
Abstract
For the search of new metal-halide perovskite solar cell materials, tolerance factors are calculated from the ionic radius of each site and are often utilized as the critical factors to expect the materials forming perovskite structure. As one of such amine hydrohalides, guanidine hydroiodide (GI) is reported not to react with PbI2. However, in this paper, we report the product of GI and SnI2 reaction, its visible light absorption, X-ray diffraction, and its solar cell operation, in spite of the more disadvantageous tolerance factor of SnI2 than PbI2. We also report the thermal stability of GI, enabling precise control of vacuum deposition, and utilization of co-evaporant induced crystallization method during the vacuum evaporation of the SnI2 film, which resulted in enlarging the SnI2 crystals and improving the short circuit current density of the solar cell.
Similar content being viewed by others


Introduction
Recently, perovskite solar cells attract much attention because of their high efficiency and ease of fabrication. In perovskite solar cells, organic halogenated perovskites are used as energy conversion materials, their compositions are generally represented as ABX3: A site is an organic amine, B site is a metal, and X site is a halogen. To make this structure stable, the size of each ion is important and the stability can be estimated from tolerance factor. For the search of new perovskite materials, tolerance factor has been calculated in various combinations1,2,3,4 of organic materials and metals. From this calculation, not only the standard organic amines of methylamine (MA) and formamidine (FA), but also various organic amines are reported3, 5 on their possibilities to form perovskite structure and to be used as solar cell materials.
One of such prospected organic amines, guanidine (G), shows tolerance factor of almost 1 by combination with PbI2 or SnI2, similar to the case of ethylamine (EA) because of their larger ionic radii than of MA and FA. For example, these values have been reported1, 3 as the followings, tolerance factors of GSnI3: 1.051 and GPbI3: 1.039 by using each ionic radius of Sn: 115 pm, Pb: 119 pm, guanidine: 278 pm, I: 220 pm. Although perovskite solar cells using EAI and PbI2 have been already reported4, in contrast, Yang et al. reported that GI and PbI2 did not form perovskite; whereas the open circuit voltage of mainly MAPbI3 composed cell was improved by adding GI6. Predicting by the tolerance factor, GI and SnI2 cannot form perovskite as well as the case of PbI2. In this paper, however, we will report that GI reacted well with SnI2 and the product absorbed wider visible light region than the reactants did. We succeeded in fabricating and operating the solar cell using only guanidine for the A site and Sn for the B site.
Results and Discussion
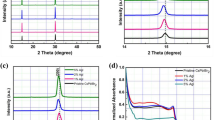
First, the reactivity of GI and inorganic material (SnI2 or PbI2) was examined by drop-coating. Films were formed from acetone solution on quartz substrates and absorbance spectra of the films were measured by an ultraviolet and visible spectrophotometer. As shown in Fig. 1a, an absorption edge around 800 nm and a shoulder around 700 nm were observed for the product of SnI2 and GI. In contrast to this clear change, no increase of visible light absorption by the reaction was observed for PbI2 and GI as same as reported6. In Fig. 1b, air stability of the product was evaluated. Although the film was preserved in the dry air, its visible absorption decreased day by day, probably because of Sn oxidization.

Characteristics of films fabricated from solution. (a) Absorbance spectra of PbI2 + GI, SnI2 + GI, SnI2, and GI films formed from acetone solutions. (b) Degradation of absorbance spectra of SnI2 + GI film. (c) X-ray diffraction pattern of SnI2 + GI film.
To distinguish whether this product is perovskite or not, we performed X-ray diffraction (XRD) measurement. As shown in Fig. 1c, the result showed clear peaks that can be attributed to Pm-3m space group of cubic perovskites and confirms that the formation of 3d -perovskite of GSnI3. Smaller peaks marked * may indicate superlattice, which imply the partial degradation of the cubic perovskite structure because of large tolerance factor or air degradation. A rough calculation using each ionic radius can reasonably explain the lattice constant of 6.23 Å. When simply supposing the sequence of AXA face-centred lattice, ABA body-centred lattice, and BXB side of the simple cubic lattice instead of ABX3, lattice constant can be calculated as 7.04, 4.54, and 6.70 Å, respectively. The average of 6.09 Å is appropriately near the experimental value of 6.23 Å, although further study is necessary to clarify more accurate crystal structure of GSnI3.
Next, we fabricated solar cell devices. We attempted fabricating the devices by vacuum deposition method. We found that GI can be normally deposited by vapor evaporation, different from the depositions of hydrohalides of MA and FA, which often has problems by pyrolysis and diffusion of the materials, such as deterioration of vacuum, serious contamination to the vacuum equipment7, and evolution of toxic hydrogen cyanide dictated on FAI8. In our vacuum system, MAI and FAI also showed contaminations to the wide area of the system. However, when we tried depositing GI, on the other hand, its vacuum deposition was stably controlled under high vacuum of 1–3 × 10−3 Pa without such problems. We consider that the reason of this higher thermal stability of GI than MAI and FAI is because methylamine without iodine ion is a gas substance at room temperature and that formamidine itself cannot be isolated from the halide, whereas guanidine is solid.
A highly efficient Sn-based perovskite device of the reverse structure type without the charge transport layer of TiO2 porous film has been reported9,10,11. Thus, we fabricated the device of the reverse structure type as depicted in Fig. 2a. The electron transport layer and hole transport layer were chosen referring to the materials used for vacuum deposition of organic solar cells12,13,14,15 as shown in Fig. 2a. All layers were successively formed by vacuum deposition on pre-patterned indium tin oxide coated glass to fabricate the solar cells. Perovskite layer was formed by two-step method: SnI2 or PbI2 layer deposition was followed by GI deposition. Throughout these perovskite layer depositions, the substrate temperature was kept at 70 °C. During all the vacuum evaporation, the degree of vacuum was 1–3 × 10−3 Pa, and the deposition rate was 0.5 Å/s for GI and 1.0 Å/s for other materials. Solar cell performances of the devices fabricated were measured in N2 filled glove box without taken out to the atmosphere.

Solar cells fabricated by two-step method of vacuum evaporation using SnI2 or PbI2 as a metal halide and GI as an amine halide. (a) Device structure, (b) picture of the films, (c) J-V curve, and (d) IPCE.
After the optimization of the perovskite layer, we reached the ratio of GI: 210 nm and SnI2: 150 nm in nominal thickness. Figure 2b,c and d show the photographs of the fabricated devices, the J-V curve of the device with 1 cm2 area measured under the AM1.5 1sun simulated illumination, and incident photon to current conversion efficiency (IPCE) of the same device but with 0.04 mm2, respectively. In Fig. 2b, the color of SnI2 + GI is brown unlike the orange color of SnI2. In Fig. 2c forward and reverse represent the direction of voltage sweep from −2 V to 2 V and from 2 V to −2 V, respectively. The device parameters of SnI2 + GI from the forward sweep were the followings: 0.60 mA/cm2 of short-circuit current density (J sc), 0.62 V of opening circuit voltage (V oc), 0.42 of fill factor (FF), and 0.16% of power conversion efficiency (PCE). On the other hand, the color of PbI2 + GI showed yellow as same as PbI2. The operation of PbI2 + GI device was unstable, and its performance was much limited to 0.12 mA/cm2 of J sc, 0.26 V of V oc, 0.36 of FF, and 0.01% of PCE. Judging from the IPCE spectra in Fig. 2d, in which the SnI2 + GI device has photo-response up to 650 nm of the long wavelength side, light absorption of the product of SnI2 + GI reaction certainly contributed to the current generation of the solar cell. On the other hand, PbI2 device has only 450–550 nm of much smaller photo-response, which might be from Alq3 or C60 in the buffer layer.
Further enhancement of the performance can be usually possible by improving device structure or post-annealing16, 17. Especially, crystallinity and particle size of the active layer have great importance on the performance of perovskite solar cells18,19,20,21,22,23,24,25,26. Here, we tried introducing co-evaporant molecules during the vacuum evaporation to induce crystallization of the film in this study. This unique method is originally an improved method of the vacuum deposition for organic films and enables crystallinity of organic films by co-evaporating liquid molecules during vacuum evaporation of the film materials27, 28. The liquid promotes surface diffusion of film material molecules, but the liquid itself does not remain on the device substrate because of substrate heating. In the case of organic photovoltaic cells, the cells whose active layers were crystallized by this method indicated improvements of their short circuit current density. Thus, we examined the utility of this unique method for perovskite solar cells, although the film materials in this study are inorganic films of SnI2 and PbI2, different from the organic materials in past studies.
We used polydimethylsiloxane (PDMS) as the co-evaporant liquid and its evaporation rate was nominally 0.2 A/s. We introduced this liquid only during the deposition of the SnI2 film. Other fabrication conditions are almost same as the above; the exception is the thickness of perovskite layer, which was thinned from the device in Fig. 2c, SnI2: 150 nm and GI: 210 nm, to the followings, SnI2: 100 nm and GI: 140 nm, because the short of the electrodes occurred with thicker devices due to the extremely rough surface formed by the co-evaporant method as shown in later. It implies that further optimization of both active layer and the buffer layer is necessary for the stable production of this type cells. As the result of the introduction of the co-evaporant liquid during the deposition of the SnI2 film, short circuit current density improved. As shown in Fig. 3a, J sc was 1.25 mA/cm2, V oc, FF, and PCE were 0.40 V, 0.46, and 0.23%, respectively from the forward sweep. Particularly, J sc is 4.3 times higher than the device without co-evaporant in Fig. 3a and 2.1 times higher than the device before thinning the film thickness in Fig. 2c. Although large hysteresis was observed, its effect on J sc was small enough to compare the difference of the devices. To confirm this performance difference and the operation of GSnI3 cells under steady-state, we added stabilized power output measurement29 under the bias application of 0.30 V as shown in Fig. 3e. J sc and PCE for the device with co-evaporant, 0.37 mA/cm2 and 0.11%, were certainly higher than 0.15 mA/cm2 and 0.045% for the device without co-evaporant, although the devices in Fig. 3e are the different batch but the same composition from them in Fig. 3a,b,c and d.

Performance improvements of SnI2 + GI solar cells by co-evaporant induced crystallization. (a) J-V curve of 1 cm2 cell, (b) IPCE, (c) absorption spectra, (d) IQE, and (e) stabilized power output under bias application of 0.30 V.
The morphological change of the SnI2 film by co-evaporant was difficult to measure because the film was easily oxidized and deteriorated in the atmosphere. Therefore, to infer the effect of co-evaporant on SnI2, the effect on PbI2 was observed by atomic force microscopy (AFM) as the substitute. Figure 4 shows an AFM image of the surface of the PbI2 film. Comparing the films with/without co-evaporant, the film without co-evaporant showed the dispersion of grain sizes, whereas the film with co-evaporant showed the obviously larger grain sizes than the former. Correspondingly, the surface roughness Sq of the film also increased from 7.30 nm to 27.9 nm. This is the first report for the effect of the co-evaporant induced crystallization of inorganic thin films.

AFM images of PbI2 films vacuum deposited (a) without co-evaporant, and (b) with co-evaporant. Sq is the root mean square height, which is standardized by ISO25178 as one of the parameters representing surface roughness.
The reason for the improvement of Jsc and corresponding IPCE in Fig. 3a and b can be partially attributed to the improvement of absorption in Fig. 3c, which suggests that the increase of roughness of SnI2 film promoted the reaction with GI. Another reason is suggested by the improvement of internal quantum efficiency (IQE) in Fig. 3d that the increase of the perovskite crystal size resulting from the grain size of SnI2 suppressed charge recombination18,19,20,21,22,23,24,25,26. As described above, we demonstrated that the co-evaporant induced crystallization is useful even for crystallization of inorganic thin films and improvement of J sc of perovskite solar cells.
Conclusions
In summary, we have revealed that, in spite of disadvantageous tolerance factor, guanidine hydro-iodide, which does not react with PbI2, reacted with SnI2 and the product of the reaction absorbed visible light much wider than the reactants do. By using this product, we succeeded in fabricating and operating the solar cells. This result confirms that the tolerance factor is not alone the key factor to form perovskite structure. Moreover, we demonstrated that GI can be thermally evaporated without problems often occurred by MAI and FAI. We succeeded in improving the short circuit current density of the vacuum deposited devices by using co-evaporant induced crystallization method during SnI2 film deposition. This is the first report of the effect of this method on inorganic materials.
Methods
Materials and substrates
To try to form perovskite, we used commercially available high purity reagents (PbI2 99.99%, GI > 97.00%: Tokyo Chemical Industry, SnI2 99.99%: AlfaAesar,) as received. C60 (Frontier Carbon) were purified by conventional train sublimation. MoO3 (99.998%, Alfa Aesar), Alq3 (Tris(8-quinolinolato)aluminum, >98%, Tokyo Chemical Industry), and LiF (Alfa Aesar) were used as is obtained. PDMS (polydimethylsiloxane, Shin-Etsu Silicones, KF96–50cs) was used as a co-evaporant. Indium tin oxide (ITO, thickness: 150 nm, resistivity: 10 Ω/cm2) coated 0.7-mm-thick glass substrates were pre-cleaned and patterned by techno Print.
Film fabrication from solution
A solution of GSnI3 was prepared at a concentration of 1.5 × 10−5 mol/ml by dissolving SnI2 and GI in acetone at a molar ratio of 1:1. In dry atmosphere (dew point was about −40 °C), the solution was dropped onto quartz glass with a surface area of 4.5 cm2 and dried at 25 °C. The solution of 0.5 ml and of 1.5 ml were used for the sample preparation of UV-Vis absorption and of XRD measurement, respectively.
Device fabrication
ITO coated glass substrates were treated by air-plasma in PIB-20 Ion-bombarder (Vacuum Devices). Vacuum deposition was carried out in a custom-designed vacuum chamber (Katagiri engineering and M&Y systems). After introducing the substrates into the vacuum chamber, entire cell fabrication was completed without exposing to the air. All vacuum instruments were connected with oil-free vacuum pumps. Film thickness and the deposition rate were observed using calibrated quartz crystal microbalances equipped with a computer monitoring system (CRTM-9000G/Depoview, Ulvac).
Device characterization
Device characterizations were performed in a glove-box connected to the vacuum chamber and completed without exposing the devices to the air after the device fabrication. The glove-box (UL-800A, Unico) was filled with N2 gas, equipped with a circulation gas purifier (CM-200, Unico). Under normal conditions, the oxygen and water contents were around 1 ppm or less. J–V curves were obtained with a custom-designed probe box (Epitec), a Precision Source/Measure Unit (2912B, Agilent), and solar cell measurement software (W32-B2900SOL4-N, Sunrise) under AM 1.5 G one sun illumination from a solar simulator (HAL-320W, Asahi Spectra). IPCE spectra were also measured with the same system under illumination of monochromatic light from a monochromator (CMS-250, Asahi Spectra) and white light bias from LED light source (SLA-100A, Sigma Koki). The temperature of the cells was kept at 25 °C using a Peltier cool plate (CP-085, Scinics). The actual area of each fabricated device was 1.21 or 0.09 cm2, and the active area during the characterization was defined as 1.00 or 0.04 cm2, respectively, by a precision aperture mask positioned directly on the substrate side of the cell.
Film characterizations
UV-vis measurement was performed after exposing the devices to the air with a UV/Vis/IR spectrophotometer (V-670, Jasco) equipped with an integrating sphere unit (ISN-723, Jasco). Atomic force microscopy image was obtained in the air by using Probe Station AFM5000II and AFM Unit AFM5100N (Hitachi High Technologies). XRD pattern was obtained in the air by using SmartLab (Rigaku). Powder diffraction pattern was solved by using Crystal Cracker software (Dr. Kurt Leinenweber).
References
Chen, Q. et al. Under the spotlight: The organic–inorganic hybrid halide perovskite for optoelectronic applications. Nano Today 10, 355–396 (2015).
Travis, W., Glover, E. N. K., Bronstein, H., Scanlon, D. O. & Palgrave, R. G. On the application of the tolerance factor to inorganic and hybrid halide perovskites: a revised system. Chem. Sci. 7, 4548–4556 (2016).
Kieslich, G., Sun, S. & Cheetham, A. K. An extended Tolerance Factor approach for organic–inorganic perovskites. Chem. Sci. 6, 3430–3433 (2015).
Im, J. H., Chung, J., Kim, S. J. & Park, N. G. Synthesis, structure, and photovoltaic property of a nanocrystalline 2H perovskite-type novel sensitizer (CH3CH2NH3)PbI3. Nanoscale Res. Lett. 7, 353–359 (2012).
Boix, P. P., Agarwala, S., Koh, T. M., Mathews, N. & Mhaisalkar, S. G. Perovskite Solar Cells: Beyond Methylammonium Lead Iodide. J. Phys. Chem. Lett. 6, 898–907 (2015).
Marco, N. D. et al. Y. Guanidinium: A Route to Enhanced Carrier Lifetime and Open-Circuit Voltage in Hybrid Perovskite Solar Cells. Nano Lett. 16, 1009–1016 (2016).
Wang, S. et al. Smooth perovskite thin films and efficient perovskite solar cells prepared by the hybrid deposition method. J. Mater. Chem. A 3, 14631–14641 (2015).
Ohki, M., Ohsawa, T., Tanaka, M. & Chihara, H. Kagaku Jiten (Dictionary of Chemistry) (Tokyo Kagaku Dojin, Tokyo, 1994) P.1381.
Marshall, K. P., Waltona, R. I. & Hatton, R. A. Tin perovskite/fullerene planar layer photovoltaics: improving the efficiency and stability of lead-free devices. J. Mater. Chem. A 3, 11631–11640 (2015).
Yang, Z. et al. Stable Low-Bandgap Pb–Sn Binary Perovskites for Tandem Solar Cells. adv. mater. 28, 8990–8997 (2016).
Liao, W. et al. Lead-Free Inverted Planar Formamidinium Tin Triiodide Perovskite Solar Cells Achieving Power Conversion Efficiencies up to 6.22%. adv. mater. 28, 9333–9340 (2016).
Mori, T. & Masumoto, Y. Effect of Organic Alloy for Suppression of Polycrystallization in BCP Thin Film. J. Photopolym. Sci. Tec. 19, 209–214 (2006).
Bartynski, A. N. et al. A Fullerene-Based Organic Exciton Blocking Layer with High Electron Conductivity. Nano Lett. 13, 3315–3320 (2013).
Xiao, X., Bergemann, K. J., Zimmerman, J. D., Lee, K. & Forrest, S. R. Small-Molecule Planar-Mixed Heterojunction Photovoltaic Cells with Fullerene-Based Electron Filtering Buffers. Adv. Energy Mater. 4, 1301557, doi:10.1002/aenm.201301557 (2014).
Kao, P. C., Chu, S. Y., Huang, H. H., Tseng, Z. L. & Chen, Y. C. Improved efficiency of organic photovoltaic cells using tris (8-hydroxy-quinoline) aluminum as a doping material. Thin Solid Films 517, 5301–5304 (2009).
Dualeh, A. et al. M. Effect of Annealing Temperature on Film Morphology of Organic–Inorganic Hybrid Perovskite Solid-State Solar Cells. Adv. funct. mater. 24, 3250–3258 (2014).
Xiao, Z. et al. Solvent Annealing of Perovskite-Induced Crystal Growth for Photovoltaic-Device Efficiency Enhancement. Adv. mater. 26, 6503–6509 (2014).
Liu, M., Johnston, M. B. & Snaith, H. J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 501, 395–398 (2013).
Chen, C. W. et al. Efficient and Uniform Planar-Type Perovskite Solar Cells by Simple Sequential Vacuum Deposition. Adv. Mater. 26, 6647–6652 (2014).
Zheng, L. et al. Morphology Control of the Perovskite Film for Efficient Solar Cells. Dalton Trans. 44(23), 10582–10593 (2015).
Liang, P. W. et al. Additive Enhanced Crystallization of Solution-Processed Perovskite for Highly Efficient Planar-Heterojunction Solar Cells. Adv. Mater. 26, 3748–3754 (2014).
Chen, Q. et al. Planar Heterojunction Perovskite Solar Cells via Vapor-Assisted Solution Process. J. Am. Chem. Soc. 136, 622–625 (2014).
Hu, H. et al. Vapour-based processing of hole-conductor-free CH3NH3PbI3 perovskite/C60 fullerene planar solar cells. RSC Adv. 4, 28964–28967 (2014).
Shao, Y., Xiao, Z., Bi, C., Yuan, Y. & Huang, J. Origin and elimination of photocurrent hysteresis by fullerene passivation in CH3NH3PbI3 planar heterojunction solar cells. Nat. Commun. 5, 5784–5790 (2014).
Nie, W. et al. High-efficiency solution-processed perovskite solar cells with millimeter-scale grains. Science 347, 522–525 (2015).
Xiao, M. et al. A Fast Deposition-Crystallization Procedure for Highly Efficient Lead Iodide Perovskite Thin-Film Solar Cells. Angew. Chem. 126, 10056–10061 (2014).
Kaji, T. et al. Co-evaporant Induced Crystalline Donor: Acceptor Blends in Organic Solar Cells. Adv. Mater. 23, 3320–3325 (2011).
Kaji, T., Nakao, S. & Hiramoto, M. Effect of Co-evaporant Induced Crystallization on Needle Growth of Phthalocyanine Thin Films. Mol. Cryst. Liq. Cryst. 578, 63–67 (2013).
Snaith, H. J. et al. Anomalous Hysteresis in Perovskite Solar Cells. J. Phys. Chem. Lett. 5, 1511–1515 (2014).
Acknowledgements
This study has been supported in part by Advanced Low Carbon Technology Research and Development Program (ALCA) of Japan Science and Technology Agency (JST), JSPS KAKENHI Grant Number 25871056, and MEXT-Supported Program for the Strategic Research Foundation at Private Universities S1411036.
Author information
Authors and Affiliations
Contributions
All Authors gave ideas on this work. Experiments were mainly done by H. Ishibashi and M. Katayama. T. Kaji and S. Tanaka guided the work. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishibashi, H., Katayama, M., Tanaka, S. et al. Hybrid perovskite solar cells fabricated from guanidine hydroiodide and tin iodide. Sci Rep 7, 4969 (2017). https://doi.org/10.1038/s41598-017-05317-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05317-w
This article is cited by
-
Determination of electrical parameters of ITO/CZTS/CdS/Ag and ITO/CdS/CZTS/Ag heterojunction diodes in dark and illumination conditions
Optical and Quantum Electronics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
