
Abstract
Mutations in the GDAP1 gene can cause Charcot-Marie-Tooth disease. These mutations are quite rare in most Western countries but not so in certain regions of Spain or other Mediterranean countries. This cross-sectional retrospective multicenter study analyzed the clinical and genetic characteristics of patients with GDAP1 mutations across Spain. 99 patients were identified, which were distributed across most of Spain, but especially in the Northwest and Mediterranean regions. The most common genotypes were p.R120W (in 81% of patients with autosomal dominant inheritance) and p.Q163X (in 73% of autosomal recessive patients). Patients with recessively inherited mutations had a more severe phenotype, and certain clinical features, like dysphonia or respiratory dysfunction, were exclusively detected in this group. Dominantly inherited mutations had prominent clinical variability regarding severity, including 29% of patients who were asymptomatic. There were minor clinical differences between patients harboring specific mutations but not when grouped according to localization or type of mutation. This is the largest clinical series to date of patients with GDAP1 mutations, and it contributes to define the genetic distribution and genotype-phenotype correlation in this rare form of CMT.
Similar content being viewed by others


Introduction
Mutations in the ganglioside-induced differentiation-associated protein 1 (GDAP1) gene cause different forms of Charcot–Marie–Tooth disease (CMT). Autosomal recessive mutations have been described in patients with axonal, intermediate and demyelinating forms of the disease, while dominantly inherited mutations cause axonal CMT1,2,3,4. These mutations are quite rare in Western countries, accounting for less than 1% of the genetically defined CMT patients in most clinical series5, 6. In contrast, in certain regions of Spain and Italy, these mutations are the most frequent cause of axonal CMT, accounting for up to 10% of the genetic diagnosis in CMT7,8,9.
The GDAP1 gene encodes a protein belonging to a glutathione S-transferase (GST) enzyme subfamily in chromosome 8q21.110. GDAP1 is localized in the outer mitochondrial membrane, and is composed of two typical GST domains at the N and C-terminal regions, two alpha loops, a single transmembrane domain, and a hydrophobic domain. Although no GST activity has been demonstrated so far, GDAP1 is involved in the regulation of mitochondrial dynamics and calcium homeostasis11, 12.
GDAP1 autosomal recessive (AR) inherited mutations cause a severe, early onset neuropathy often leading to wheelchair-dependency in the second or third decade. Most of these patients develop unilateral or bilateral vocal cord paresis, and diaphragmatic weakness in the latter stages of the disease13. It has been suggested that recessive mutations which cause truncating proteins develop a more severe phenotype, while missense mutations may be associated with a slightly milder course14.
On the other hand, autosomal dominant (AD) inherited mutations cause a much milder phenotype, characterized by adult onset, predominantly distal involvement, and slow progression, most of these patients remaining ambulant throughout their lives15, 16. Certain clinical characteristics, like dysphonia or dysautonomia, have been described in isolated patients, but no clear genotype-phenotype correlation has been established in AD inherited mutations9, 17.
The aim of the study is to describe the distribution of patients with GDAP1 mutations across Spain and to expand the knowledge of the clinical course and the genotype-phenotype correlation.
Results
We identified 99 patients from 46 different families harboring causative GDAP1 mutations. There was no sex predominance in the series and ages ranged from 3–79 years. There were three patients in which only genetic information was available. Nerve conduction studies were performed in 75 patients while other ancillary tests were less frequently performed: leg muscle magnetic resonance imaging in 22 cases and sural nerve pathology in 15.
Genotype
The genetic information is recorded in Table 1. Thirteen different mutations were detected, 3 inherited in an autosomal dominant fashion and the other 10 were recessive. All had been previously reported. The two most common mutations were AD p.R120W, and AR p.Q163X. The type of mutations was quite diverse including 6 missense, 4 nonsense, 2 frameshift mutations and one splice-site variant. They were localized along exons 2–6 and affected the 2 GST domains and the α-loop predominantly. A schematic diagram of the GDAP1 protein and the mutations found in this series is reproduced in Fig. 1.

Localization in the GDAP1 gene of the mutations detected. AD: Autosomal dominant, AR: Autosomal recessive, in blue: missense mutations, in red: truncating mutations.
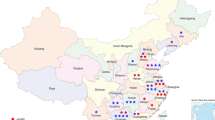
The AD p.R120W was detected predominantly in families near the Mediterranean coast, while p.R226del was found only in two families in the region of Galicia, in the Northwest of Spain (Fig. 2). The AR patients were distributed throughout Spain, but there existed a cluster of affected individuals in the North of Spain, especially in the Basque country and neighboring regions.

Patient distribution throughout Spain AD: Autosomal dominant, AR: Autosomal recessive, light blue diamond: patient with the AD p.R120W mutation, dark blue diamond: patient with the AD p.R226del mutation, medium blue diamond: patient with the AD p.T157P mutation, red square: patient with AR mutations. The map was created with SimpleMappr, an online tool to produce publication-quality point maps. [Retrieved from http://www.simplemappr.net. May 24, 2017]; Shorthouse, David P. 2010.
Clinical characteristics
The clinical features of the series are summarized in Table 2. Patients were separated between AD and AR inherited mutations because all the severity scores and certain clinical features were different between the groups.
Patients with AD inherited mutations had a mild-moderate neuropathy with variable age of onset (4–65 years). The first symptoms most commonly reported were distal lower leg weakness and cramps (83%). Foot deformities (pes cavus, Achilles tendon contracture or hammer toes) were quite common (85.2%), but only one patient presented scoliosis and none dysphonia or respiratory failure. Severity scores were usually between the mild-moderate range, and 28.6% considered themselves asymptomatic. There was important clinical variability regarding severity scores even between family members as can be seen in the pedigree of Fig. 3. Three patients died during follow-up, but the cause of death was not related with the neuropathy.

Clinical variability in a family harboring the AD p.R120W mutation. yrs: years, LL: Lower limbs, CMTNS: Charcot-Marie-Tooth neuropathy score, CMTES: Charcot-Marie-Tooth examination score.
Patients with AR inherited mutations suffered a severe early onset neuropathy (0–12 years) causing a great disability. Most of the patients were wheelchair-bound before age 20, and only three patients older than 40 years remained ambulant. Dysphonia was present in more than half of the AR patients and appeared usually in the second decade (9–35 years). Respiratory dysfunction was also quite common, and 19.5% of the patients required non-invasive ventilation (29–47 years). Intra-familiar variability was not prominent, although most were sporadic patients. Six patients died, two because of cardiovascular events, one of hepatic cirrhosis, and three due to infections that could be related to the severe disability inherent to the disease course.
Nerve conduction studies
The electrophysiological findings are detailed in Table 3. In AD patients there was a decrease of CMAP and SNAP amplitudes with normal conduction velocities, corresponding to a mild axonal motor and sensory neuropathy. Of the 12 clinically asymptomatic patients that underwent nerve conduction studies, in 8 we detected a decrease in SNAP of the sural nerve and/or CMAP of the peroneal nerve. The other 4 asymptomatic patients had normal nerve conduction studies, but 3 of them had detectable abnormalities in needle electromyography (large polyphasic motor unit action potentials in distal lower limb muscles with no spontaneous muscular activity), while it was not performed in the other patient.
In AR patients the decrease in CMAP and SNAP was much more marked, as were the number of nerves that were unexcitable when explored. These findings were more severe in the lower limb nerves; in fact all patients over 25 years had unexcitable sural and peroneal nerves. Conduction velocities were generally not reduced except in nerves with very low amplitudes (Fig. 4b). In these cases, when conduction velocity was measured to proximal muscles, it was always within the normal range.

(a) Dispersion chart of the CMTNS scores and ages in the first examination of patients with AD and AR inherited mutations. (b) Dispersion chart of the CMAP of the median nerve the motor conduction velocity in patients with AD and AR inherited mutations. AD: Autosomal dominant, AR: Autosomal recessive, CMTNS: Charcot-Marie-Tooth neuropathy score, CMAP: Compound muscle action potential.
Muscle magnetic resonance imaging
Lower limb muscle magnetic resonance imaging findings are detailed in Table 4, and representative images are shown in the supplementary Figure S1. It was performed in 22 patients, 19 of them harboring the AD p.R120W mutation. In those patients the findings were in concordance with that previously reported including fatty infiltration with a distal to proximal gradient16. In 3/5 clinically asymptomatic patients, there was detectable fatty substitution in the intrinsic foot muscles and in 2/5 also in the muscles of the calf. In the 2 asymptomatic patients with normal muscle MRI, the muscles of the feet were not studied. In the calf, the muscles of the posterior compartment (soleus > gastrocnemius) were affected earlier and more severely than those of the anterolateral compartment.
MRI was performed in two patients with the AD p.R226del mutation. In one symptomatic patient there were detectable abnormalities consisting in fatty infiltration with a distal to proximal gradient, and the same posterior > anterolateral pattern in the calf. The other was asymptomatic, although minor abnormalities could be detected in the clinical examination. The muscle MRI in this patient was normal in the calf and thigh muscles, but the feet were not studied.
One patient with an AR p.Q163X/p.L344R mutation also underwent muscle MRI testing. He was the AR patient with the less severe phenotype (CMT neuropathy score - CMTNS: 12, Functional disability score - FDS: 2, age 49 at the time of the MRI) and there was complete fatty substitution of the muscles in the feet and posterior compartment of the calf, but in the anterolateral muscles of the calf and the thigh muscles, fatty infiltration was only partial.
Nerve pathology
Sural nerve biopsy was performed in 15 patients, 8 with AR and 7 with AD inherited mutations. Representative images are shown in the supplementary Figure S1. The pathologic characteristics of 7 of these patients had been previously been reported, and are similar to the other 8 patients1, 16, 18. The main findings were concordant with an axonal neuropathy (depletion of myelinated fibers, myelin thinning, and rather frequent regenerative clusters) with minor myelin abnormalities (abnormal myelin foldings and occasional onion bulb-like formations). The fiber loss was especially prominent in AR patients.
Genotype-phenotype correlation
The most important genetic factor that influenced the phenotype was the mode of inheritance. Certain clinical features like the presence of dysphonia and respiratory dysfunction were only present in patients with AR inheritance and scoliosis was more frequent in patients with AR inheritance (p < 0.001) (Table 2). In this group, 75% of the patients were wheelchair-bound at a median age of 15 years. By contrast none in the group of AD inheritance lost ambulation with a median follow-up of nearly 7 years. The presence of any kind of neuropathic symptoms was more frequent in patients with AR inheritance, as were higher severity scores in any of the scales employed (p < 0.05). In Fig. 4a the CMTNS scores of the first examination in both groups can be compared. With a logistic regression model, the only co-variables that could independently predict a higher CMTNS score were the mode of inheritance: OR 38, CI 95% [4–474.2] and the disease duration OR 1.1 CI 95% [1.05, 1.19].
Regarding the patients with AD inheritance, we detected minor clinical differences between the two most frequent mutations. The members of the two families with the p.R226del mutation had a slightly milder disease course. Also, in the p.R120W mutation ankle extensors and flexors were affected to a similar degree (3.9 and 3.7 mean MRC scores respectively) while in those with the p.R226del mutation weakness was more marked in ankle extension (4.3 and 4.7 mean MRC scores).
In the patients with AR inherited mutations no clinical differences could be detected when grouping patients according to localization of the mutation or mutation type. Neither could we detect any statistically significant difference in age of onset, years to wheelchair, or other severity scores between patients with ‘truncating’ mutations and those with two missense mutations. In patients with the same mutation, clinical variability regarding severity was not prominent. Regarding specific mutations, the only two patients with the homozygous p.R282C mutation seemed to have a milder clinical course, as did the only patient compound heterozygous for the p.Q163X and p.L344R mutations.
Discussion
This multicentric cross-sectional study provides information about 99 patients with CMT caused by GDAP1 mutations. In Spain the relative frequency of GDAP1 mutations has been reported to be as high as 13% of the genetically defined CMT in certain regions7. Patients in this series were distributed throughout Spain, and in most centers comprised the first cause of CMT2, excluding GJB1 mutations. This contrasts with the scarcity of these mutations in most Western Countries, accounting for less than 2% of the genetically defined patients in series from the United Kingdom, Germany, United States, and also in the cohort of patients from the Inherited Neuropathies Consortium5, 6, 19, 20. On the other hand, there have been reports of regional Italian clinical CMT series that found frequencies of GDAP1 mutations of > 7%, and after analyzing a group of patients referred to the Medical Genetics Unit of the University of Genoa, the authors conclude that GDAP1 mutations should be the first genetic diagnosis to be considered in an Italian patient with CMT28, 9. Further information about the relative frequency of GDAP1 mutations in the South of France and other countries of the Mediterranean coastline (Greece, Turkey, Morocco, etc.) will be needed to clarify the geographical distribution of these mutations.
In Spain, the high frequency of GDAP1 mutations among CMT2 patients is in part due to the high frequency of two mutations. The p.R120W substitution accounted for 81% of AD patients and was detected mostly in the Mediterranean basin, while p.Q163X was detected either as a homozygous or compound heterozygous mutation in 73.2% of AR patients, especially in the North/Northwest of Spain. Both of these mutations had been studied previously with haplotype analysis and postulated as having a founder effect in the region, although they have been detected in families in other European and American countries1, 16, 21.
Regarding the phenotype, the most important factor that influences the clinical characteristics of these patients is the mode of inheritance (Table 2). The patients with AR inherited mutations have a more severe course and certain clinical features like dysphonia and respiratory dysfunction were exclusively found in this group13, 18. In fact when the CMTNS scores of the series were modeled with a logistic regression analysis, the only factor that could independently predict higher CMTNS scores apart from disease evolution was AR inheritance. Although AR patients had a more severe course as a group, there existed a certain overlap between the severity scores when grouped by the inheritance pattern (Fig. 4a).
For example, there were two AR patients with a milder phenotype than the rest of the cohort (CMTNS scores of 12 and 16 at ages 49 and 46 respectively). The latter is still ambulant with crutches at 56 years of age, and harbors the homozygous missense p.R282C change. This mutation has only been detected in him and his sister, who has a slightly more severe phenotype and became wheelchair-bound only at age 52. The other patient was a compound heterozygous for p.Q163X and p.L344R, and remained ambulant with orthosis until his death due to hepatic cirrhosis at age 55. Even taking into account these outliers, when the clinical characteristics or severity scores were compared according to the localization of the mutation or mutation type, no differences could be established. It has been reported that patients with two ‘truncating’ mutations have a more severe phenotype than those with two missense mutations, but when we compared age of onset, years to wheelchair or other impairment scores we could not replicate these findings14.
In patients with AD inheritance, phenotypic variability regarding severity was prominent, even within affected members of the same family. There are three AD patients with a severe neuropathy according to CMTNS, one of them needing a walker for ambulation; while 28.6% considered themselves asymptomatic, two of them older than 70 years. The factors that influence this variability are largely unknown, but the search for genetic modifiers in this disease led to the detection of a concomitant junctophilin-1 (JPH1) change which modified GDAP1 function in one of the families with the p.R120W mutation included in this series22. JPH1 substitutions have been detected in two other families, one of them is represented in Fig. 3 where the moderately affected siblings have inherited a change in JPH1 from their mother, and the p.R120W GDAP1 mutation from their practically asymptomatic father (in press). In any case, important clinical variability can be found in at least 6 other families with the p.R120W mutation and no concomitant changes in JPH1 have been detected.
Minor differences could be established between the phenotype in the p.R226del and p.R120W mutations but these have to be interpreted in the context of the clinical variability inherent to this disease. In the p.R120W mutation ankle flexion weakness could be detected as soon as ankle extension weakness, and fatty substitution predominated in the posterior compartment of the calf, while in the p.R226del mutation the MRI findings were similar in the symptomatic patient, but in the clinical examination ankle extension weakness predominated. In any case the description of more patients will be necessary to confirm these findings.
Autosomal dominant GDAP1 mutations have been described as having an incomplete penetrance, but in this series all asymptomatic patients had detectable abnormalities in clinical examination or ancillary testing except one. Of the 16 asymptomatic patients 9 had abnormalities in the examination, 11/12 in the electrophysiological studies and 3/6 in the MRI. The only patient that had normal examination and ancillary tests did not undergo needle electromyography or feet MRI, which are the most sensitive tests in this series. This is in keeping with the findings in the Italian p.Arg120Gly mutation where the phenotypic variability mimicked a reduced penetrance, but electrophysiological studies were unequivocally abnormal in affected patients23.
These findings enhance the complexity of the clinical phenotype and of predicting the clinical course in GDAP1 mutations. This last can be especially challenging, but is necessary for the development of future clinical trials. In this regard there are several considerations that are important in this CMT subtype. In AR patients the severity of the phenotype determined that the scales employed could not adequately assess disease progression except in the first or second decade, as afterwards there was a clear ceiling effect in both the CMTNS and FDS scales. In these patients ancillary testing could contribute scarcely as MRI of the lower limbs was affected to a great degree and the rate of change in these patients would be difficult to interpret. In AD patients, apart from the intra-familiar variability, the analysis of the available longitudinal information shows that the change rate of CMTNS per year in one same patient can be quite variable (data not shown). The responsiveness to change probably could be improved if the CMTNS version 2, or the Rasch-wheighted CMTNS version 2 scores had been used, but this was impossible as radial nerve conduction studies were available for only a minority of the patients24. In any case, taking into account the rarity of this CMT subtype, and the intra-familiar variability, more objective measures of disease progression will be necessary, like the MRI biomarkers that have already been developed for CMT1A25.
This is the largest clinical series of CMT patients with GDAP1 mutations and it contributes to expand the knowledge of the genetic distribution and genotype-phenotype correlation of this disease.
Methods
Subjects
This cross-sectional retrospective observational study included all CMT patients with causative GDAP1 mutations evaluated at 14 centers throughout Spain during the 2000–2016 timeframe. 74 Patients were selected from a nationwide register of hereditary neuropathies including 1405 patients, 307 of them defined as CMT2. This registry was developed in 2012 being part of the Spanish Registry of Neuromuscular Diseases project (NMD-ES). After a call for patients in the Neuromuscular Work Group of the Spanish Neurology Society (SEN), 25 other patients were identified. All patients included have signed an informed consent form specific for the NMD-ES registries which was approved by the Ethics Committee of the Hospital de la Santa Creu i Sant Pau. Genetic, demographic, clinical, electrophysiological, and pathologic information was evaluated. Only genetically confirmed patients were included. All experiments were performed in accordance with relevant guidelines and regulations.
Mutational analysis
Mutation analysis of the GDAP1 gene (NM_018972.2) was performed mostly in the same center as previously described using an Applied Biosystems 3730xl DNA analyzer (Foster City, CA, USA)1. To investigate the novelty of the identified variants, the following databases were consulted: dbSNP (http://www.ncbi.nlm.nih.gov/SNP), ESP6500 (http://evs.gs.washington.edu/EVS/), ExAC (http://exac.broadinstitute.org/), and CSVS (http://csvs.babelomics.org/). In silico analysis was performed to predict the pathogenicity of the novel variants, using SIFT (http://sift.bii.a-star.edu.sg/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) algorithms for missense changes and NNSPLICE v0.9 (http://www.fruitfly.org/seq_tools/splice.html) for splicing alterations. Whenever possible, segregation analysis was performed.
Clinical assessments
A standardized history and symptom questionnaire was employed to collect the basal information. Clinical examinations included strength using the standard Medical Research Council (MRC) scale, pinprick and vibratory sensory loss, reflexes, as well as a general and neurologic examination. Severity of the neuropathy was evaluated with the CMT neuropathy score version 1 (CMTNSv1) as a significant proportion of these patients had not undergone the neurophysiologic radial nerve testing which is mandatory for the CMTNS version 226, 27. The CMT examination score (CMTES) was used for patients without nerve conduction studies. The functional disability scale (FDS), which grades the functional impairment from 0–8, was used to measure the disability status28. Respiratory dysfunction was defined by standard spirometry values in each center.
Nerve conduction studies
Motor and sensory nerve conduction velocities (NCVs) were performed using standard techniques. Temperature was controlled during the procedure and kept at more than 32 °C. Compound muscle action potential amplitudes (CMAP), sensory nerve action potential amplitudes (SNAP), conduction velocities and distal latencies were recorded.
Muscle magnetic resonance imaging (MRI)
MRI was performed on the feet and distal legs. The protocol employed was the same as described previously, and fatty substitution was graded from 0 to 4 as follows: 0, no fat signal in muscle; 1, some fatty streaks; 2, fat occupying a minor part of muscle; 3, similar amount of fat and muscle tissue; 4, fat occupying the greater part of muscle16, 29. The muscles in the calf were grouped according to 4 anatomical compartments: anterior compartment (tibialis anterior, extensor hallucis longus, and extensor digitorum longus), lateral compartment (peronei longus and brevis), superficial posterior compartment (soleus and gastrocnemius), and deep posterior compartment (tibialis posterior, flexor digitorum longus, and flexor hallucis longus).
Nerve pathology
Sural nerve biopsy was performed before the genetic diagnosis was confirmed, or for investigational reasons after specific informed consent. Sections were evaluated under a light microscope, and morphometric data was analyzed when available18.
Statistical studies
Patient characteristics, data from clinical examination, electrophysiological, examination and physical disability were analyzed using descriptive statistics with the program R (version 3.3.3), the ordinal package (version 2015.6–28) and brglm (0.5–9). A logistic regression model was developed to determine the co-variables that could predict a higher CMTNS score.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Cuesta, A. et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot–Marie–Tooth type 4A disease. Nat. Genet. 30, 22–25 (2002).
Senderek, J. et al. Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot–Marie–Tooth neuropathy. Brain. 126, 642–649 (2003).
Baxter, R. V. et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot–Marie–Tooth disease type 4A/8q21. Nat. Genet. 30, 21–22 (2002).
Claramunt, R. et al. Genetics of Charcot–Marie–Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 42, 358–365 (2005).
Fridman, V. et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry. 86, 873–878 (2015).
Gess, B., Schirmacher, A., Boentert, M. & Young, P. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul. Disord. 23, 647–651 (2013).
Sivera, R. et al. Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology. 81, 1617–1625 (2013).
Manganelli, F. et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 19, 292–298 (2014).
Pezzini, I. et al. GDAP1 mutations in Italian axonal Charcot-Marie-Tooth patients: Phenotypic features and clinical course. Neuromuscul. Disord. 26, 26–32 (2016).
Marco, A., Cuesta, A., Pedrola, L., Palau, F. & Marín, I. Evolutionary and structural analyses of GDAP1, involved in Charcot–Marie–Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol. 21, 176–187 (2004).
Niemann, A., Ruegg, M., La Padula, V., Schenone, A. & Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot–Marie–Tooth disease. J. Cell Biol. 70, 1067–1078 (2005).
Pla-Martın, D. et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 55, 140–151 (2013).
Sevilla, T. et al. Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain. 131, 3051–3061 (2008).
Cassereau, J. et al. Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp. Neurol. 227, 31–41 (2011).
Zimoń, M. et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology. 77, 540–548 (2011).
Sivera, R. et al. Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 15, 334–344 (2010).
Auranen, M. et al. Dominant GDAP1 founder mutation is a common cause of axonal Charcot-Marie-Tooth disease in Finland. Neurogenetics. 14, 123–132 (2013).
Sevilla, T. et al. Clinical, electrophysiological and morphological findings of Charcot-Marie-Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain. 126, 2023–2033 (2003).
Murphy, S. M. et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry. 83, 706–710 (2012).
Saporta, A. S. et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 69, 22–33 (2011).
Martí Massó, J. F., Zarranz, J. J., Otaegui, D. & López de Munain, A. Neurogenetic disorders in the Basque population. Ann. Hum. Genet. 79, 57–75 (2015).
Pla-Martín, D. et al. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 24, 213–229 (2015).
Manganelli, F. et al. A novel autosomal dominant GDAP1 mutation in an Italian CMT2 family. J. Peripher. Nerv. Syst. 17, 351–355 (2012).
Sadjadi, R. et al. Psychometrics evaluation of Charcot-Marie-Tooth Neuropathy Score (CMTNSv2) second version, using Rasch analysis. J. Peripher. Nerv. Syst. 19, 192–196 (2014).
Morrow, J. M. et al. MRI biomarker assessment of neuromuscular disease progression: a prospective observational cohort study. Lancet Neurol. 15, 65–77 (2016).
Shy, M. E. et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology. 64, 1209–1214 (2005).
Murphy, S. M. et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 16, 191–198 (2011).
Birouk, N. et al. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. 120, 813–823 (1997).
Chung, K. W. et al. Different clinical and magnetic resonance imaging features between Charcot-Marie-Tooth disease type 1A and 2A. Neuromuscul. Disord. 18, 610–618 (2008).
García-Sobrino, T. et al. Phenotypical features of a new dominant GDAP1 pathogenic variant (p.R226del) in axonal Charcot-Marie-Tooth disease. Neuromuscul. Disord. S0960–8966, 10.1016 (2017).
Nelis, E. et al. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 59, 1865–1872 (2002).
Cortez, A. et al. Clinical, electrophysiological and genetic finding in three patients with CMT4A. Neuromuscul. Dis. 19, 565–566 (2009).
Kabzińska, D. et al. Charcot-Marie-Tooth disease type 4C caused by a novel Pro153Leu substitution in the GDAP1 gene. Acta. Myol. 26, 108–111 (2007).
Bouhouche, A. et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): phenotype-genotype correlations in 13 Moroccan families. Brain. 130, 1062–1075 (2007).
Acknowledgements
This collaborative joint project is awarded by IRDiRC and funded by the Instituto de Salud Carlos III (ISCIII) - Subdirección General de Evaluación y Fomento de la Investigación within the framework of the National R + D + I Plan [Grants no. IR11/TREAT-CMT, PI12/00453, PI12/00946, PI15/00187], co-funded with FEDER funds. Dr Espinos has a “Miguel Servet” contract funded by the ISCIII and CIPF [Grant no. CPII14/00002]. Roberto Fernandez-Torron recieved a Rio Hortega Grant from Instituto de Salud Carlos III. The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is an initiative from the ISCIII. National Registry of Hereditary Neuropathy (NMD-ES Project) is partially funded by Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER).We would also like to acknowledge the effort of Victoria Fornés from the Statistics department in Hospital La Fe.
Author information
Authors and Affiliations
Contributions
R.S. Collection of relevant clinical information, manuscript elaboration. M.F. Collection of relevant clinical information, manuscript elaboration. V.L. Genetic and molecular studies, figure elaboration, manuscript revision. T.G.S. Collection of relevant clinical information, manuscript revision. P.B.A. Genetic and molecular studies, collection of relevant clinical information. J.P. Collection of relevant clinical information, manuscript revision. R.F. Collection of relevant clinical information, manuscript revision. A.L.M. Collection of relevant clinical information, manuscript revision. C.M. Collection of relevant clinical information, manuscript revision. L.V. Collection of relevant clinical information, manuscript revision. P.C. Collection of relevant clinical information, manuscript revision. R.R. Collection of relevant clinical information, manuscript revision. S.S. Collection of relevant clinical information and database coordination. I.I. Collection of relevant clinical information, manuscript revision. A.L.F. Collection of relevant clinical information, manuscript revision. A.N. Collection of relevant clinical information, manuscript revision. C.C. Collection of relevant clinical information, manuscript revision. M.G.R. Collection of relevant clinical information, manuscript revision. S.I.P. Collection of relevant clinical information, manuscript revision. A.L.P.N. Collection of relevant clinical information, manuscript revision. J.B. Collection of relevant clinical information, manuscript revision. A.G. Collection of relevant clinical information, manuscript revision. C.C. Collection of relevant clinical information, manuscript revision. A.C. Collection of relevant clinical information, manuscript revision. J.E. Collection of relevant clinical information, manuscript revision. M.J.C. Collection of relevant clinical information, manuscript revision. M.B. Collection of relevant clinical information and coordination. C.D. Collection of relevant clinical information, manuscript revision. F.P. Collection of relevant clinical information, manuscript revision. J.J.V. Collection of relevant clinical information, manuscript revision. C.E. Genetic and molecular studies, manuscript revision. T.S. Manuscript elaboration and revision, study coordination.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sivera, R., Frasquet, M., Lupo, V. et al. Distribution and genotype-phenotype correlation of GDAP1 mutations in Spain. Sci Rep 7, 6677 (2017). https://doi.org/10.1038/s41598-017-06894-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06894-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
