMS Microbiol. Lett. 363, fnw190 (2016).
Fortier, L. C. & Moineau, S. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 73, 7358–7366 (2007).
Wang, J. B, Lin, N. T., Tseng, Y. H. & Weng, S. F. Genomic characterization of the novel Aeromonas hydrophila phage ahp1 suggests the derivation of a new subgroup from phiKMV-Like family. PLoS One 11 (2016).
Moineau, S., Durmaz, E., Pandian, S. & Klaenhammer, T. R. Differentiation of two abortive mechanisms by using monoclonal antibodies directed toward lactococcal bacteriophage capsid proteins. Appl. Environ. Microbiol. 59, 208–212 (1993).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 5–8, doi:10.1093/bioinformatics/btu661 (2014).
Wan, Y., Renner, D. W., Albert, I. & Szpara, M. L. VirAmp: a galaxy-based viral genome assembly pipeline. Gigascience 4, 19 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Koboldt, D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly (Austin). 6, 80–92 (2012).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–14 (2014).
Suzuki, R. & Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170 (2014).
Kosakovsky Pond, S. L., Frost, S. D. W. & Muse, S. V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (2005).
McArthur, A. G. et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357 (2013).
Mao, C. et al. Curation, integration and visualization of bacterial virulence factors in PATRIC. Bioinformatics 31, 252–258 (2015).
Acknowledgements
We would like to thank Alexander Culley (U. Laval) for editorial assistance. We also would like to thank André Savard and Jeff Gauthier (U. Laval) for their help during the water sampling campaign and Bryan Merrill (Brigham Young University) for providing curated terminase sequences and advices regarding DNA packaging strategies. We acknowledge funding from the Ministère de l’agriculture, des pêcheries et de l’alimentation du Québec (INNOVAMER Program), the Natural Sciences and Engineering Research Council of Canada (NSERC) and Ressources Aquatiques Québec (RAQ). A.T.V. received an Alexander Graham Bell Canada Graduate Scholarships from the NSERC. S.J.C. is a research scholar from the Fonds de Recherche du Québec en Santé (FRQS). S.M. holds a Tier 1 Canada Research Chair in Bacteriophages.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: A.T.V., V.E.P., D.M.T., S.M., and S.J.C. Performed the experiments: A.T.V., V.E.P., A.B., and D.M.T. Analyzed the data: A.T.V., V.E.P., A.B., D.M.T., S.M., S.J.C. Contributed reagents/materials/analysis tools: A.T.V., V.E.P., and D.M.T. Wrote the paper: A.T.V., V.E.P., S.M., and S.J.C. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vincent, A.T., Paquet, V.E., Bernatchez, A. et al. Characterization and diversity of phages infecting Aeromonas salmonicida subsp. salmonicida . Sci Rep 7, 7054 (2017). https://doi.org/10.1038/s41598-017-07401-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07401-7
This article is cited by
-
Centroid of the bacterial growth curves: a metric to assess phage efficiency
Communications Biology (2024)
-
Isolation of vB_AsaM_LPM4 reveals the dynamics of Prophage 3 in Aeromonas salmonicida subsp. salmonicida
Archives of Virology (2023)
-
Complete genome sequence analysis and phylogenetic classification of the novel Aeromonas phage AHP-1, a potential member of the genus Tequatrovirus
Archives of Virology (2022)
-
Characterization of Novel Bacteriophage AhyVDH1 and Its Lytic Activity Against Aeromonas hydrophila
Current Microbiology (2021)
-
Characterization of bacteriophage T7-Ah reveals its lytic activity against a subset of both mesophilic and psychrophilic Aeromonas salmonicida strains
Archives of Virology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
