
Abstract
Protein-protein interactions networks (PPINs) are known to share a highly conserved structure across all organisms. What is poorly understood, however, is the structure of the child interface interaction networks (IINs), which map the binding sites proteins use for each interaction. In this study we analyze four independently constructed IINs from yeast and humans and find a conserved structure of these networks with a unique topology distinct from the parent PPIN. Using an IIN sampling algorithm and a fitness function trained on the manually curated PPINs, we show that IIN topology can be mostly explained as a balance between limits on interface diversity and a need for physico-chemical binding complementarity. This complementarity must be optimized both for functional interactions and against mis-interactions, and this selectivity is encoded in the IIN motifs. To test whether the parent PPIN shapes IINs, we compared optimal IINs in biological PPINs versus random PPINs. We found that the hubs in biological networks allow for selective binding with minimal interfaces, suggesting that binding specificity is an additional pressure for a scale-free-like PPIN. We confirm through phylogenetic analysis that hub interfaces are strongly conserved and rewiring of interactions between proteins involved in endocytosis preserves interface binding selectivity.
Similar content being viewed by others
Introduction
Interface interaction networks (IINs), also referred to as structural interaction networks1, 2, domain-domain interaction networks3, 4, or structurally annotated pathways5, are a map of the binding sites proteins use for various interactions. Such a map can be used to model how competition modulates signal transduction4, 6; predict the effects of domain mutations on disease2, 7,8,9 and the immune response10, predict dosage sensitivity by identifying linear motifs and promiscuous regions11, and study the structure and dynamics of multi-protein complexes12. For example, Actin can form long fibers because it has a “barbed” end that binds to a “pointed” end of another Actin protein. On a typical protein-protein interaction network (PPIN) map, this interaction would appear as a self-edge, whereas more accurately, they are two distinct binding sites with their own share of possible partners.
We ask four major questions in this work. First, is the structure of IINs conserved across PPINs? Second, does this structure reflect any selective constraints on protein interactions? Third, do the presence of hubs in the PPIN network affect the types of IIN structures possible? And fourth, do hubs in the PPIN provide an advantage (relative to random networks) in producing selective interface interactions with minimal interfaces, suggesting a new benefit for scale-free PPINs? The answer is yes in all cases.
We analyze the structure of four PPINs with IINs defined: two smaller manually curated networks (621 total interactions) and two larger automatically constructed networks (6,893 interactions). Little work has been done on IIN structure, in large part due to the paucity of experimental and crystallography data identifying where proteins bind to one another. The protein data bank13 provides the optimal resource for having a computer automatically assign interfaces. However, with limited crystal structures of proteins in complex, homology modeling5, 14, 15 is needed to help infer domains and interfaces used for interactions. Interfaces assigned through homology modeling are only putative, however, as this approach is limited in accuracy. The binding sites discovered will depend on the experimental templates used, and even if the sites have similar sequence there is no guarantee of an interaction15. Stein et al., using known PPIs from six organisms including humans, estimated that less than 30% have a template for comparative modeling16. The Interactome3D approach uses several criteria to improve accuracy in predicting binding interfaces, but recovered acceptable models for only ~64% of interactions in their database5. Homology modeling will also miss many short linear motif (SLiM)-mediated interactions17, both due to their rapid evolution18 and low affinity, which has hindered experimental detection19. As we see below, limited accuracy in automatically predicted interfaces significantly alters the structure of the IIN, although major features are still visible.
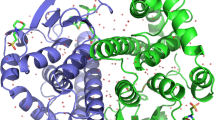
With manual curation, in contrast, putative interfaces can be refined, corrected, or rejected, and the many protein interactions that lack homology models can be assigned based on detailed biochemical approaches, functional studies, and analysis of disordered regions and SLiMs. So far, two such IINs have been constructed to this gold standard: the clathrin-mediated endocytosis network in yeast20, and the ErbB signaling network in humans4 (Figure 1). Despite being independently constructed by different research groups, the two share similar features: fragmentation into multiple components, little clustering, and a high frequency of square and hub motifs. With the exception of the presence of hubs, these features differ from their parent PPINs, and thus display a unique topology that we show results due to different selective forces.

PPINs all contain hub proteins and their IINs have distinctive topologies. We analyze the PPINs of the manually curated yeast endocytosis (a) and human ErbB networks (b) with all domains and interfaces identified and shown here colored by domain type (Table S2). The resulting interface interaction networks (IINs) in (c) and (d), respectively, have highly distinct topologies that reflect the needs of interfaces to achieve strong functional binding and minimize non-functional interactions. Both IINs break into multiple components with a selection of hub interfaces, and they contain an abundance of hub and square motifs with a minimal (or zero) number of triangle motifs.
For the second question, we propose that one of the selective forces shaping IIN structure is the need to maintain high binding specificity. Due to the chemical nature of binding sites, occasionally nonspecific misinteractions will occur. Avoiding these misinteractions has been demonstrated to be a fundamental force limiting the number of distinct proteins in an organism21, 22, protein expression levels11, 23, 24, binding strengths25, and interface interaction motifs21, 26. In regards to IIN motifs, it was found via an amino acid residue optimization model that specific motifs (and not others) and a fragmented IIN structure were needed to optimally design protein interfaces for high specificity21, 26. We first compare IIN structures to randomized versions, to demonstrate the biological networks’ clear departure from the statistically most probable IIN structure. We then construct a trainable fitness function to reproduce the observed biological IIN. This fitness function favors network motifs that have been shown to improve the sequence-based binding selectivity of interfaces26, and also penalizes high interface diversity. Hence we do not optimize amino-acid sequences, as has been done previously, but rather the network motifs shown to correspond with highly selective sequences21, 26. Limiting total interface numbers both lowers the number of possible misinteractions that must be optimized against (order of n2)22 and mimics the limited size of proteins, which cannot harbor unlimited interfaces. Because the search space for possible IINs of a given PPIN is enormous (quantified below), we used a Monte Carlo sampling algorithm combined with a fitness function (Methods) to find the optimal IIN at various parameterizations, similar to previous work optimizing spatial networks27.
Because the automatically constructed1, 2 IINs contained systematic errors, largely due to missing SLiMs as binding partners and incorrect replacements, we restricted our training and sampling procedure to the two manually curated networks. However, this outcome highlighted a powerful advantage of visualizing the IINs: the network motifs can be used to identify erroneous domain-domain interaction predictions. Disagreements over the evolution of proteins and their networks can often be attributed to variability and poor overlap in PPIN datasets28. Boosting domain assignment accuracy by identifying errors in automatically constructed networks using network motifs, as we demonstrate here, improves these crucial resources for understanding protein function and evolution.
To address the third question and learn how the presence of protein hubs affects the IIN sampling space, we combined both analytical and computational sampling approaches to characterize the structure of IINs as a function of varying PPIN structure. PPINs feature a degree distribution that is approximately power-law or “scale-free”, meaning (loosely speaking) that a few proteins act as hubs, while the majority of proteins are specialized to only a few interaction partners29. This same basic structure describes airport networks, and is the optimal structure for maximizing transport with minimal costs30. By considering the possibility of a random PPIN, we can then compare whether this alternative structure is different and possibly worse than a scale-free PPIN in terms of IINs possible. For example, a well-known advantage of scale-free PPINs relative to random networks is their ability to maintain connectivity under attack31. Because IINs have not been studied in the context of their parent PPIN, we first establish how the whole domain of possible IINs varies with PPIN structure, showing that hubs do alter the space of IINs in specific ways.
For our fourth question, we sought to test whether the real PPINs were any better for developing selective binding than the random PPINs. We applied our data-trained fitness function at its optimal parameters to sample IINs for scale-free versus random PPINs of the same size. Random PPINs proved more difficult to optimize, requiring the evolution of significantly more interfaces (penalized in our fitness function) in order to achieve the same level of binding complementarity encoded in the IIN motifs. This runs counter to the parsimonious use of domains across species, where new domain combinations rather than new domains drive functional divergence32. Ultimately our result suggests an additional pressure for a scale-free-like PPIN. It is a cheaper (fewer interfaces) design for maintaining a multitude of selective binding interactions.
Our model emphasizes that selectivity in interface binding is critically conserved across IINs, and that hubs in the PPIN provide an advantage in this regard, largely because they may contain hub interfaces. As a final analysis we use phylogenetic analysis to test whether interface binding selectivity is conserved as protein-protein interactions are rewired throughout evolution33. We use this analysis to test whether, despite this rewiring, hub interfaces are nonetheless conserved, providing a new physico-chemical argument supporting the conservation of hub proteins.
Results
IINs for the biological PPINs have highly specialized features sensitive to rewiring
To determine if IIN structure is conserved across PPINs, we first characterize the manually curated PPINs from yeast and humans shown in Fig. 1a,b (Table S2), which involve different protein sets but both exhibit scale-free-like topologies. Analysis of both their IINs (Fig. 1c,d, Table S2) demonstrates that they both share highly similar features to one another and are topologically unique. They have fragmented structure, almost no triangle motifs (low Cglobal), a higher fraction of hub versus chain motifs, and a significant fraction of square motifs (Table 1). In contrast, expected values for these features, calculated by randomly rewiring the interface interactions while keeping the PPIN structure intact, have no similarities (Table 1). Rewired IINs organized into a giant component with many chains, increased triangles (higher clustering coefficient Cglobal), and minimal squares (Table 1, Fig. S1, Table S6). The lack of hub interfaces in these rewired IINs is reflected by the low preferential attachment exponent (P.A.E.), which varies from 0 for random networks to ~1 for scale-free networks (Methods).
The structure of the two automatically constructed IINs1, 2 was in some ways similar to the manually curated IINs, but they are closer on the spectrum towards a randomly rewired network. Similar to the manually curated networks, they have a large PAE, indicating hub interfaces in the network, and a similar fraction of square motifs (Table 1). They also have correspondingly more hub motifs in the network than would be observed in a random network. A significant difference is the degree of fragmentation. The manually curated networks are nearly fully connected at the PPIN level, and yet the IINs contain a largest connected component of only 23–35% of nodes. In contrast, Human SIN2 is already fragmented at the PPIN level (43% of nodes in the largest component), and the IIN fragmentation is therefore more strongly driven by the PPIN fragmentation. The Yeast SIN1 is even more dramatic. The reason for the higher connectivity in these IINs is the larger ratio of chain to hub motifs (Table 1), as chain motifs prevent fragmentation into many distinct modules (Fig. S2). The number of triangle motifs, which is directly quantified by the clustering coefficient Cglobal, is also significantly higher in these networks than in the manually curated networks (Table 1). Does the increased randomness of these IIN connections occur due to mis-identification of interaction interfaces? By following up on this implication by investigating the many unexpected triangles in the automatically curated IINs, we found this was true (Fig. S2 and Supplementary Text S5).
We found mis-assignments of interface interactions can be largely attributed to a lack of linear motifs included as potential binding partners, and a permissive decision-making algorithm. Application of the INstruct website34 to predicting CME protein interface interactions produces only 44 interactions (versus 206 for the manually curated network of Fig. 1a 20). Of these 44 predicted interactions, only 1 defines the correct domains (Fig. S2). This method predicts a disproportionate abundance of homo-dimers. Many interactions are predicted to be SH3-SH3 interactions (including in the Human SIN2 (Fig. S2)), but even in the crystal structures, SH3 domains form homo-dimers only in special cases when mediated by a ligand (such as a PRD)35. We also note that some structured domains (such as kinase domains) must be recognized as containing multiple protein binding interfaces. Many kinase domains, for example, form dimers through distinct interfaces and can still perform catalysis36.
Network motifs in the IINs indicate suppression of nonfunctional interactions
For our second question, we connect the special conserved structure of the biological IINs (Fig. 1 and Table 1) to constraints on binding selectivity. In previous work, using Monte Carlo based optimization of amino acid sequences in small networks, it was shown that when interface interactions were mediated by hub or pair motifs, and not chain motifs, the binding selectivity (measured via the energy of binding interactions) of the interfaces was significantly higher21. Thus the level of achievable binding complementarity and selectivity is encoded in these basic motifs, which include hubs, pairs, and chains. Subsequently, it was shown that IINs were also more selective if they were highly fragmented into modules26. In both cases this is because it is easier to optimize the interface sequences for both strong specific interactions, and against non-functional mis-interactions. All of these trends are clearly present in the biological IINs, and not the random IINs (Table 1). In Fig. 2 we further illustrate how, for the same reason, square motifs are beneficial to selectivity, and triangle motifs are detrimental. While it is perfectly possible to design interfaces that will bind strongly in any motif configurations, the real challenge is to simultaneously suppress the nonfunctional interactions possible for those motifs. For the chain motif, the challenge is preventing the interaction between the two ends of the chains. For the triangle motif, in order for all three distinct domains to attract one another, they must all be similar to one another. If an interface binds a very similar interface to itself, it will likely also bind to itself. Thus, triangle motifs are only consistent with high-selectivity optimization if their interfaces are also self-binding. We found that for the one triangle present in the ErbB IIN, this was indeed the case. Two kinase domains form not only a heterodimer with a shared target, they also both form homo-dimers36, and hence we added these previously undefined self-interactions to the network.

Motifs uncommon in the biological IINs due to poor interface binding selectivity. (a) IIN motifs that confer high selectivity. Binding partners may achieve structural and chemical complementarity with few constraints. (b) IIN motifs with poor selectivity. (c,d) For the motifs with poor selectivity, (chain and triangle motifs) functional interactions are indicated by solid lines and interfaces should be optimized to stably form these complexes. However, the possible non-functional interactions between the interfaces (red dashed lines) are difficult to simultaneously optimize for much weaker binding because they share structural and chemical complementarity to the functional complexes. For the chain motif, illustrated with SH3 and PRD domains, the top interaction (S2-P1) is hard to prevent. For the triangle motif, illustrated with kinase domain interfaces that form side-by-side dimers (B-raf, Raf-136, Ksr1) the homo-dimer binding interaction is hard to prevent. In fact, these kinase interfaces really do form both hetero- and homo-dimers, so the biological system has no challenge for optimizing selectivity. Binding surfaces are colored by residue as non-polar (white), polar (green), acidic (red), and basic (blue). Example structures from 2RPN, 2LCS, 1UWH, 3OMV.pdb. Truly non-functional interactions (i.e. PRD-PRD) are just illustrations.
The space of possible interface networks for a PPIN is enormous and varies with protein degrees
Our third question considers how the PPIN structure might constrain the IINs accessible. While a PPIN and its interface interaction network (IIN) must evolve together, it is not obvious how one constrains the other, given that a protein can use one or many interfaces for its various partners. To illustrate properties of IINs constrained to a PPIN, in Fig. 3 we enumerate the 8 possible IINs for the simple PPIN of three proteins binding. The total number of possible interface networks is determined by the number of interactions (degree, k) per protein and quantified through the Bell number Bk. Bell numbers grow rapidly and hence high-degree hub nodes can dramatically increase the number of possible IINs, meaning a scale-free PPIN will have significantly more IINs possible than a random PPIN because of its hubs. We calculate 10166 IINs for the clathrin-mediated endocytosis (CME) PPIN in Fig. 1a, and 10143 for a similarly sized random PPIN (more than the number of atoms in the universe!) (Table S1, Supplementary Text S4). Both types of PPINs produce IINs with an expected degree distribution that is random, not scale-free. This is because configurations that create hub interfaces, which are necessary to produce a scale-free IIN, are rare. However, hub proteins do cause several subtle shifts in the properties of the IINs possible, including slightly fewer expected interfaces, more 4-node motifs (tetramers) and more hub interfaces (Table S1, Supplementary Text S4). Since these are the features important in the biological IINs, this is an indication that the hub proteins found in scale-free PPINs may promote more selective IINs.

Each PPIN has many possible IINs, and only some are good for promoting selectivity. For the simple PPIN with three interacting proteins, there are 8 possible IINs with either 3, 4, 5 or 6 interfaces (blue squares). Because each IIN has different motifs present, only a subset will be favored in biological networks. The top row contains IINs with chain motifs or a triangle motif (red box), which are bad for promoting selectivity and less common in biological IINs. The bottom row contains favorable motifs, and in the green box is the only IIN that allows a true protein trimer to form. IINs with 4 or 5 interfaces are most common, as counted in the histogram. The same trend holds for much larger PPINs, with the sparse and dense IINs becoming increasingly rare, and hub interfaces less common.
Strong motif biases are needed to reproduce biological IINs
To answer question four, and address whether the PPIN structure influences the ability to produce biologically optimal IIN structures, we first needed to be able to sample biologically realistic IINs given a PPIN. To do so we created a fitness function and trained it to reproduce the networks of Fig. 1. Due to the inaccuracies of the automatically constructed IINs (Table 1), we did not include them to avoid training the fitness functions towards erroneous network structures. The fitness function is biologically motivated to penalize features that promote mis-interactions, to not penalize features that promote strong interactions, and to capture physical size constraints of proteins. We therefore included a bias against triangle subgraphs without self loops (parameterized by β) and chain subgraphs (parameterized by κ), which are difficult to optimize for structural and chemical complementarity as explained above (Fig. 2). These two separate terms resolved a problem we found with our previous fitness function26: this earlier approach did penalize chain subgraphs, but it also ended up penalizing biologically realistic square subgraphs. Our current fitness function does not penalize squares. We introduced a third parameter, μ, to penalize having large numbers of interfaces in the network, both because this increased diversity leads to more possible misinteractions22 and because proteins have limited volume for extra interfaces. Finally, in the biological IINs, protein pairs can interact through multiple domains, resulting in a significant increase in edges from the PPIN to the IIN (Table 1). Our fourth and final term thus allowed new duplicate edges in the IIN but limited their growth by a parameter ω. All four parameters are dimensionless and weight topological properties of the network (see Methods for details and illustrations).
We had to optimize the four parameters of our fitness function to locate the biological IINs out of the enormous space of possible IINs (e.g. 10166), where all parameters were greater than or equal to zero, and setting a parameter to zero effectively turned off that fitness pressure on the IINs (Fig. S3). All four parameters were needed. We found that the key to generating realistic IIN features required a balance of creating new fragmented modules without introducing too many interfaces. To do so required re-using interfaces that would generate either isolated star hubs (e.g. turquoise nodes in Fig. 1c) or hubs connected in square clusters (e.g. orange and pink nodes in Fig. 1c,d). In Fig. 4 we show how the most important parameters for simultaneously capturing these dominant features of the IINs were κ and μ. The parameter κ penalizes chains and μ penalizes the creation of new interfaces, and together they exhibit the most sensitive control over the IIN structure (Methods). Star hubs, like squares, result from pressure to avoid chains and hence are also positively selected for with increasing κ (Fig. S3). Our trained fitness function samples IINs with very close agreement to the observed CME network (Fig. 4d, Fig. S3) with parameters κ = 2, μ = 0.42, β = 4 and ω = 0.1. Comparable parameters applied to the ErbB PPIN (κ = 2.3, μ = 0.45, β = 4) except we lowered ω to 0.02 to account for the much greater frequency of edge duplication. In the discussion we consider ways to further improve the agreement.

Learning how to select biologically realistic IINs for a PPIN using a parameterized fitness function. Because biological IINs are so distinct from a randomly generated IIN, we needed a four parameter fitness function to bias the sampling towards the correct: (a) number of interfaces (b) size of the largest module/fragment (c) Frequency of square motifs in the IINs, as well as other properties (Fig. S3). The results were most sensitive to variation in the parameters κ and μ (on the axes) that regulated the square-to-chain ratios and number of interfaces, respectively, in the fitness function. White stars on color bars indicate observed values of the CME PPIN (Fig. 1a). (d) By training the fitness function, we achieved very good agreement between the properties of the sampled IINs and observed CME IIN with optimal fitness parameters κ = 2, μ = 0.42, β = 4 and ω = 0.1. Comparable parameters applied to the ErbB PPIN (κ = 2.3, μ = 0.45, β = 4) except we lowered ω to 0.02 to account for the much greater frequency of edge duplication.
PPINs need hubs to minimize new domain interfaces
We used our trained fitness function to sample IINs for a variety of PPIN topologies and sizes and therefore answer our fourth question. We compared the CME and ErbB PPINs with PPINs of the same size but a random degree distribution, and performed the same experiment for new PPINs both more and less densely connected than these (Fig. S4). Regardless of the size of the PPINs, we found that because random PPINs lack hub proteins, they cannot produce selective domain modules without significant addition of new interfaces (Fig. 5). Thus random PPINs do have a disadvantage, as evolving more interfaces is a more costly way of mediating protein-protein interactions than re-using domains already optimized for selectivity.

Scale-free PPINs produce fitter IINs than random PPINs. We performed fitness sampling for selective IINs on the ErbB scale-free like PPIN (top) and a random network with the same number of proteins and PPIs (bottom). For the scale-free like PPIN (top) fewer interfaces (n = 290) were needed to produce selective motifs, including 2000 squares (in green circled modules). Without hub proteins, the random PPIN (bottom) produced only 12 squares, and introduced many additional interfaces (n = 356) in order to maintain selective motifs. The same trends held with the CME PPIN (Fig. S1). IINs discovered with random PPINs were also less fit than those found with scale-free PPINs (see Table S7). Nodes with >9 partners are shown in blue.
The main advantage of hub proteins in a PPIN is that they are capable of more highly connected hub interfaces in the IIN. Although hub interfaces are still possible for a random PPIN of sufficient density (Figs S4 and S5A), the reduced size and frequency of these hubs limits how many square motifs can form (Fig. S5B). Square cluster components are a prominent feature of the biological IINs and they are critical for maintaining selectivity with a minimum number of interfaces. Without access to these motifs, random PPINs require more interface splitting to instead produce selective star hubs. These results were robust to changes in the fitness function that allowed larger fluctuations in interfaces per protein (Supplementary Text S1, Fig. S1). Ultimately, our results suggest that a scale-free-like PPIN is beneficial to evolving specificity in interface binding interactions.
Network rewiring maintains selectivity
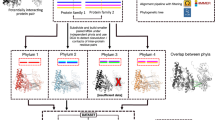
Our results imply that selectivity in interface interactions is highly conserved across various protein networks. Therefore, if we compare IINs across evolution, we should find that rewiring of interactions between species is not random (as they are treated in growth models) but correlated and constrained to maintain this selectivity. Orthologous proteins with similar domain sets may change protein interactions but should preserve domain partners, as has been experimentally observed in SH3 domain interactions between worms and yeast37. By comparing the yeast CME PPIN with a human CME PPIN constructed (Methods) from 64 proteins with recognized functional homology38 (Table S4), we find that rewiring events are highly correlated and attributable to specific binding domains (Fig. 6). From yeast to humans, about half of the interactions are conserved. Of those that are lost, 39% are due to lack of a homologous protein, and 98% of the remainder involved at least one domain that retained no interaction partners (Fig. 6C, Table S5). A major source of divergence was domains targeting the linear motif proline rich domains (PRDs) and phospho-sites (Fig. 6B). SH3-PRD interactions accounted for over half the losses from yeast to humans. The divergence of these interactions can be attributed to the biological distinctions between yeast and metazoan CME: in yeast the actin cytoskeleton is required to deform the stiffer cell membrane and the SH3 containing proteins link the cytoskeleton to the clathrin-coated vesicle38. New interactions gained within the human PPIN were concentrated in a few proteins, most significantly in the AP-2 complex (Fig. S6, Table S5). The source of these new interactions is an added appendage domain to the human AP-2 complex that interacts with a range of diverse binding partners39. Without this hub domain, the yeast AP-2 complex evolved with few binding partners, accounting for the minimal interaction conservation between the homologs.

Network rewiring between yeast and human CME networks is correlated and controlled by specific domains. (a) Comparison of the CME interactome of 56 yeast proteins with that of their 64 human homologs reveals the majority of interactions are either conserved or lost from yeast to humans due to a missing homolog in the human network. Analysis of changes from the human to the yeast interactome in Fig. S6. (b) Of the interactions lost from yeast to human, they were highly correlated, with most being lost due to a full protein homolog being lost, or a domain losing all binding partners. (c) Most lost interactions involved SH3 and proline rich domain (PRD) interactions, or kinase-phosphosite interactions, highlighting the fluidity of linear motif driven interactions. (d) Some yeast proteins conserved almost no interactions with human counterparts, and these proteins contain SH3 domains.
Hub interfaces in the CME and ErbB networks are strongly conserved
Our results also emphasize the importance of hub interfaces to avoid the need for new domain innovation. We thus predict hub interfaces should be preferentially conserved throughout evolution. With all the domain information available for the two manually curated networks (Fig. 1), we can isolate the contribution of hub interfaces to hub protein evolution. Hub proteins may evolve more slowly40, and one (among other41) rationale is that it is harder to change with so many binding partners. However, a conflicting observation is that hub proteins also have more disordered regions42, which evolve more rapidly43. Furthermore, a distinction between evolutionary rates of different hub types (date vs party hubs) may actually be attributable to expression levels44, 45, which, along with number of translational events46 are the strongest predictors of evolutionary rates47. Our analysis (Methods and Table S3) of residue conservation demonstrates that hub interfaces (defined in two independent ways) are significantly more likely to be conserved than other binding interfaces, with almost 90% being strongly conserved, compared with 70% of non-hub interfaces (Table S8). Because we evaluate conservation on both hub and non-hub interfaces of the very same proteins, the effects of protein expression level variation towards conservation are automatically accounted for. Whether a protein has high or low expression, its hub interfaces are more strongly conserved than its non-hub interfaces. It is the interfaces that bind to the hub interfaces that are more likely to have weaker conservation (Table S8), hence facilitating the growth and rewiring to hub interfaces. This analysis thus directly explains how many hub proteins can participate in more rewiring events33, but still evolve slowly: the partners are the ones evolving to achieve binding.
Hub proteins and disordered regions
Lastly, to better characterize the hub proteins in our network, some of which do not contain hub interfaces, we assessed the role of disordered regions in hub proteins for mediating interactions. We found that hub proteins with few interfaces, and thus more highly connected hub interfaces, were less likely to use disordered regions to mediate interactions (Fig. S7). This is expected because hub interfaces are highly conserved (Table S8), and are thus unlikely to be disordered regions such as PRDs, which have low conservation (Table S8). Examples from our two networks are the kinase PRK1, which uses its kinase domain for ~83% of interactions, and the ErbB proteins MAPK1 and PIK3R1. Conversely, hub proteins with many interfaces, and thus without hub interfaces, used disordered regions to mediate a significantly larger fraction of their interactions. Examples include the hubs LAS17 and ABP1, which use disordered interfaces for 78% and 46% of interactions, respectively. But many hub proteins fall in between, existing on a stratum between having several unstructured binding regions and having a few highly connected structured binding domains (Fig. S2).
Discussion
PPINs feature a scale-free-like topology. Much like airport networks, a few proteins act as hubs, while the majority of proteins are specialized to only a few interaction partners. Stochastic growth models48,49,50 provide a simple explanation for how protein networks acquire a scale-free topology. Hubs are generated via protein genes duplicating and diverging48, 51, where at least one of the duplicated proteins retains an original interaction as they sub-functionalize52, 53. While gene duplication and divergence is undoubtedly a source of evolutionary changes to protein interactions, the network growth models of duplication and divergence have an unrealistic portrayal of rewiring, usually performing only one rewiring per duplication event, and without incorporating any physico-chemical or evolutionary basis for the rewiring. Rewiring happens on a much faster evolutionary timescale than gene duplication: the human interactome has been estimated to rewire 1000 times per million years33, 51, whereas gene duplication is estimated to occur at a rate of 2 to 30 events per million years54, 55 (assuming 20,000 genes), with the majority of these duplications being deleted by natural selection56. Orthologous proteins between species are often highly rewired, as a recent study comparing the yeast and worm SH3 interactome found37. Additionally, growth models ignore homo-dimers despite their prevalence57 and influence on evolving new interactions58.
Biological rewiring is capable of abolishing the majority of interactions from one species to another59, and creating and destroying transcription factor60 and protein hubs such as AP-261 between species38. If the rewiring were random, it would destroy any scale-free structure created by gene duplication. Yet scale-free topology is conserved, and this suggests rewiring is not random and hubs are preferentially conserved51. A scale-free topology is known to provide benefits relative to a random network in that it fortifies communication across networks by centralizing connections into hubs31. We propose that our results provide another advantage of hubs in PPINs: they improve binding selectivity and avoidance of misinteractions. This selection pressure is of molecular origin and reflects directly on the primary physico-chemical requirements of proteins to fold into stable structures and bind to other molecules. Hub proteins allow the creation of hub interfaces, which facilitates chemical and structural complementarity and selectivity with the fewest number of interfaces needed.
We note that the actual IINs were not the most optimal solutions in any fitness landscape. Raising the temperature allowed us to sample more randomized versions of the optimal solutions, but the real IINs departed from the optimum in specific, rather than random ways, suggesting additional selective pressure acting on the network structure. Firstly, the real IINs had a smaller number of isolated modules. Each large module corresponds to a particular binding mode; e.g. SH3 to PRD or Ras to GEF interactions. Cells have a limited number of domain/interface types to work with, but our model only limited total interface numbers and not types, as we did not assign types to interface nodes. However, one way we could capture this selection pressure on interface types is by applying selection pressure in our sampling against the total number of modules. The same motif structure in fewer modules would better match the observed biological IIN structure and also mimic the limited number of domain types used by proteins. Secondly, our fitness function applied selection pressure against motifs that were sub-optimal in terms of binding selectivity, but in some cases, these interactions may be optimal in terms of function without truly sacrificing selectivity. How? They can be essentially turned on or off by regulation such as phosphorylation or allostery. This is especially true of “bridge” interfaces that connect otherwise separate modules. The ARC40 subunit of the ARP2/3 complex acts as a bridge node in the CME IIN that can be inhibited from binding actin62. However, it is difficult to select for functional constraints without knowing the true function of every protein in the network, and even then function is not a generic constraint; it would have to be selected for in a targeted way. It is noteworthy however that we are able to reproduce key features of the IINs without the need for incorporating protein function.
Finally, it is estimated that at least 40% of proteins bind to themselves, and the majority of these interactions involve a homo-dimer using the same interface57. In networks, however, these interactions produce self-loops that are often ignored when calculating network properties and simulating network growth, despite providing a justification for frequent paralog interactions in growth models58. They are ignored because having another unique edge type increases the combinatorial complexity of network structures, but we found here that they are critical in correctly capturing motif selectivities. This is best illustrated by the triangle motif in Fig. 2 that switches from low to high specificity with the introduction of multiple self-interactions. The optimal selectivity for a self-binding interface is as an isolated node, or as part of a pair of hetero-dimer forming homo-dimer interfaces, as is clearly evident in the CME IIN (Fig. 1a). Self-binding nodes are least selective as hub interfaces because suppressing non-functional interactions grows more difficult with more partners that are not self-binding. These distinctive motif preferences for self-binding interfaces present another important consideration for curating domain assignments in PPINs, in this case suggesting both potential mis-assignments and missing assignments.
Methods and Models
Fitness function to sample IINs on a PPIN
Given a fixed PPIN, we used Monte Carlo sampling in the space of IIN structures with networks structures accepted or rejected via the Boltzmann weight \({e}^{-({f}_{new}-{f}_{old})/{k}_{B}T}\). The four parameter (ω, β, κ, μ) fitness function given by
controlled the numbers of interfaces N int and edges M IIN in the IINs, as well as the triangle motifs and square-to-chain motif ratio via the local clustering and grid coefficients63, C i,3 and C i,4s.
where k i is the degree of node “i”, k i 2nd is the number of nodes two steps away from “i”, and N triangle,i and N square,i are respectively the number of triangles and squares which pass through “i”. A dummy square (+ 1 term in numerator and denominator) in the grid coefficient is used to penalize having a high number of chains even when N square,i equaled zero. Triangles on which at least two of the nodes had self-edges were ignored, since this is not a constraint against high specificity. The fitness function penalizes having a high clustering coefficient (many triangles), a low grid coefficient (many chains), a high number of interfaces, and it penalizes duplicating too many edges (Fig. 7).

Interface networks for a given protein network can be sampled via Monte Carlo methods with or without bias. (a) Inputs and parameters for our stochastic IIN sampling model for a given PPIN that is not altered. (b) Monte Carlo reversible move sets (5 moves possible) to transition between IIN structures. (c) A two protein network with 2 PPIs can be enumerated into 31 distinct IINs when one extra edge is allowed. Moves between states were enumerated as a Markov chain to determine the factors necessary for detailed balance. (d) Proof of detailed balance in the toy model (C). The probability of being in a given state is proportional to its propensity e −f/kBT, where “f” is the assigned fitness penalty (low “f” = more fit) and kBT is set to 2. The blue line is the theoretical stationary distribution based on propensities, and the red circles are the MC sampled results.
Monte Carlo sampling of networks
We first initialized the IIN structure to either the dense extreme (one interface per protein), the sparse extreme (new interface per each edge), or the known IIN structure. Moves (illustrated in Fig. 7) were accepted or rejected based on the Boltzmann criteria, where we were careful to ensure detailed balance given the different probabilities of generating forwards and reverse moves (p gen) via the acceptance probability:
where f is the fitness of the IIN defined in Eq. 1, and k B T is the effective temperature. We verified our implementation for a small test network in Fig. 7. The entire space of possible IINs could be sampled by setting kBT = ∞. For the fitness sampled IINs, we found a range of kBT = 0.1–1 to be optimal. Modified versions of sampling to test the robustness of our network properties are described in Supplementary Text S1.
Simulations were allowed to equilibrate for the first 1/5 of the total number of iterations, (usually ~1 million iterations) after which the statistics of each network sampled was recorded so as to record average statistics favored by the fitness function. The best-fit (lowest fitness penalty) network discovered was also recorded.
Optimizing fitness function parameters
We tested a wide range of values for our four fitness parameters ω, β, κ, and μ (Fig. 7) to identify the optimal set for describing the biological IINs. We systematically set each parameter set to zero, to completely remove that selection pressure from the fitness function, with results summarized in Fig. S3. All four were needed. As discussed, κ and μ were the most important and the most tightly coupled for capturing the detailed local structure of the IINs (Fig. 4, Fig. S3). We initially varied μ over 0.2–2 and κ from 1–10, later decreasing the optimal search range to values of μ from 0.2–0.6, and κ from 1–3. Results were least sensitive to β, which we varied from 1 to 6, although past values of 2 there was not a significant change in resulting triangle motifs. β was least important because for sparse networks such as the IINs, triangle motifs are already relatively rare to sample. Results were sensitive to ω, which was varied over 0.001 to 1. However, the dependence on ω was straightforward to interpret, as it only controlled the number of edges and was largely uncoupled from other network features.
Quantifying network degree distributions
We generated the spectrum of networks ranging from homogenous to scale-free using a single parameter (α) via the method of Goh et al.64. We term this parameter α the “preferential attachment exponent” (PAE) of the network. A PAE = 0 corresponds to a Poisson (random) network with λ = 〈k〉, and PAE = 1 roughly corresponds to a power-law (scale-free) network with γ = 2. We reverse fit the degree distribution of our sampled IINs by generating networks with specific P.A.E.s for comparison. Degree distributions for 11 values of the P.A.E. (0, 0.1, 0.2 … 1) were generated by building 30 networks (per P.A.E.) with the same number of nodes and edges as the IIN. Least χ2 distance was used to choose the best-fit P.A.E. for the degree distribution of the given IIN. We had to modify the algorithm of Goh et al.64 to generate networks that did not contain orphan nodes, and this procedure is detailed in the Supplementary Text S3.
Statistic for identifying ‘date’ vs ‘party’ hubs
The distribution of interfaces for a protein is calculated by normalizing the Stirling numbers of the second kind (see Supplementary Text S1 for definitions). We use this probability distribution to generate a statistic for identifying proteins with an unusually high (party hubs) or an unusually low (date hubs) number of interfaces. For a protein with degree k and U interfaces, we can calculate a p-value using a two-tailed test, given by
where t can take only integer values [1:k]. If U = (k + 1)/2, p-value \(\equiv 1\).
In Table S1 we report these p-values per protein, indicating which proteins have an unusually small or large number of interfaces.
Generation of alternate PPIN structures
Five variations of the CME network20 were used to test PPIN constraints on IIN sampling: a “dense” network with the same P.A.E. where 186 edges were added to the existing CME network, a “sparse” network also with a comparable P.A.E. where 93 edges were deleted, and a random version of each of the preceding three networks with the same number of proteins and PPIs using the Erdos-Renyi algorithm. Finally, a random version of the ErbB PPIN4 was also used.
Phylogenetic analysis of yeast CME proteins and human ErbB proteins
To determine the evolutionary conservation of domains in the 56 yeast CME proteins and 127 human ErbB proteins, we collected orthologs of each protein, ran multiple sequence alignments with MAFFT65, and analyzed residue conservation with the ConSurf66 rate4site program (or web-server). To assign a conservation score to each domain, the average over all residues in the domain were taken (Table S3). Orthologs were constructed from BLAST67 searches against the UniRef90 clustered sequence database with an E-value cutoff of 0.0001. This approach to use BLAST searches against UniRef90 to identify orthologs across all species is the same as used in other conservation calculation approaches66, 68. Consistent with these approaches68, we kept only sequences that were similar in length to the query sequence (25% longer or shorter) and shared sequence identity of 35%-95% before performing the multiple sequence alignment (MSA).
Hub interfaces were defined in two independent ways: firstly, as any interface with 5 or more interactions (results shown in Table 2). Secondly, we used the statistic defined in Eq 5 to identify proteins with an unusually low number of interfaces given their connectivity, implying the presence of hub interfaces. The statistics were almost identical, with 89% and 71% of hub and non-hubs, respectively, being more conserved than average.
Network rewiring between yeast CME proteins and human CME proteins
We constructed the CME interaction network for human homologs of the yeast proteins using the review of Weinberg et al.38 as a guide to functional homologs in metazoans. Most human homologs were identified directly from this review38, and in a few cases we supplemented this with human orthologs identified from the EggNOG database69, which were confirmed by BLAST searches of the yeast proteins against exclusively human proteins. Nine yeast proteins lacked human homologs (as was previously documented38) and the remaining 45 yeast proteins were matched with 64 human homologs, as compiled in Table S4. Interactions between these 64 proteins were then extracted from BioGRID. We also added 9 interactions involving actin or the Arp2/3 complex and removed 11 involving the Arp2/3 complex to be consistent with the publications used to make the interface assignments in yeast20 that involved crystal structures of metazoan homologs.
The yeast CME network contained 18 PPIs that were mediated through multiple duplicate binding modes (Fig. S6A). These interactions were found to be slightly more conserved than single binding mode interactions, with 9 conserved interactions, 4 lost due to a lost homolog, and 5 lost despite both proteins retaining homologs and domains.
Code for network sampling and analysis is available from github https://github.com/mjohn218/network_sampling_MC.
References
Kim, P. M., Lu, L. J., Xia, Y. & Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 314, 1938–1941, doi:10.1126/science.1136174 (2006).
Wang, X. et al. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nature biotechnology 30, 159–164, doi:10.1038/nbt.2106 (2012).
Bjorkholm, P. & Sonnhammer, E. L. Comparative analysis and unification of domain-domain interaction networks. Bioinformatics 25, 3020–3025, doi:10.1093/bioinformatics/btp522 (2009).
Kiel, C., Verschueren, E., Yang, J.-S. & Serrano, L. Integration of protein abundance and structure data reveals competition in the ErbB signaling network. Science signaling 6, ra109, doi:10.1126/scisignal.2004560 (2013).
Mosca, R., Ceol, A. & Aloy, P. Interactome3D: adding structural details to protein networks. Nat Methods 10, 47–53, doi:10.1038/nmeth.2289 (2013).
Chylek, L. A. et al. Rule-based modeling: a computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip Rev Syst Biol Med 6, 13–36, doi:10.1002/wsbm.1245 (2014).
Park, J., Lee, D. S., Christakis, N. A. & Barabasi, A. L. The impact of cellular networks on disease comorbidity. Mol Syst Biol 5, 262, doi:10.1038/msb.2009.16 (2009).
Watkins, A. M. & Arora, P. S. Structure-based inhibition of protein-protein interactions. Eur J Med Chem 94, 480–488, doi:10.1016/j.ejmech.2014.09.047 (2015).
Kumar, A., Butler, B. M., Kumar, S. & Ozkan, S. B. Integration of structural dynamics and molecular evolution via protein interaction networks: a new era in genomic medicine. Curr Opin Struct Biol 35, 135–142, doi:10.1016/j.sbi.2015.11.002 (2015).
Ozbabacan, S. E. A., Gursoy, A., Nussinov, R. & Keskin, O. The Structural Pathway of Interleukin 1 (IL-1) Initiated Signaling Reveals Mechanisms of Oncogenic Mutations and SNPs in Inflammation and Cancer. Plos Comput Biol 10, doi:10.1371/journal.pcbi.1003470 (2014).
Vavouri, T., Semple, J. I., Garcia-Verdugo, R. & Lehner, B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138, 198–208, doi:10.1016/j.cell.2009.04.029 (2009).
Deeds, E. J., Krivine, J., Feret, J., Danos, V. & Fontana, W. Combinatorial complexity and compositional drift in protein interaction networks. PLoS One 7, e32032, doi:10.1371/journal.pone.0032032 (2012).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res 28, 235–242 (2000).
Webb, B. & Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci 86, 2 9 1–2 9 37, doi:10.1002/cpps.20 (2016).
Szilagyi, A. & Zhang, Y. Template-based structure modeling of protein-protein interactions. Curr Opin Struct Biol 24, 10–23, doi:10.1016/j.sbi.2013.11.005 (2014).
Stein, A., Mosca, R. & Aloy, P. Three-dimensional modeling of protein interactions and complexes is going ‘omics. Curr Opin Struct Biol 21, 200–208, doi:10.1016/j.sbi.2011.01.005 (2011).
Dinkel, H. et al. ELM 2016-data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44, D294–D300, doi:10.1093/nar/gkv1291 (2016).
van der Lee, R. et al. Classification of intrinsically disordered regions and proteins. Chem Rev 114, 6589–6631, doi:10.1021/cr400525m (2014).
Weatheritt, R. J., Luck, K., Petsalaki, E., Davey, N. E. & Gibson, T. J. The identification of short linear motif-mediated interfaces within the human interactome. Bioinformatics 28, 976–982, doi:10.1093/bioinformatics/bts072 (2012).
Johnson, M. E. & Hummer, G. Interface-resolved network of protein-protein interactions. PLoS computational biology 9, e1003065, doi:10.1371/journal.pcbi.1003065 (2013).
Johnson, M. E. & Hummer, G. Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks. Proceedings of the National Academy of Sciences of the United States of America 108, 603–608, doi:10.1073/pnas.1010954108 (2011).
Zhang, J., Maslov, S. & Shakhnovich, E. I. Constraints imposed by non-functional protein-protein interactions on gene expression and proteome size. Molecular systems biology 4, 210, doi:10.1038/msb.2008.48 (2008).
Heo, M. Y., Maslov, S. & Shakhnovich, E. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proceedings of the National Academy of Sciences of the United States of America 108, 4258–4263, doi:10.1073/pnas.1009392108 (2011).
Yang, J.-R., Liao, B.-Y., Zhuang, S.-M. & Zhang, J. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proceedings of the National Academy of Sciences of the United States of America 109, E831–840, doi:10.1073/pnas.1117408109 (2012).
Levy, E. D., De, S. & Teichmann, S. A. Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. Proceedings of the National Academy of Sciences of the United States of America 109, 20461–20466, doi:10.1073/pnas.1209312109 (2012).
Johnson, M. E. & Hummer, G. Evolutionary pressure on the topology of protein interface interaction networks. The journal of physical chemistry. B 117, 13098–13106, doi:10.1021/jp402944e (2013).
Gastner, M. T. & Newman, M. E. Optimal design of spatial distribution networks. Phys Rev E Stat Nonlin Soft Matter Phys 74, 016117, doi:10.1103/PhysRevE.74.016117 (2006).
Levy, E. D. & Pereira-Leal, J. B. Evolution and dynamics of protein interactions and networks. Curr Opin Struc Biol 18, 349–357, doi:10.1016/j.sbi.2008.03.003 (2008).
Yook, S. H., Oltvai, Z. N. & Barabasi, A. L. Functional and topological characterization of protein interaction networks. Proteomics 4, 928–942, doi:10.1002/pmic.200300636 (2004).
Scott McShan, R. W. The implications of hub-and-spoke routing for airline costs and competitiveness. Logistics and Transportation Review 25, 209–230 (1989).
Albert, R., Jeong, H. & Barabasi, A. L. Error and attack tolerance of complex networks. Nature 406, 378–382, doi:10.1038/35019019 (2000).
Orengo, C. A. & Thornton, J. M. Protein families and their evolution-a structural perspective. Annu Rev Biochem 74, 867–900, doi:10.1146/annurev.biochem.74.082803.133029 (2005).
Beltrao, P. & Serrano, L. Specificity and evolvability in eukaryotic protein interaction networks. Plos Comput Biol 3, 258-267, doi:ARTN e2510.1371/journal.pcbi.0030025 (2007).
Meyer, M. J., Das, J., Wang, X. J. & Yu, H. Y. INstruct: a database of high-quality 3D structurally resolved protein interactome networks. Bioinformatics 29, 1577–1579, doi:10.1093/bioinformatics/btt181 (2013).
Kaneko, T., Li, L. & Li, S. S. C. The SH3 domain - a family of versatile peptide- and protein-recognition module. Front Biosci 13, 4938–4952, doi:10.2741/3053 (2008).
Rushworth, L. K., Hindley, A. D., O’Neill, E. & Kolch, W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol 26, 2262–2272, doi:10.1128/MCB.26.6.2262-2272.2006 (2006).
Xin, X. F. et al. SH3 interactome conserves general function over specific form. Molecular systems biology 9, doi:ARTN 65210.1038/msb.2013.9 (2013).
Weinberg, J. & Drubin, D. G. Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol 22, 1–13, doi:10.1016/j.tcb.2011.09.001 (2012).
Schmid, E. M. et al. Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly. Plos Biol 4, 1532-1548, doi:ARTN e26210.1371/journal.pbio.0040262 (2006).
Fraser, H. B., Hirsh, A. E., Steinmetz, L. M., Scharfe, C. & Feldman, M. W. Evolutionary rate in the protein interaction network. Science 296, 750–752, doi:10.1126/science.1068696 (2002).
Manna, B., Bhattacharya, T., Kahali, B. & Ghosh, T. C. Evolutionary constraints on hub and non-hub proteins in human protein interaction network: Insight from protein connectivity and intrinsic disorder. Gene 434, 50–55, doi:10.1016/j.gene.2008.12.013 (2009).
Haynes, C. et al. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. Plos Computational Biology 2, 890–901, doi:ARTN e10010.1371/journal.pcbi.0020100 (2006).
Brown, C. J. et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol 55, 104–110, doi:10.1007/s00239-001-2309-6 (2002).
Batada, N. N. et al. Stratus not altocumulus: A new view of the yeast protein interaction network. Plos Biol 4, 1720–1731, doi:ARTN e31710.1371/journal.pbio.0040317 (2006).
Batada, N. N. et al. Still stratus not altocumulus: Further evidence against the date/party hub distinction. Plos Biol 5, 1202-1206, doi:ARTN e15410.1371/journal.pbio.0050154 (2007).
Drummond, D. A. & Wilke, C. O. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10, 715–724, doi:10.1038/nrg2662 (2009).
Pal, C., Papp, B. & Lercher, M. J. An integrated view of protein evolution. Nature Reviews Genetics 7, 337–348, doi:10.1038/nrg1838 (2006).
Vázquez, A., Flammini, A., Maritan, A. & Vespignani, A. Modeling of Protein Interaction Networks. Complexus 1, 38–44 (2003).
Eisenberg, E. & Levanon, E. Y. Preferential attachment in the protein network evolution. Physical Review Letters 91, doi:ARTN 13870110.1103/PhysRevLett.91.138701 (2003).
Kim, W. K. & Marcotte, E. M. Age-Dependent Evolution of the Yeast Protein Interaction Network Suggests a Limited Role of Gene Duplication and Divergence. Plos Comput Biol 4, doi:ARTN e100023210.1371/journal.pcbi.1000232 (2008).
Wagner, A. How the global structure of protein interaction networks evolves. P Roy Soc B-Biol Sci 270, 457–466, doi:10.1098/rspb.2002.2269 (2003).
Middendorf, M., Ziv, E. & Wiggins, C. H. Inferring network mechanisms: The Drosophila melanogaster protein interaction network. Proceedings of the National Academy of Sciences of the United States of America 102, 3192–3197, doi:10.1073/pnas.0409515102 (2005).
Navlakha, S. & Kingsford, C. Network archaeology: uncovering ancient networks from present-day interactions. PLoS Comput Biol 7, e1001119, doi:10.1371/journal.pcbi.1001119 (2011).
Cotton, J. A. & Page, R. D. Rates and patterns of gene duplication and loss in the human genome. Proc Biol Sci 272, 277–283, doi:10.1098/rspb.2004.2969 (2005).
Pan, D. & Zhang, L. Quantifying the major mechanisms of recent gene duplications in the human and mouse genomes: a novel strategy to estimate gene duplication rates. Genome Biol 8, R158, doi:10.1186/gb-2007-8-8-r158 (2007).
Katju, V. & Bergthorsson, U. Copy-number changes in evolution: rates, fitness effects and adaptive significance. Front Genet 4, 273, doi:10.3389/fgene.2013.00273 (2013).
Levy, E. D., Pereira-Leal, J. B., Chothia, C. & Teichmann, S. A. 3D complex: A structural classification of protein complexes. Plos Comput Biol 2, 1395-1406, doi:ARTN e15510.1371/journal.pcbi.0020155 (2006).
Ispolatov, I., Yuryev, A., Mazo, I. & Maslov, S. Binding properties and evolution of homodimers in protein-protein interaction networks. Nucleic Acids Res 33, 3629–3635, doi:10.1093/nar/gki678 (2005).
Matthews, L. R. et al. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res 11, 2120–2126, doi:10.1101/gr.205301 (2001).
Madan Babu, M., Teichmann, S. A. & Aravind, L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol 358, 614–633, doi:10.1016/j.jmb.2006.02.019 (2006).
Schmid, E. M. & McMahon, H. T. Integrating molecular and network biology to decode endocytosis. Nature 448, 883–888 (2007).
Ti, S. C., Jurgenson, C. T., Nolen, B. J. & Pollard, T. D. Structural and biochemical characterization of two binding sites for nucleation-promoting factor WASp-VCA on Arp2/3 complex. Proc Natl Acad Sci USA 108, E463–471, doi:10.1073/pnas.1100125108 (2011).
Cardarelli, L., Maxwell, K. L. & Davidson, A. R. Assembly mechanism is the key determinant of the dosage sensitivity of a phage structural protein. P Natl Acad Sci USA 108, 10168–10173, doi:10.1073/Pnas.1100759108 (2011).
Goh, K. I., Kahng, B. & Kim, D. Universal behavior of load distribution in scale-free networks. Phys Rev Lett 87, 278701, doi:10.1103/PhysRevLett.87.278701 (2001).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066 (2002).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350, doi:10.1093/nar/gkw408 (2016).
Camacho, C. et al. BLAST+: architecture and applications. Bmc Bioinformatics 10, 421, doi:10.1186/1471-2105-10-421 (2009).
Chica, C., Labarga, A., Gould, C. M., Lopez, R. & Gibson, T. J. A tree-based conservation scoring method for short linear motifs in multiple alignments of protein sequences. Bmc Bioinformatics 9, doi:Artn 22910.1186/1471-2105-9-229 (2008).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44, D286–D293, doi:10.1093/nar/gkv1248 (2016).
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the NIH under Award No. R00GM098371 to M.E.J. The research used NSF XSEDE resources, the Homewood high-performance cluster at JHU and the Maryland Advanced Research Computing Center (MARCC).
Author information
Authors and Affiliations
Contributions
D.O.H., B.H.S., P.X., and M.E.J. analyzed data, D.O.H., B.H.S. and M.E.J. developed software, M.E.J. designed research, D.O.H. and M.E.J. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holland, D.O., Shapiro, B.H., Xue, P. et al. Protein-protein binding selectivity and network topology constrain global and local properties of interface binding networks. Sci Rep 7, 5631 (2017). https://doi.org/10.1038/s41598-017-05686-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05686-2
This article is cited by
-
Single residue modulators of amyloid formation in the N-terminal P1-region of α-synuclein
Nature Communications (2022)
-
Integrating protein copy numbers with interaction networks to quantify stoichiometry in clathrin-mediated endocytosis
Scientific Reports (2022)
-
Building protein-protein interaction networks for Leishmania species through protein structural information
BMC Bioinformatics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
