
Abstract
Maternal high-fat diet (HFD) alters hypothalamic developmental programming and disrupts offspring energy homeostasis in rodents. 17β-estradiol (E2) also influences hypothalamic programming through estrogen receptor (ER) α. Therefore, we hypothesized that females lacking ERα would be more susceptible to maternal HFD. To address this question, heterozygous ERα knockout (WT/KO) dams were fed a control breeder chow diet (25% fat) or a semi-purified HFD (45% fat) 4 weeks prior to mating with WT/KO males or heterozygous males with an ERα DNA-binding domain mutation knocked in (WT/KI) to produce WT, ERα KO, or ERα KIKO females lacking ERE-dependent ERα signaling. Maternal HFD increased body weight in WT and KIKO, in part, due to increased adiposity and daytime carbohydrate utilization in WT and KIKO, while increasing nighttime fat utilization in KO. Maternal HFD also increased plasma leptin, IL-6, and MCP-1 in WT and increased arcuate expression of Kiss1 and Esr1 (ERα) and liver expression of G6pc and Pepck in WT and KIKO. Contrary to our hypothesis, these data suggest that loss of ERα signaling blocks the influence of maternal HFD on energy homeostasis, inflammation, and hypothalamic and liver gene expression and that restoration of ERE-independent ERα signaling partially reestablishes susceptibility to maternal HFD.
Similar content being viewed by others


Introduction
Because maternal influences can impact physiological trait expression, the consequences of the obesity epidemic in reproductive-age women are borne by the next generation through alterations in maternal programming of fetal and neonatal development. Indeed, it is estimated that ~35–40% of reproductive-age women are obese or overweight in the USA1. The current idea of intergenerational links between maternal nutrition and health began with the “Thrifty Gene” hypothesis proposed by David J.P. Barker2. Stated simply, a poor nutritional environment during pregnancy, lactation, and early infancy predisposes offspring whose adult nutritional environment is richer than the gestational diet to chronic diseases including ischemic heart disease, stroke, hypertension, Type II DM, and obesity2.
Recent studies on maternal obesity or the effect of maternal high-fat diet (HFD) have demonstrated similar effects on offspring energy homeostasis. These studies found lower birth weights in treated offspring compared to control offspring followed by a catch-up weight gain, adult obesity, and insulin resistance, especially on an obesogenic diet3,4,5. Other studies showed higher birth and adult weights in offspring of diet-induced obesity (DIO) dams compared to control offspring6. Although the molecular mechanisms underlying the effects of maternal HFD are still being explored, changes in hypothalamic gene expression, melanocortin circuitry, neurogenesis, and neuroinflammation have emerged as central mediators of pathogenesis7,8,9,10,11. For example, maternal HFD stimulates hypothalamic neurogenesis of orexigenic neuropeptide Y (NPY) neurons and suppresses anorexigenic proopiomelanocortin (POMC) neurons in male offspring12, which favors hyperphagia. Maternal HFD also hypermethylates the POMC promoter in the hypothalamus of female offspring, which potentially reduces expression of the gene, leading to an increase in food intake and a reduction in energy expenditure13.
The reproductive steroid 17β-estradiol (E2) regulates various aspects of energy homeostasis through both peripheral actions and central mechanisms. The key brain regions that mediate the effects of E2 on energy homeostasis are the hypothalamus and the hindbrain14,15,16,17 wherein E2 suppresses feeding and augments energy expenditure and activity primarily through estrogen receptor (ER) α18, 19. Indeed, ERα knockouts (KO) exhibit an obese phenotype with increased visceral adiposity and decreased energy expenditure20, 21.
ERα signaling functions through nuclear-initiated and membrane-initiated signaling. To control gene expression, nuclear-initiated ERα signaling binds to DNA directly through the estrogen response elements (ERE) or through ERE-independent mechanisms, such as protein-protein interactions with other transcription factors22. ERα can also activate membrane-initiated signaling cascades (MAPK, PLC, PI3K) to modulate cell physiology and control gene expression23,24,25,26,27,28. The restoration of ERE-independent signaling (both membrane- and nuclear-initiated) in ERα KO female mice normalizes energy homeostasis. These females, called ERα KIKO, express an ERα that does not bind to ERE but retains nuclear-initiated tethered transcriptional regulation and membrane-initiated activation of signaling cascades. Adult KIKO females do not become obese or glucose intolerant, suggesting that ERE-independent ERα signaling is sufficient for the normal development and maintenance of energy and glucose homeostasis29, 30. Thus, a potential basis for the disruption in energy homeostasis in KO females is the loss of ERE-independent ERα signaling during neurogenesis31,32,33 and the proliferation and differentiation of neural stem cells34.
Because the loss of ERα and the influence of maternal HFD alters hypothalamic developmental programming leading to dysregulation of energy homeostasis, we hypothesized that the total loss of ERα would make female mice more susceptible to the effects of maternal HFD. Furthermore, because ERE-independent ERα signaling restores normal energy homeostasis, we also hypothesized that ERE-independent ERα signaling would be protective against the effects of maternal HFD. To address these hypotheses, we employed a standard maternal HFD paradigm using heterozygous dams mated to heterozygous males and followed their WT, KO, and KIKO female offspring into adulthood.
Results
Body weight and body composition
By week 5 (peripubertal), females from HFD-fed dams of each genotype weighed more than their counterparts from control (Con)-fed dams (Fig. 1a). WT from Con-fed dams (n = 11) weighed 15.9 ± 0.12 g, and WT from HFD-fed dams (n = 11) weighed 17.4 ± 0.2 g (P < 0.05). KIKO from Con-fed dams (n = 9) weighed 15.3 ± 0.6 g, and KIKO from HFD-fed dams (n = 9) weighed 17.6 ± 0.4 g (P < 0.01). KO from Con-fed dams (n = 9) weighed 16.5 ± 0.4 g, and KO from HFD-fed dams (n = 12) weighed 19.5 ± 0.5 g (P < 0.001). However, this effect of maternal HFD was lost in KO females by week 9 (post-puberty) while WT and KIKO females from HFD-fed dams were slightly heavier than WT and KIKO from Con-fed dams throughout the study (data not shown).

Body weight and body composition of adult females. (a) Body weights at week 5 in all genotypes from Control-fed and HFD-fed dams. (b) Body weights at week 25 in all genotypes after 20 weeks of a low-fat chow diet. (c) Percent body fat (fat mass/body mass) of female mice from all groups. (d) Percent lean mass (lean mass/body mass) of female mice from all groups. Control = maternal control diet and HFD = maternal HFD. Data were analyzed by two-way ANOVA with post-hoc Newman-Keuls test. Sample sizes were 9 to 12 per genotype per treatment and data are expressed as mean ± SEM. Capped lines denote comparison between maternal diets within genotypes. Asterisks (*) denote comparison to WT within the same diet group. The pound sign (#) denotes comparison of KIKO and KO within the diet group. (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
After 23 weeks on a standard (low-fat) chow diet (Fig. 1b), WT from Con-fed dams (n = 11) weighed 24.6 ± 0.7 g, and WT from HFD-fed dams (n = 11) weighed 28.5 ± 1.2 g (P < 0.05). KIKO from Con-fed dams (n = 9) weighed 24.5 ± 0.9 g, and KIKO from HFD-fed dams (n = 9) weighed 29.0 ± 1.5 g (P < 0.05). KO from Con-fed dams (n = 9) weighed 29.9 ± 1.6 g, and KO from HFD-fed dams (n = 12) weighed 30.5 ± 1.3 g (ns). In summary, KO from Con-fed dams weighed more than their WT and KIKO counterparts. However, maternal HFD increased body weight in WT and KIKO and not in KO females. Collectively, these data suggest that the loss of ERE-dependent signaling in KO abrogates the effects of maternal HFD.
Body fat accumulation (% fat mass) in the Con-fed females was similar to our previous study (32) with KO fatter than WT (P < 0.5) but not KIKO. Maternal HFD increased body fat in WT (P < 0.01) and KIKO (P < 0.05; Fig. 1c), indicating that increased deposition of adipose tissue underlies the increase in body weight for WT and KIKO from HFD-fed dams. KO from Con-fed females had less lean mass than WT (P < 0.01) and KIKO (P < 0.05), which also had less lean mass than WT (P < 0.05; Fig. 1d). WT from HFD-fed dams had more lean mass than both KIKO (P < 0.05) and KO (P < 0.01).
Food intake was measured for the Con-fed females in all genotypes for 1 week in single-housing cages. As previously reported32, WT consumed more food than KIKO or KO during the weeklong trial. Average food intake for the week was 22.7 ± 1.8 g in WT, 18.3 ± 0.7 g in KIKO (P < 0.05), and 18.1 ± 1.3 g in KO (P < 0.05), which corroborates our previous findings35 (data not shown). However, we observed a loss of body weight in KIKO and KO during the week, most likely due to the stress of single housing. Therefore, all control females were placed back in group-housed cages and allowed to recover body weights prior to glucose and insulin tolerance testing. We did not examine food intake in females from HFD-fed dams due to concerns that the short-term feeding studies would be confounded by the stress of single housing.
Metabolic parameters
To determine the effects of maternal HFD on energy expenditure, substrate utilization, and activity, all females were transferred to a Comprehensive Lab Animal Monitoring System (CLAMS) unit for 48 h using data only from the last 24 h to calculate metabolic parameters and activity36. V.O2 was affected by genotype and maternal HFD (Fig. 2a). During the day, V.O2 was elevated by maternal HFD in WT (P < 0.0001) and KO (P < 0.01), and during the night, V.O2 was elevated by maternal HFD only in WT (P < 0.05). WT females from HFD-fed dams exhibited higher nighttime V.O2 than their KIKO (P < 0.05) and KO (P < 0.01) counterparts. Maternal HFD eliminated the differences between nighttime and daytime V.O2 in KIKO and KO females.

Metabolic and activity parameters in females from all genotypes after 20 weeks of adult chow diet determined using the CLAMS. (a) V.O2 (ml/min/kg); (b) V.CO2 (ml/min/kg); (c) Respiratory exchange ratio (RER) (V.CO2 /V.O2); (d) Energy expenditure (kCal/hr/lean mass (g)); (e) X-plane activity (counts); and (f) Z-plane activity (counts). Data were analyzed by a multi-factorial ANOVA (genotype, maternal diet, time) with post-hoc Newman-Keuls test. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
V.CO2 was affected by genotype, maternal HFD, and time (Fig. 2b). Maternal HFD elevated daytime V.CO2 in WT (P < 0.0001) but not in KIKO or KO. V.CO2 was elevated at nighttime compared to daytime in all genotypes (WT: P < 0.0001; KIKO: P < 0.05; KO: P < 0.01) from Con-fed dams. Similar to V.O2, maternal HFD eliminated this elevation during the night in KIKO and KO. Daytime V.CO2 in KIKO from Con-fed dams was elevated compared to WT (P < 0.05), and nighttime V.CO2 in KO from HFD-fed dams was lower than WT (P < 0.01).
Respiratory exchange ratio (RER) was affected by genotype, time, and the interaction of genotype and maternal HFD (Fig. 2c). Daytime RER was elevated by maternal HFD in WT (P < 0.05) but not in KIKO or KO. As we have previously reported, daytime RER in KIKO from Con-fed dams was higher than in WT (P < 0.001) or KO (P < 0.05), indicating that KIKO females preferentially utilize carbohydrates during the day compared to both WT and KO. Hence, nighttime RER was not higher in KIKO from Con-fed dams as was found in WT and KO. Interestingly, RER in KO was generally lower than both WT and KIKO except for nighttime RER in KO from Con-fed dams. Because body weight can influence metabolism, RER was analyzed by an analysis of covariance (ANCOVA) with body weight as a covariate and plotted as a function of body weight to illustrate these effects (Supplemental Figure S2a). Overall, neither genotype nor maternal HFD affected the relationship of body weight and RER.
Heat production (energy expenditure) normalized to lean body mass was affected by genotype, maternal diet, time, and interactions of genotype and maternal diet and maternal diet and time (Fig. 2d). In both maternal diets, heat was elevated in the nighttime compared to the daytime in WT and KIKO, but only KO from Con-fed dams. Unlike V.O2, V.CO2, and RER, maternal HFD elevated heat production in WT and KIKO during both time periods, but only during the daytime in KO. Finally, daytime heat production in KIKO and KO from Con-fed dams was higher compared to WT (P < 0.05 for both). Elevation of heat production indicates higher metabolic rates, thus maternal HFD augmented metabolic rates only during the daytime and independent of activity in WT and KIKO. We also analyzed daytime and nighttime heat by an analysis of covariance (ANCOVA) with body weight as a covariate (Supplemental Figure S2b and c). As expected, maternal HFD affected the relationship of body weight and heat during the day (P < 0.0001) and night (P < 0.0001), although there was an interaction between genotype and maternal diet (P < 0.05) during the daytime.
Both X-plane and Z-plane activity were affected by genotype, time, and the interactions between genotype and time, but only X activity was affected by maternal HFD (Fig. 2e and f). X-plane activity was higher in the nighttime than the daytime in all genotypes, regardless of maternal diet. However, both KIKO and KO females were less active in the nighttime compared to WT, regardless of maternal diet, as previously reported35. Interestingly, there was a subtle but significant increase in daytime activity in WT due to maternal HFD (P < 0.05). Z-plane activity was higher in the nighttime than the daytime for all genotypes, regardless of diet. However, maternal HFD reduced daytime Z-plane activity in KO and reduced nighttime Z-plane activity in KIKO (P < 0.01) and KO (P < 0.001) compared to WT. These data suggest that ERE-dependent ERα signaling is necessary for the maintenance of normal activity in female mice.
Glucose and insulin tolerance
To determine the effects of maternal HFD on glucose homeostasis, we conducted glucose and insulin tolerance tests on all females. For the GTT, all mice were fasted overnight (1700h – 0900 h). Fasting glucose levels, an indicator of a diabetic-like state, were not affected by genotype (Fig. 3a). There was no effect of maternal HFD on terminal blood triglycerides (non-fasted) (data not shown). Glucose tolerance was determined over 180 min following an ip injection of glucose (2 g/kg). Glucose clearance was slower in KO from Con-fed dams compared to WT and KIKO females at 60, 90, 120, and 180 min (Fig. 3b) and in KO from HFD-fed dams compared to WT and KIKO females at 30, 60, 90, and 120 min (Fig. 3c). Maternal HFD did not alter glucose clearance in any genotype (Fig. 3d), although maternal HFD augmenteded glucose clearance in WT at 30 min (P < 0.05, comparison not shown). Integral analysis of the area under the curve (AUC) illustrates the influence of genotype on glucose clearance (Fig. 3d). KO exhibited slower glucose clearance compared to WT, regardless of diet treatment (P < 0.05, P < 0.01, respectively). Insulin tolerance was measured over 120 min after an ip injection of insulin. In all genotypes, insulin-induced glucose clearance was not altered by maternal HFD (Fig. 4a–c; A comparison of all groups for the GTT and ITT is presented in Supplemental Figure S3). Therefore, the primary driver behind the inhibition of glucose clearance is the loss of ERE-independent actions by ERα, which has recently been elucidated in an adult HFD study with the same transgenic strains37.

Fasting glucose levels and glucose tolerance test (GTT) in adult females from all genotypes after 20 weeks of adult chow diet. (a) Fasting glucose levels. Results from the GTT from (b) all genotypes from Control-fed dams and (c) all genotypes from HFD-fed dams. (d) Area under the curve (AUC) analysis for all genotypes from both maternal diets. (a and d) Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls test. (b and c) Data were analyzed by repeated-measures, multi-factorial ANOVA with post-hoc Newman-Keuls test. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).

Insulin tolerance test (ITT) in adult females from all genotypes after 20 weeks of adult chow diet. Results from the ITT from (a) all genotypes from Control-fed dams and (b) all genotypes from HFD-fed dams. (c) AUC analysis for all genotypes from both maternal diets. (a and b) Data were analyzed by repeated-measures, multi-factorial ANOVA with post-hoc Newman-Keuls test. (c) Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls test. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
Hormones and inflammatory cytokines
To determine if maternal HFD alters endogenous E2 production, we measured E2 in all groups. E2 levels were not affected by maternal HFD and were higher in KO compared to WT and KIKO in both groups (Fig. 5a). To determine the effects of maternal HFD on leptin, insulin, and inflammatory cytokines, we analyzed plasma samples using multiplex assays. Maternal HFD did not alter plasma insulin levels in WT or KIKO. In contrast, maternal HFD produced hyperinsulinemia in KO, which expressed four times the plasma insulin as KO from Con-fed dams (P < 0.01; Fig. 5b), suggesting that ERE-independent signaling is protective against the effects of maternal HFD on insulin production. Maternal HFD increased plasma leptin in WT (P < 0.05), and plasma leptin in KO from HFD-dams were lower than WT (P < 0.01; Fig. 5c).

Peripheral peptide hormones and inflammatory cytokines from all genotypes after 20 weeks of adult chow diet. (a) Plasma levels of 17β-estradiol (pg/ml). (b) Plasma levels of insulin (ng/ml). (c) Plasma levels of leptin (ng/ml). (d) Plasma levels of IL-6 (pg/ml). (e) Plasma levels of MCP-1 (pg/ml). (f) Plasma levels of TNFα (pg/ml). Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls test. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
The selected inflammatory cytokines IL-6, MCP-1, and TNFα are all implicated in obesity38. Plasma IL-6 levels were primarily affected by genotype (Fig. 5d). Plasma IL-6 levels in KO were higher than WT and KIKO in control (P < 0.0001) and HFD (P < 0.01) groups, and IL-6 was elevated by maternal HFD in WT (P < 0.05). Plasma MCP-1 expression was affected by genotype and maternal HFD (Fig. 5e). KO from Con-fed dams expressed more MCP-1 compared to WT (P < 0.001) and KIKO (P < 0.01) and KO from HFD-fed dams expressed less MCP-1 compared to WT (P < 0.0001) and more than KIKO (P < 0.05). MCP-1 was also lower in KIKO from HFD-fed dams compared to WT (P < 0.0001). However, maternal HFD increased the levels of plasma MCP-1 in WT (P < 0.0001) and decreased plasma MCP-1 in KO (P < 0.05). Plasma TNFα levels were not affected by either genotype or maternal HFD (Fig. 5 f). Elevated levels of IL-6 and MCP-1 in KO females and in WT females from HFD-dams indicate chronic obesity and suggest that ERE-independent ERα signaling (KIKO) protects against systemic inflammation.
Arcuate gene expression
To determine if maternal HFD had a differential impact on ARC gene expression, we analyzed expression of selected genes involved in energy balance, including neuropeptides and hormone receptors39. For the anorectic neuropeptide, POMC, maternal HFD increased expression only in KO (Fig. 6a). Pomc expression was also dependent on genotype. Specifically, Pomc expression in WT from HFD-fed dams was higher than in KIKO (P < 0.05) and lower than in KO (P < 0.05). Pomc was also lower in KIKO from HFD-fed dams than KO (P < 0.001). Cart expression was not altered by genotype or maternal HFD (Fig. 6b). Expression of the orexigenic neuropeptide, NPY, was affected by genotype (Fig. 6c), with KO from HFD-dams expressing more Npy than both WT (P < 0.001) and KIKO (P < 0.05). Agrp expression was affected by genotype and maternal diet (Fig. 6d). Maternal HFD reduced Agrp expression in WT females. WT females from Con-fed dams also expressed more Agrp than both KIKO (P < 0.0001) and KO (P < 0.01). Conversely, KO females from HFD-fed dams expressed more Agrp than WT (P < 0.01) and KIKO (P < 0.01) females. Expression of Kiss1, a gene that has dual roles in reproduction and energy homeostasis40, 41, was augmented by maternal HFD in both WT (P < 0.05) and KIKO (P < 0.05) but not in KO (Fig. 6e). Kiss1 expression in KO from HFD-fed dams was lower than both WT and KIKO (P < 0.05 for both). Expression of the ERα gene, Esr1, was dependent on genotype and maternal diet (Fig. 6 f). Receptor expression was reduced in KO compared to both WT (Con: P < 0.01; HFD: P < 0.0001) and KIKO (Con: P < 0.05; HFD: P < 0.0001) and was augmented by maternal HFD in WT females (P < 0.0001), as previously reported42, and in KIKO (P < 0.0001). Maternal HFD reduced arcuate expression of the insulin receptor (Insr) in WT (P < 0.05) (Supplemental Table S2) and KO from Con-fed dams expressed less Insr than WT (P < 0.001). Arcuate expression of the leptin receptor (Lepr) was augmented by maternal HFD in KIKO (P < 0.01) and was differentially expressed between the genotypes from HFD-fed dams (Supplemental Table S2).

Arcuate gene expression in all genotypes after 20 weeks of adult chow diet. (a) Pomc; (b) Cart; (c) Npy; (d) Agrp; (e) Kiss1; and (f) Esr1 (ERα) expression normalized to WT from Control-fed dams. Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls test within each genotype. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
Liver gene expression
Because the effects of maternal HFD can also occur in peripheral organs that are involved in energy and glucose homeostasis43,44,45, we examined liver gene expression. Glucose-6-phosphatase (G6pc) expression, which controls hepatic glucose production46, was elevated by maternal HFD in WT (P < 0.0001) and KIKO (P < 0.05) females (Fig. 7a). Expression was dependent on genotype as both KIKO and KO expressed less G6pc than their WT counterparts. Phosphoenolpyruvate carboxykinase (Pepck), which is essential for gluconeogenesis, was differentially expressed between the genotypes and augmented by maternal HFD in WT (P < 0.0001) and KIKO (P < 0.05; Fig. 7b). KIKO and KO from HFD-fed dams expressed less Pepck than WT (P < 0.001 and P < 0.0001, respectively). Diacylglycerol O-acyltransferase 2 (Dgat2), which is an essential enzyme in the production of triglycerides47, was not affected by genotype or maternal HFD (Fig. 7c). Fatty acid synthase (Fas), which controls fatty acid production48, was augmented by maternal HFD in WT (P < 0.01), with lower expression in KIKO (P < 0.05) and KO (P < 0.01) from HFD-fed dams than WT (Figure 9d). Sterol regulatory element-binding protein 1 (Srebp1), a regulator of liver transcription for glucose, fatty acid, and lipid production49, was not altered by maternal HFD but was expressed less in KO than in WT (P < 0.05 for both; Fig. 7e). Esr1 expression was elevated by maternal HFD in KIKO females (P < 0.05; Fig. 7f). Esr1 was expressed at lower levels in KIKO (P < 0.001) and KO (P < 0.0001) from Con-fed dams compared to WT and at lower levels in KO from HFD-dams compared to WT (P < 0.0001) and KIKO (P < 0.01). Maternal HFD increased liver Insr expression in WT (P < 0.05) and KIKO (P < 0.01) and was expressed at lower levels in KIKO (P < 0.05) and KO (P < 0.01) from HFD-fed dams than WT. Maternal HFD reduced Lepr expression in WT (P < 0.01) and was expressed at lower levels in both KIKO (P < 0.0001) and KO (P < 0.0001) from Con-fed dams compared to WT (Supplemental Table S2).

Liver gene expression in all genotypes after 20 weeks of adult chow diet. (a) G6pc; (b) Pepck; (c) Dgat2; (d) Fas; (e) Srebp1; and (f) Esr1 (ERα) expression normalized to WT from Control-fed dams. Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls test within each genotype. See Fig. 1 for information on treatment categories, sample sizes, and statistical comparisons (a/*/# = P < 0.05; b/**/## = P < 0.01; c/***/### = P < 0.001; d/****/#### = P < 0.0001).
Discussion
Understanding the impact of maternal HFD on the development of central and peripheral mechanisms controlling energy homeostasis is key to addressing obesity and other metabolic diseases. Many studies in the field of maternal programming have examined male offspring mostly to avoid complications from the influence of circulating E2 on energy homeostasis in females during the estrous cycle, which is largely mediated by ERα. The role of ERα in the development of the reproductive functions of hypothalamus has previously been examined50, 51, yet its role in the development of energy homeostasis is largely unknown. Therefore, we set out to identify the importance of ERα in the development of female energy homeostasis by testing the hypothesis that females lacking ERα (KO) are more susceptible to the effects of maternal HFD. Instead, we found that KO from HFD-fed dams were not heavier than KO from Con-fed dams. This suggests that the disruption caused by the loss of ERα produces a “ceiling” effect and reduces the influence of maternal HFD. As previously reported, the ERE-independent ERα signaling present in KIKO females was sufficient to restore normal energy and glucose homeostasis compared to KO females. We found that it was also sufficient to restore the susceptibility to maternal HFD29, 35 because KIKO females, similar to the WT, were heavier after maternal HFD when fed a control diet. These data suggest that ERE-independent ERα signaling during development partially restores sensitivity to maternal HFD.
Recently, in an unpublished study, Flowers and colleagues52 (2014) presented evidence that KIKO and KO females are especially sensitive to diets low in phytoestrogens, which may confound the interpretation of our data. In our study, the dietary constituents both in the maternal and adult diets are not fully consistent, especially in regards to phytoestrogens. The control maternal diet used in the current study contains soy and an unknown concentration of phytoestrogens. In a 2007 study, phytoestrogens were measured at ~120 μg/g (ppt) chow in the same diet53, which is higher than the phytoestrogens in the HFD used in our study (Research Diets, personal communication). Furthermore, a previous study demonstrated that a lack of phytoestrogens in a diet fed to both dam and offspring produced heavier males and females at PND90, with more body fat and higher serum leptin levels, and a reduction in glucose clearance only in males54. In comparison, the adult diet used in our study was low in phytoestrogens (<75 ppm), but maternal HFD did increase body fat in WT and KIKO and plasma leptin levels in WT females.
In female rodents, E2 controls adipose deposition by decreasing visceral fat deposition primarily through an ERα-mediated mechanism55. In our study, the difference in fat mass between WT and KO was eliminated by maternal HFD as WT from HFD-fed dams were fatter than WT from Con-fed dams. These data suggest that the total loss of ERα reduces the developmental programming effects of maternal HFD on adipogenesis, which is restored by ERE-independent ERα signaling. However, ERα is not the only membrane-associated ER that has been implicated in the control of adiposity. GPER1 controls adiposity in females during DIO and may underlie some of the effects on adiposity found in the KIKO and KO females56.
Maternal HFD altered metabolism and activity by augmenting daytime V.O2, V.CO2, RER, and heat production (energy expenditure) in WT and heat production in KIKO and KO. These data suggest that the mechanisms of substrate utilization and energy expenditure are influenced during development, in part, by ERE-dependent and ERE-independent ERα signaling in females. The loss of ERα blocks the increase in carbohydrate utilization caused by maternal HFD during the day, which consequently blocks the nighttime increase in carbohydrate utilization. Thus, these effects on KO substrate utilization may play a role in the “ceiling effect” on obesity due to maternal HFD. Furthermore, the increase in energy expenditure after maternal HFD, which is found in the daytime in all genotypes, but only in WT and KIKO in the nighttime, may be a consequence of body weight gain in WT and KIKO36, 57. This suggests that the loss of ERE-independent signaling (in KO) during development results in an inhibition of the compensatory response in energy expenditure in heavier females.
Conversely, maternal HFD reduced activity in KO, widening the already prominent genotypic differences in activity. A recent study found that maternal HFD reduced exploratory behaviors and voluntary activity and increased anxiogenic behaviors in HFD-fed male and female mice58. We hypothesize that many of these effects are due to the mechanisms that ERα controls in the hypothalamus, both developmentally and during adulthood. In fact, selective deletion of ERα in neurons throughout the mouse brain produced an obese phenotype with an increase in food intake, a reduction in energy expenditure, increased adiposity, and suppressed activity59. In the same study, specific deletion of ERα in POMC neurons increased body weight, heat production, and activity59. Thus, the loss of ERα in select neurons during development produces phenotypes similar to those phenotypes produced by maternal HFD in KIKO and KO.
While activation of ERα in the liver is a primary pathway of E2 to control glucose production and insulin sensitivity, ERα also acts in adipose tissue and skeletal muscle60, 61. In our study, glucose clearance was reduced by the total loss of ERα signaling, as has been previously reported29, 37, maternal HFD did not have an impact on glucose clearance. Presumably, KIKO mice, like WT, retain the ability to shuttle glucose from the circulation due, in part, to the membrane-initiated ERα mechanisms that regulate glucose transporter type 4 (GLUT4) expression and insulin-induced trafficking to the membrane in skeletal muscle60, 62, 63. GLUT4 expression is increased through ERα activation in the extensor digitorum longus63, despite the lack of a consensus ERE in the GLUT4 promoter region, suggesting that ERE-independent signaling is key.
Similar to other maternal studies64,65,66, glucose homeostasis is not disrupted by maternal HFD in WT female offspring due to the protective effects of circulating estrogens activating both membrane-initiated and nuclear-initiated ERα signaling. However, the loss of total ERα signaling did not induce greater susceptibility as originally hypothesized. Likewise, maternal HFD did not alter insulin tolerance in any genotype, despite hyperinsulinemia in KO from HFD-fed dams, indicating that maternal HFD does induce insulin intolerance in the peripheral organs involved in glucose clearance. Interestingly, E2 replacement, both systemically and centrally (intracerebroventricular), in ovariectomized female rodents controls energy homeostasis, hepatic glucose production, and insulin sensitivity67, 68. Thus, we cannot ignore the potential role of ERα signaling in the hypothalamus when discussing the effects of maternal HFD on insulin and glucose homeostasis.
While maternal HFD did not have a clear effect on glucose homeostasis (fasting levels and glucose clearance), maternal HFD increased liver expression of G6pc and Pepck in WT and KIKO females. Elevated levels of these gluconeogenic enzymes suggest that hepatic glucose production is elevated in these genotypes from HFD-fed dams, which would require hyperinsulinemic-euglycemic clamp measurements. Interestingly, these genes were not upregulated in KO which may be evidence of protective hepatic glucose metabolism and contribute to the lower blood glucose levels. Furthermore, these genes were differentially expressed between the genotypes (WT expressed more than both KIKO and KO) and may produce a phenotype more susceptible to the effects of diet-induced obesity in adulthood.
Low-grade, elevated inflammation is a result of obesity due to increased production of inflammatory cytokines by adipose tissue. These cytokines are transported to organs that control metabolic processes e.g., liver, brain, and muscle69,70,71 and contribute to the developmental programming of maternal HFD9, 72, 73. In our study, maternal HFD augmented the peripheral inflammatory signals MCP-1 and IL-6 only in WT while MCP-1 and IL-6 was elevated in every KO group compared to WT and KIKO. Thus, the response to maternal HFD in WT includes an increase in cytokine production and may be a result of the increase in adiposity. However, this response in cytokine production to maternal HFD is lost in female mice that lack ERE-dependent ERα signaling despite increased adiposity. Furthermore, E2, through an ERα-mediated mechanism, enhances the HFD-induced increase in plasma IL-6 and TNFα levels in OVX female mice74. In our study, IL-6 was elevated in KO females, which were not insulin intolerant, from both Con-fed and HFD-fed dams. The elevation of IL-6, without other inflammatory signals, may promote glucose-stimulated insulin secretion from the pancreas75,76,77,78 and protect these females from further disruption to insulin homeostasis by maternal HFD.
ERα-mediated control of ARC gene expression is a primary mechanism to modulate hypothalamic and homeostatic functions79. Many studies have found that ARC neuropeptides are not altered by maternal HFD or obesity in adult male mice and rats5, 7, 80, while other studies have shown that maternal HFD stimulates and/or suppresses Npy and Pomc expression81,82,83. Due to the role that these ARC neuropeptides have in hypothalamic control of energy homeostasis, we hypothesized that maternal HFD would augment Npy/Agrp and suppress Pomc/Cart. However, we found elevated expression of the anorexigenic neuropeptide, Pomc, in KO females due to maternal HFD, which may result in a suppression in food intake. Conversely, expression of the orexigenic neuropeptide, Agrp, was reduced by maternal HFD in WT females, which may also result in a suppression in food intake. Interestingly, Kiss1 expression, which has recently been implicated in the control of energy homeostasis in rodents40, 84, was elevated in WT and KIKO females by maternal HFD and may play a role in the effects of maternal HFD in these genotypes. Collectively, these data would indicate that the “ceiling effect” found in KO females may be due to an elevated anorexigenic gene expression profile and that both anorexigenic and orexigenic neuropeptides are impacted by maternal HFD, dependent on the availability of ERα signaling mechanisms.
Little is known about the interactions of maternal HFD and ERα on ARC gene expression, although hypothalamic ERα (and ERβ) protein expression is increased in female offspring from dams fed a HFD enriched with high levels of n-6 PUFA42. Our data are consistent with these findings, showing a two- to three-fold increase in Esr1 expression in the ARC in WT and KIKO due to maternal HFD. The effect of these elevated levels of Esr1 on energy homeostasis and on hypothalamic development is unknown but may be involved in ameliorating the effects of maternal HFD on neuroinflammation72, 85. Furthermore, because ERα mediates the actions of E2 on food intake and energy expenditure in the hypothalamus, the increase in ERα expression may be protective against the effects of maternal HFD in the WT and partially in the KIKO.
In conclusion, our study suggests that both ERE-dependent or ERE-independent ERα signaling during development influences the effects of maternal HFD on offspring energy and glucose homeostasis, inflammation, and gene expression. Presumably, the effects on energy expenditure and activity are central in origin, although further investigation is required. One potential mechanism is the epigenetic regulation of ERα in the brain by maternal HFD, which has previously been demonstrated with maternal behaviors and endocrine disruptors86, 87. Furthermore, ERα signaling regulates DNA methylation through the control of DNMT genes and other epigenetic factors in a variety of tissues88,89,90. The loss of ERα-induced epigenetic modifications along with the modulation of neurogenesis31,32,33 and neural stem cell proliferation and differentiation34 during development may abrogate the effects of maternal HFD. However, these data would indicate that at least some of these mechanisms involve ERE-independent ERα signaling since KIKO mice are susceptible to maternal HFD.
Materials and Methods
Animals
All animal treatments were in accordance with institutional guidelines based on National Institutes of Health standards and were performed with Institutional Animal Care and Use Committee approval at Rutgers University. Female wild-type (WT C57BL/6 J), ERα KO (KO), and ERα KIKO (KIKO) transgenic mice (provided by Dr. Ken Korach, NIEHS)91, 92 were selectively bred in-house and maintained under controlled temperature (23 °C) and photoperiod conditions (12/12 h light/dark cycle) with food and water ad libitum. WT/KO heterozygous males and females were mated to produce ERα KO females. Non-classical ERα knock-in heterozygous males (WT/KI) and WT/KO heterozygous females were crossed to generate KIKO females. WT females were generated from both colonies and used with their KIKO and KO littermates. At weaning, females were tagged and ear-clipped for genotyping. Genotype was determined by PCR of extracted DNA using previously published protocols91, 92.
Maternal HFD Experimental Design
To determine the effects of maternal high-fat diet on energy homeostasis in female offspring, we modeled our experiment after a previous study that compared the effects of two maternal diets: a standard chow diet and a semi-purified high-fat diet93. Breeding WT/KO (n = 12/maternal diet) dams were fed either a standard breeder chow diet (Con, 25% fat kCal, 3.83 kcal/g, Lab Diet 5015; Lab Diet, St. Louis, MO, USA) or a high-fat diet (HFD, 45% fat kCal, 4.73 kcal/g, D12451; Research Diets, New Brunswick, NJ, USA) for 4 weeks prior to breeding with an untreated WT/KO or WT/KI male. Pregnant dams continued on the same diet for the duration of gestation and lactation (~10 weeks). HFD-fed dams gained more weight than the Con-fed dams prior to breeding (data not shown) but were not metabolically characterized during gestation or lactation to reduce the impact of stress on developmental programming94 and specifically on neuronal ERα expression95. After parturition, male pups were culled by postnatal day (PND) 4 to reduce the influence of litter size on offspring energy homeostasis. The average litter size was 9.1 ± 0.2 pups (n = 24) for Con-fed WT/KO dams and 8.8 ± 0.2 for HFD-fed WT/KO dams (n = 24). The average number of female pups per litter was 4.4 ± 0.2 for Con-fed WT/KO dams and 4.5 ± 0.2 for HFD-fed WT/KO dams. At PND 21, female pups from each litter were weaned and genotyped. Offspring were weaned onto a standard chow (13% kCal fat, 3.48 kcal/g, Lab Diet 5V75; low phytoestrogen, < 75 ppm) as the maternal control diet is specifically made to accommodate the high energetic needs of breeding females. At 5 weeks, all identified WT, KIKO, and KO females were weighed. Females were group-housed by genotype to reduce the social stress of single housing per IACUC protocols.
Adult Offspring Experimental Design
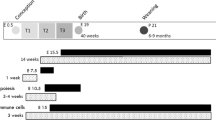
From 5 to 25 weeks of age, females were weighed weekly. We did not monitor the estrous cycle as neither KO nor KIKO exhibit a normal estrous cycle, which makes it difficult to compare to WT29, 30. At the end of 25 weeks, body composition was measured in each female using an EchoMRI 3-in-1 Body Composition Analyzer (Echo Medical Systems, Houston, TX, USA) followed by a 48 h run in a Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments, Columbus, OH, USA) to measure metabolic parameters and activity (X and Z plane). Females were then housed alone for one week to measure daily food intake. Afterward, a glucose tolerance test (GTT) was performed on each female. Females were fasted overnight (1700 h–0900 h) in a new cage. At the start of the test and 30 min after local anesthetizing of the tail with lidocaine, mice were placed in Plexiglass restrainers and tails were nicked to collect a baseline (time = 0) glucose reading using a glucometer (AlphaTRAK2). Immediately after baseline, females were injected intraperitoneally (ip) with a bolus of glucose (2.0 g/kg body weight) and individually housed in clean cages. Tail blood samples were collected at 15, 30, 60, 90, 120, and 180 min post-injection. After 180 min, all mice were returned to their home cages with ad libitum access to water and food. After sufficient recovery (~3 d), an insulin tolerance test (ITT) was performed after a 5 h fast in a similar manner as the GTT with an ip injection of insulin (0.75 units/kg). Blood samples were collected from the tail in individual cages at 15, 30, 60, 90, and 120 min post-injection. See Supplemental Figure S1 for a graphical illustration of the maternal and adult experimental design.
Brain and Body Dissections
After sufficient recovery from the ITT (~1 week), females were decapitated after sedation with ketamine (100 µl of 100 mg/ml, ip) at 1000 h. Trunk blood was collected in a K+ EDTA collection tube and analyzed for triglyceride levels using a CardioChek (Polymer Technology Systems, Indianapolis, IN, USA). Plasma was prepared for peptide hormone and inflammatory cytokine analysis by adding a protease inhibitor, 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, 1 mg/mL, Sigma-Aldrich, St. Louis, MO, USA), to each collection tube. Samples were maintained on ice until centrifugation at 3,000 rpm for 10 min at 4 °C. Plasma was stored at −80 °C until analysis. Insulin, leptin, interleukin 6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor α (TNFα) were determined by multiplex assay (MMHMAG-44K, EMD Millipore, Billerica, MA, USA).
Abdominal cavity was dissected for liver tissue (secondary lobe). Liver tissue was fixed in RNAlater (Life Technologies, Grand Island, NY, USA) and stored at –80 °C. Liver RNA was extracted using a standard TRIzol® extraction (Life Technologies) coupled with Macherey-Nagel NucleoSpin® RNA extraction and DNase-1 kit (Bethlehem, PA, USA). The brain was immediately extracted from the skull and rinsed in ice-cold Sorensen’s buffer for 30 sec. The brain was cut using a brain matrix (Ted Pella, Redding, CA, USA) into 1-mm thick coronal rostral and caudal blocks corresponding to Plates 42 to 47 and Plates 48 to 53, respectively, from The Mouse Brain in Stereotaxic Coordinates (Paxinos & Franklin 2008, 3rd Edition)96. Blocks of the basal hypothalamus (BH) were transferred to RNAlater (Life Technologies) and stored overnight at 4 °C. The rostral and caudal parts of the arcuate nucleus were dissected from slices using a dissecting microscope. Dissected tissue was stored at –80 °C. Total RNA was extracted from the combined rostral and caudal arcuate nucleus using Ambion RNAqueous-Micro Kits (Life Technologies) per the manufacturer’s protocol. Total RNA was treated with DNase I using the extraction kit protocol at 37 °C for 30 min to minimize any genomic DNA contamination. Liver and arcuate RNA quantity and quality were determined using a NanoDrop ND-2000 spectrophotometer (ThermoFisher, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer and RNA Nano Chips (Agilent Technologies, Santa Clara, CA, USA). Only samples with RNA Integrity Number (RIN) > 7 were used.
Analysis of gene expression used standard protocols for quantitative real-time PCR (qPCR) as previously published35. Briefly, complementary DNA (cDNA) was synthesized using a standard Superscript III reverse transcriptase (Life Technologies) protocol: 5 min at 25 °C, 60 min at 50 °C, and 15 min at 70 °C. All primers were designed to span exon-exon junctions and synthesized by Life Technologies, using Clone Manager 5 software (Sci Ed Software, Cary, NC, USA). See Supplemental Table S1 for a listing of all the primer sequences used for quantitative real-time PCR (qPCR). Primers for Esr1 were designed between exon 1 and 2, which is not deleted in the Ex3a ERα KO. qPCR amplification followed standard protocols for either PowerSYBR Green (Life Technologies) or Sso Advanced SYBR Green (BioRad, Hercules, CA, USA) master mixes on CFX-Connect Real-time PCR instrument (BioRad). All efficiencies were between 90–110%. The relative mRNA expression was calculated using the ΔΔCT method utilizing a calibrator of diluted (1:20) cDNA from liver or BH of an untreated male. The geometric mean of the reference genes Actb, Hprt, and Gapdh was used to calculate δCq values. Quantification values were generated only from samples showing a single product at the expected melting point. All gene expression data were expressed as an n-fold difference relative to the calibrator97.
Statistical Analysis
All data were expressed as mean ± SEM. Due to the occurrence of female WT, KIKO, and KO in each litter (~1 WT and 1 transgenic female/litter), each female represents one litter and all data were analyzed as such. All data were analyzed using Statistica 7.1 software (StatSoft, Tulsa, OK, USA) and by a two-way (maternal diet, genotype) or multi-factorial (maternal diet, genotype, time) ANOVA followed by a post-hoc Newman-Keuls test. GTT and ITT data were analyzed using repeated-measures, two-way ANOVA with a post-hoc Newman-Keuls test. All gene expression data were normalized to WT Control group for comparison across genotypes. All ANOVA statistics are presented in Supplemental Tables S3–S5. In all experiments, effects were considered significant at α ≤ 0.05.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Flegal, K. M. et al. Trends in Obesity Among Adults in the United States, 2005 to 2014. Jama 315, 2284 (2016).
Barker, D. J., Bull, a. R., Osmond, C. & Simmonds, S. J. Fetal and placental size and risk of hypertension in adult life. BMJ 301, 259–262 (1990).
Howie, G. J., Sloboda, D. M., Kamal, T. & Vickers, M. H. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol 587, 905–915 (2009).
Howie, G. J., Sloboda, D. M., Reynolds, C. M. & Vickers, M. H. Timing of maternal exposure to a high fat diet and development of obesity and hyperinsulinemia in male rat offspring: same metabolic phenotype, different developmental pathways? J Nutr Metab 2013, 517384 (2013).
Giraudo, S. Q. et al. Maternal high fat feeding and gestational dietary restriction: Effects on offspring body weight, food intake and hypothalamic gene expression over three generations in mice. Pharmacol Biochem Behav 97, 121–129 (2010).
Samuelsson, A. M., Matthews, P. a., Jansen, E., Taylor, P. D. & Poston, L. Sucrose feeding in mouse pregnancy leads to hypertension, and sex-linked obesity and insulin resistance in female offspring. Front Physiol 4, 1–11 (2013).
Vogt, M. C. et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509 (2014).
Le Foll, C., Irani, B. G., Magnan, C., Dunn-Meynell, A. & Levin, B. E. Effects of maternal genotype and diet on offspring glucose and fatty acid-sensing ventromedial hypothalamic nucleus neurons. Am J Physiol Regul Integr Comp Physiol 297, R1351–R1357 (2009).
Sanders, T. R., Kim, D. W., Glendining, K. a. & Jasoni, C. L. Maternal obesity and IL-6 lead to aberrant developmental gene expression and deregulated neurite growth in the fetal arcuate nucleus. Endocrinology 155, 2566–2577 (2014).
Fahrenkrog, S. et al. Cross-fostering to diabetic rat dams affects early development of mediobasal hypothalamic nuclei regulating food intake, body weight, and metabolism. J Nutr 134, 648–654 (2004).
Bilbo, S. D. & Tsang, V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J 24, 2104–2115 (2010).
Breton, C. The hypothalamus-adipose axis is a key target of developmental programming by maternal nutritional manipulation. J Endocrinol 216 (2013).
Marco, A., Kisliouk, T., Tabachnik, T., Meiri, N. & Weller, A. Overweight and CpG methylation of the Pomc promoter in offspring of high-fat-diet-fed dams are not ‘reprogrammed’ by regular chow diet in rats. FASEB J 1–10 doi:10.1096/fj.14-255620 (2014).
Santollo, J., Torregrossa, A. M. & Eckel, L. a. Estradiol acts in the medial preoptic area, arcuate nucleus, and dorsal raphe nucleus to reduce food intake in ovariectomized rats. Horm Behav 60, 86–93 (2011).
Asarian, L. & Geary, N. Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-a-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 148, 5656–5666 (2007).
Thammacharoen, S., Lutz, Ta, Geary, N. & Asarian, L. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 149, 1609–1617 (2008).
Musatov, S. et al. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA 104, 2501–2506 (2007).
Roepke, T. A. Oestrogen modulates hypothalamic control of energy homeostasis through multiple mechanisms. J Neuroendocrinol 21, 141–150 (2009).
Asarian, L. & Geary, N. Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci 361, 1251–1263 (2006).
Heine, P., Taylor, J., Iwamoto, G., Lubahn, D. & Cooke, P. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA 97, 12729–12734 (2000).
Naaz, a et al. Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): a potential role for estrogen receptor beta (ERbeta). Horm Metab Res 34, 758–763 (2002).
Hammes, S. R. & Levin, E. R. Extranuclear steroid receptors: Nature and actions. Endocr Rev 28, 726–741 (2007).
Qiu, J. et al. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci 26, 5649–5655 (2006).
Roepke, T. A. et al. Genes associated with membrane-initiated signaling of estrogen and energy homeostasis. Endocrinology 149, 6113–6124 (2008).
Roepke, T. A. et al. Contribution of a membrane estrogen receptor to the estrogenic regulation of body temperature and energy homeostasis. Endocrinology 151, 4926–4937 (2010).
Levin, E. R. Integration of extranuclear and nuclear actions of estrogen. Mol Endocrinol 19, 1951–1959 (2005).
Vasudevan, N. & Pfaff, D. W. Membrane-initiated actions of estrogens in neuroendocrinology: Emerging principles. Endocr Rev 28, 1–19 (2007).
Ronnekleiv, O., Malyala, A. & Kelly, M. Membrane-Initiated signaling of estrogen in the brain. Semin Reprod Med 25, 165–177 (2007).
Park, C. J. et al. Genetic rescue of nonclassical ERα signaling normalizes energy balance in obese Erα-null mutant mice. J Clin Invest 121, 604–612 (2011).
Jakacka, M. et al. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol 16, 2188–2201 (2002).
Couse, J. F. & Korach, K. S. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr Rev 20, 358–417 (1999).
Küppers, E., Krust, A., Chambon, P. & Beyer, C. Functional alterations of the nigrostriatal dopamine system in estrogen receptor-α knockout (ERKO) mice. Psychoneuroendocrinology 33, 832–838 (2008).
Semaan, S. J. & Kauffman, A. S. Sexual differentiation and development of forebrain reproductive circuits. Curr Opin Neurobiol 20, 424–431 (2010).
Brannvall, K. Estrogen-Receptor-Dependent Regulation of Neural Stem Cell Proliferation and Differentiation. Mol Cell Neurosci 21, 512–520 (2002).
Mamounis, K. J., Yang, J. A., Yasrebi, A. & Roepke, T. A. Estrogen response element-independent signaling partially restores post-ovariectomy body weight gain but is not sufficient for 17 b -estradiol’ s control of energy homeostasis. Steroids 81, 88–98 (2014).
Lam, Y. Y. & Ravussin, E. Indirect calorimetry: an indispensable tool to understand and predict obesity. Eur J Clin Nutr 1–5 doi:10.1038/ejcn.2016.220 (2016).
Yasrebi, A., Rivera, J. A., Krumm, E. A., Yang, J. A. & Roepke, T. A. Activation of Estrogen Response Element-independent ERα signaling protects female mice from diet-induced obesity. Endocrinology 158, 319–334 (2017).
Ye, J. & McGuinness, O. P. Inflammation during obesity is not all bad: evidence from animal and human studies. Am J Physiol Endocrinol Metab 304, E466–77 (2013).
Coll, A. P., Farooqi, I. S. & O’Rahilly, S. The Hormonal Control of Food Intake. Cell 129, 251–262 (2007).
Nestor, C. C. et al. Optogenetic stimulation of arcuate nucleus Kiss1 neurons reveals a steroid-dependent glutamatergic input to POMC and AgRP neurons in male mice. Mol Endocrinol 30, 630–644 (2016).
Padilla, S. L. et al. AgRP to Kiss1 neuron signaling links nutritional state and fertility. Proc Natl Acad Sci 114, 201621065 (2017).
Cabanes, A., De Assis, S., Gustafsson, J. A. & Hilakivi-Clarke, L. Maternal high n-6 polyunsaturated fatty acid intake during pregnancy increases voluntary alcohol intake and hypothalamic estrogen receptor alpha and beta levels among female offspring. Dev Neurosci 22, 488–493 (2000).
Attig, L. et al. Dietary Alleviation of Maternal Obesity and Diabetes: Increased Resistance to Diet-Induced Obesity Transcriptional and Epigenetic Signatures. PLoS One 8, (2013).
Ashino, N. G. et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem 23, 341–348 (2012).
Borengasser, S. J. et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology 154, 4113–4125 (2013).
Von Wilamowitz-Moellendorff, A. et al. Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes 62, 4070–4082 (2013).
Zammit, V. a. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem J 451, 1–12 (2013).
Leonhardt, M. & Langhans, W. Fatty acid oxidation and control of food intake. Physiol Behav 83, 645–651 (2004).
Strable, M. S. & Ntambi, J. M. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol 45, 199–214 (2010).
Kumar, D., Periasamy, V., Freese, M., Voigt, A. & Boehm, U. In utero development of kisspeptin/GnRH neural circuitry in male mice. Endocrinology 156, 3084–3090 (2015).
Walker, D. M., Kirson, D., Perez, L. F. & Gore, A. C. Molecular profiling of postnatal development of the hypothalamus in female and male rats. Biol Reprod 87, 129 (2012).
Flowers, M., Sanek, N. & Levine, J. Maternal Phytoestrogen Consumption Programs Body Weight Regulation By Non Classical Estrogen Receptor Alpha Signaling in Female Offspring. Progr 96th Annu Meet Endocr Soc Abstract MON-0932 http://press.endocrine.org/doi/abs/10.1210/endo-meetings.2014.OABA.18.MON-0932 (2014).
Jensen, M. N. & Ritskes-Hoitinga, M. How isoflavone levels in common rodent diets can interfere with the value of animal models and with experimental results. Lab Anim 41, 1–18 (2007).
Ruhlen, R. L. et al. Low phytoestrogen levels in feed increase fetal serum estradiol resulting in the ‘fetal estrogenization syndrome’ and obesity in CD-1 mice. Environ Health Perspect 116, 322–328 (2008).
Shi, H., Seeley, R. J. & Clegg, D. J. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 30, 396–404 (2009).
Wang, A. et al. GPR30 regulates diet-induced adiposity in female mice and adipogenesis in vitro. Sci Rep 6, 34302 (2016).
Speakman, J. R. Measuring energy metabolism in the mouse - theoretical, practical, and analytical considerations. Front Physiol 4, 34 (2013).
Johnson, S. et al. Effects of a maternal high-fat diet on offspring behavioral and metabolic parameters in a rodent model. J Dev Orig Health Dis 8, 75–88 (2017).
Xu, Y. et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 14, 453–465 (2011).
Barros, R. P. a. & Gustafsson, J.-Å. Estrogen receptors and the metabolic network. Cell Metab 14, 289–299 (2011).
Mauvais-Jarvis, F., Clegg, D. J. & Hevener, A. L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34, 309–338 (2013).
Jelenik, T. & Roden, M. How estrogens prevent from lipid-induced insulin resistance. Endocrinology 154, 989–992 (2013).
Gorres, B. K., Bomhoff, G. L., Morris, J. K. & Geiger, P. C. In vivo stimulation of oestrogen receptor α increases insulin-stimulated skeletal muscle glucose uptake. J Physiol 589, 2041–54 (2011).
Dunn, Ga & Bale, T. L. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152, 2228–2236 (2011).
King, V. et al. Maternal obesity has little effect on the immediate offspring but impacts on the next generation. Endocrinology 154, 2514–2524 (2013).
Yokomizo, H. et al. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic β-cell function in adult offspring with sex differences in mice. Am J Physiol Endocrinol Metab 306, E1163–75 (2014).
Yonezawa, R. et al. Central versus peripheral impact of estradiol on the impaired glucose metabolism in ovariectomized mice on a high-fat diet. Am J Physiol Endocrinol Metab 303, E445–56 (2012).
Liu, J. et al. Intrahypothalamic estradiol regulates glucose metabolism via the sympathetic nervous system in female rats. Diabetes 62, 435–443 (2013).
Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860–867 (2006).
Shoelson, S. E., Lee, J. & Goldfine, A. B. Inflammation and insulin resistance. J Clin Invest 116, 1793–1801 (2006).
Ribas, V. et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ER -deficient mice. Am J Physiol Endocrinol Metab 298, 304–319 (2010).
Rother, E. et al. Hypothalamic JNK1 and IKKβ activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology 153, 770–781 (2012).
Grayson, B. E. et al. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 151, 1622–1632 (2010).
Riant, E. et al. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 150, 2109–2117 (2009).
Krause, M. da, S. et al. Physiological concentrations of interleukin-6 directly promote insulin secretion, signal transduction, nitric oxide release, and redox status in a clonal pancreatic β-cell line and mouse islets. J Endocrinol 214, 301–311 (2012).
Suzuki, T. et al. Interleukin-6 enhances glucose-stimulated insulin secretion from pancreatic β-cells: Potential involvement of the PLC-IP3-dependent pathway. Diabetes 60, 537–547 (2011).
Ellingsgaard, H. et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17, 1481–1489 (2011).
Allen, T. L., Whitham, M. & Febbraio, M. A. IL-6 muscles in on the gut and pancreas to enhance insulin secretion. Cell Metab 15, 8–9 (2012).
Yang, J., Mamounis, K. J., Yasrebi, A. & Roepke, T. A. Regulation of gene expression by 17β-estradiol in the arcuate nucleus of the mouse through ERE-dependent and ERE-independent mechanisms. Steroids 107, 128–138 (2016).
Gorski, J. N., Dunn-Meynell, A. a. & Levin, B. E. Maternal obesity increases hypothalamic leptin receptor expression and sensitivity in juvenile obesity-prone rats. Am J Physiol Regul Integr Comp Physiol 292, R1782–R1791 (2007).
Page, K. C., Malik, R. E., Ripple, J. a. & Anday, E. K. Maternal and postweaning diet interaction alters hypothalamic gene expression and modulates response to a high-fat diet in male offspring. Am J Physiol Regul Integr Comp Physiol 297, R1049–R1057 (2009).
Chen, H., Simar, D. & Morris, M. J. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: Interaction with postnatal nutritional environment. PLoS One 4 (2009).
Rajia, S., Chen, H. & Morris, M. J. Maternal overnutrition impacts offspring adiposity and brain appetite markers-modulation by postweaning diet. J Neuroendocrinol 22, 905–914 (2010).
Mittelman-Smith, M. A. et al. Arcuate kisspeptin/neurokinin B/dynorphin (KNDy) neurons mediate the estrogen suppression of gonadotropin secretion and body weight. Endocrinology 153, 2800–2812 (2012).
Brown, C. M., Mulcahey, T. a., Filipek, N. C. & Wise, P. M. Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors alpha and beta. Endocrinology 151, 4916–4925 (2010).
Peña, C. J., Neugut, Y. D. & Champagne, F. a. Developmental timing of the effects of maternal care on gene expression and epigenetic regulation of hormone receptor levels in female rats. Endocrinology 154, 4340–4351 (2013).
Kundakovic, M. et al. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci USA 110, 9956–61 (2013).
Ariazi, E. A. et al. A New Role for ERα: Silencing via DNA Methylation of Basal, Stem Cell, and EMT Genes. Mol Cancer Res 15, 152–164 (2016).
Ung, M., Ma, X., Johnson, K. C., Christensen, B. C. & Cheng, C. Effect of estrogen receptor α binding on functional DNA methylation in breast cancer. Epigenetics 9, 523–532 (2014).
Wu, Z. et al. 17b-oestradiol enhances global DNA hypomethylation in CD4-positive T cells from female patients with lupus, through overexpression of oestrogen receptor-α-mediated downregulation of DNMT1. Clin Exp Dermatol 39, 525–532 (2014).
Hewitt, S. C. et al. Novel DNA Motif Binding Activity Observed In Vivo With an Estrogen Receptor α Mutant Mouse. Mol Endocrinol 28, 899–911 (2014).
Hewitt, S. C. et al. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J 24, 4660–4667 (2010).
Sun, B. et al. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 61, 2833–2841 (2012).
Dunn, G. a., Morgan, C. P. & Bale, T. L. Sex-specificity in transgenerational epigenetic programming. Horm Behav 59, 290–295 (2011).
Pallarés, M. E. et al. Age-dependent effects of prenatal stress on the corticolimbic dopaminergic system development in the rat male offspring. Neurochem Res 38, 2323–2335 (2013).
Paxinos, G. & Franklin, K. B. J. The Mouse Brain in Stereotaxic Coordinates, Compact, Third Edition: The coronal plates and diagrams. (Academic Press, 2008).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108 (2008).
Acknowledgements
The authors must thank Dr. Sara Campbell for the use of the EMD Millipore MAGPIX® Multiplex® System, Dr. Judith Storch for the use of the Comprehensive Lab Animal Monitoring System and the EchoMRI 3-in-1 Body Composition Analyzer, and the many undergraduate students who assisted in genotyping and weighing the mice. This research was supported by funds from USDA-NIFA NJ06107 and from National Institutes of Health R00DK083457, R00DK083457-S1, and P30ES005022.
Author information
Authors and Affiliations
Contributions
T.A.R. contributed to conceptualization and to methodology; T.A.R., A.Y., J.A.Y., A.V., and K.J.M. contributed to formal analysis; A.Y., A.V., E.A.K., J.A.Y., K.J.M., and T.A.R. contributed to investigation; T.A.R. contributed to writing, review, and editing and contributed to funding acquisition and supervised the research.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roepke, T.A., Yasrebi, A., Villalobos, A. et al. Loss of ERα partially reverses the effects of maternal high-fat diet on energy homeostasis in female mice. Sci Rep 7, 6381 (2017). https://doi.org/10.1038/s41598-017-06560-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06560-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
