
Abstract
Chemical weathering consumes atmospheric carbon dioxide through the breakdown of silicate minerals and is thought to stabilize Earth’s long-term climate. However, the potential influence of silicate weathering on atmospheric pCO2 levels on geologically short timescales (103–105 years) remains poorly constrained. Here we focus on the record of a transient interval of severe climatic warming across the Toarcian Oceanic Anoxic Event or T-OAE from an open ocean sedimentary succession from western North America. Paired osmium isotope data and numerical modelling results suggest that weathering rates may have increased by 215% and potentially up to 530% compared to the pre-event baseline, which would have resulted in the sequestration of significant amounts of atmospheric CO2. This process would have also led to increased delivery of nutrients to the oceans and lakes stimulating bioproductivity and leading to the subsequent development of shallow-water anoxia, the hallmark of the T-OAE. This enhanced bioproductivity and anoxia would have resulted in elevated rates of organic matter burial that would have acted as an additional negative feedback on atmospheric pCO2 levels. Therefore, the enhanced weathering modulated by initially increased pCO2 levels would have operated as both a direct and indirect negative feedback to end the T-OAE.
Similar content being viewed by others


Introduction
The chemical weathering of rocks constitutes a negative and stabilizing feedback to Earth’s long-term (108–109 yr) climate by consuming atmospheric CO2, modulating the greenhouse effect and, in turn, global temperatures1,2,3. On these timescales, chemical weathering is dominantly regulated by tectonics, atmospheric pCO2, temperature, the lithology of materials being weathered, and the strength of the hydrological cycle3. Although the influence of weathering on long-term climate is well established3, much less is known about how this process potentially operates and influences climate on shorter times scales (<106 yr)4.
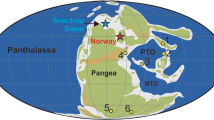
The T-OAE of the Early Jurassic Period constituted an ephemeral interval of global warming, perturbations in the global carbon cycle5, widespread oceanic anoxia6, and elevated marine extinction rates7. These environmental and ecological changes have been linked to the emplacement of the Karoo-Ferrar Large Igneous Province (LIP) and subsequent injection of greenhouse gases into the atmosphere8 (Fig. 1). Specifically, the addition of mantle-derived CO2 and thermogenic CH4 derived from the emplacement of the LIP9,10,11 and subsequent releases of CH4 from marine clathrates12, 13 and terrestrial environments14, 15 to the oceans and atmosphere are the proposed drivers of the T-OAE warming and carbon cycle perturbations. These perturbations are now recorded in sedimentary successions as pronounced negative carbon isotope excursions (CIEs), which occurred during a long-term trend to more positive carbon isotope values. This negative excursion is followed by a positive CIE thought to be the result of enhanced organic matter burial under anoxic conditions in marine and lacustrine environments5, 6. Collectively, these two carbon isotope excursions are used to stratigraphically define the T-OAE interval.

Global palaeogeography of the Early Toarcian (modified from ref. 71). Star represents this study’s location. Arrows point to the UK study locations20, 23, which are geographically close to one another. Hatched outline in southern Pangaea (present-day southern Africa and Antarctica) represents location and known extent of Karoo-Ferrar Large Igneous Province. Dark grey represents landmasses, light blue represents shallow seas, and dark blue represents open oceans. CPM = Central Pangaean Mountains. See ref. 15 for a list of locations that document the T-OAE CIE.
Under the enhanced greenhouse effect triggered by elevated levels of atmospheric greenhouse gases during the T-OAE, global temperatures would have increased and the hydrological cycle would have strengthened5. Rising pCO2, global temperatures, and precipitation rates would have led to accelerated weathering rates3. To investigate the proposition of accelerated weathering during the T-OAE, we have utilized osmium isotope (187Os/188Os) stratigraphy to reconstruct the 187Os/188Os composition of seawater over the event (see Supplemental Information).
The 187Os/188Os composition of seawater (187Os/188Ossw) reflects the sources of osmium to the ocean: rivers that drain continents (187Os/188Oscont ≈ 1.4) and aeolian dust (187Os/188Osaeol ≈ 1.04) represent a radiogenic end-member, and alteration of juvenile ocean crust or from the mantle (187Os/188Osm ≈ 0.12) and cosmic dust/bolides (187Os/188Oscos ≈ 0.12) represent an unradiogenic end-member16 (SI Fig. 1 and Supplemental Information). The flux of cosmic and aeolian dust represents a small fraction of the global input of osmium to the oceans and does not readily dissolve in seawater, and therefore does not appreciably affect the ocean’s 187Os/188Ossw composition16, 17. The present-day 187Os/188Ossw (~1.06) reflects the relatively greater input of continental-derived osmium to the ocean as compared to mantle-sourced osmium. Importantly, the short residence time of osmium in the oceans (~103–104 yr)18 permits the osmium isotope system to record ephemeral changes in global weathering patterns on the order of 103 to 105 years in the geological record19.
The 187Os/188Os compositions of organic-rich sediments are known to record the 187Os/188Os composition of contemporaneous seawater19, and serve as an archive of the past marine osmium isotope compositions. A previous osmium isotope study of the T-OAE interval from a sedimentary succession in the Cleveland Basin of Yorkshire, United Kingdom indicates that, during the event, there was a concomitant, transient increase of 187Os/188Ossw values by 0.720 (Fig. 2). This record was originally interpreted to be the result of an increase in continental weathering rates of 400 to 800%20. However, it has been suggested that these data reflect regional climatic changes where enhanced local runoff influenced the 187Os/188Ossw composition of the European epicontinental sea, which the Cleveland Basin was part of (Fig. 1), and therefore the 187Os/188Os record does not reflect a global weathering signal21. Key to this dispute is whether the Cleveland Basin was significantly hydrographically restricted so the local 187Os/188Ossw signal could be modified21, 22. A recently published osmium isotope record across the T-OAE from the Mochras borehole23, located in nearby Wales, displays a much less pronounced excursion of 0.4 during the T-OAE interval (Fig. 2), which further suggests that geochemical changes recorded in the Cleveland Basin were likely influenced by regional climatic and oceanographic dynamics18, 24, 25.

Records of the osmium isotope excursion across the T-OAE CIE from Yorkshire, United Kingdom20 and the Mochras borehole23. The Yorkshire dataset was originally interpreted to represent a 400–800% increase in continental weathering rates20; however, other interpretations suggests that the radiogenic values during the exaratum ammonite subzone were caused by hydrographic restriction21, 22. The close palaeogeographic proximity between these two sites, coupled with their significantly different 187Os/188Osi values suggests a regional influence on 187Os/188Ossw values in the European epicontinental seaway during the T-OAE.
To resolve whether the transient increases in 187Os/188Os observed across the T-OAE were indeed a global signal, we have investigated the osmium isotope record from the Lower Jurassic Fernie Formation of the Western Canada Sedimentary Basin located in present-day western Alberta (Fig. 1). This new location was situated on the eastern margin of the ocean of Panthalassa and therefore was located in a different ocean basin from the previously studied Yorkshire and Mochras sites (Figs 1 and 2). Ammonite biostratigraphy and carbon isotope stratigraphy of the Fernie Formation at East Tributary of Bighorn Creek has identified the upper Pliensbachian to middle Toarcian interval and the T-OAE CIEs15, 26,27,28. Zircon U-Pb dates from two bentonites located near the base of the section also provide temporal constraint and an age model for the section (Fig. 3; see Methods and Supplementary Data). Importantly, the entire interval of the East Tributary succession contains organic-rich strata (2–8% TOC; Figs 1 and 3)15, and thus represents an ideal location to reconstruct the global 187Os/188Ossw over the T-OAE interval (see Supplemental Information).

Chemostratigraphy of the Lower Jurassic Fernie Formation from East Tributary of Bighorn Creek, Alberta. δ13Corg = organic carbon isotopic compositions from ref. 15. 187Os/188Osi = initial osmium isotopic composition of organic-rich sediments. Lithostratigraphic members of the Fernie Formation, Stages of the Jurassic, and ammonite zonations for both northwestern Europe and western North American shown to the left of the stratigraphic column (refer to ref. 15 for the details of their placements). Vertical gray line in 187Os/188Osi record is the end-member 187Os/188Osm value of ~0.12. We report new single zircon U-Pb CA-ID-TIMS ages of 188.58 ± 0.17 (0.25) [0.32] Ma in the bentonite at −1.9 meters and 185.49 ± 0.16 (0.25) [0.32] Ma in the bentonite at 2.35 meters, located in the margaritatus Zone of NW Europe or the kunae Zone of western NA (see Methods and SI Data 2).
Results
187Os/188Osi record from North America
The high-resolution initial 187Os/188Os (187Os/188Osi) record of the East Tributary succession (see Supplemental Information) displays extremely unradiogenic values (187Os/188Osi ≈ 0.25) in the Pliensbachian and Lowest Toarcian, followed by a prominent radiogenic excursion (187Os/188Osi ≈ 0.6) during the Toarcian CIEs (Fig. 3). The 187Os/188Osi values decrease after the Toarcian CIE and asymptotically approach ~0.4 (Fig. 3; see Supplemental Information). Locally at East Tributary, aluminum and titanium concentrations increase 3-fold during the 187Os/188Osi excursion and remain high for the rest of the record (see Fig. 3 and SI Dataset 1), which suggests a local increase in the contribution of continentally derived materials during the event. However, their concentrations remain high as 187Os/188Osi values decrease after the Toarcian CIE, which suggests a minimal influence of a detrital component of rhenium and osmium to the osmium isotopic signature (see Fig. 3, Methods, and SI Dataset 1).
Discussion
Comparison of Early Jurassic 187Os/188Osi records
Other marine 187Os/188Osi records from the Lower Jurassic (Hettangian through Toarcian stages) generally show unradiogenic values20, 23, 29, 30. These are likely related to relatively elevated inputs of unradiogenic osmium from the weathering of the Central Atlantic Magmatic Province (CAMP) and the alteration of juvenile oceanic lithosphere or direct injection of mantle-derived osmium from initial opening of the North Atlantic31. The Upper Pliensbachian portion of our record from northeastern Panthalassa has broadly similar values to those observed in the European epicontinental sea20, 23, 29, which suggests they are representative of the global 187Os/188Ossw values, and indicative of a well-mixed Early Jurassic ocean. Further, the East Tributary 187Os/188Osi record shows a similar pattern to the other available records during the interval that contains the T-OAE20, 23. All the sites record an excursion to higher 187Os/188Osi values that follow the falling limb of the Toarcian negative CIE. This trend is followed by a return to lower 187Os/188Osi values after the rising limb of the negative CIE. However, in all cases 187Os/188Osi declines to values slightly higher than those observed before the excursion.
While all the 187Os/188Osi records display a similar overall pattern, their 187Os/188Osi values differ. The Yorkshire and East Tributary datasets have similar 187Os/188Osi values before and after the T-OAE (~0.3 and ~0.4, respectively); however, the Yorkshire dataset shows an excursion to significantly more radiogenic values (187Os/188Osi ≈ 1) during the T-OAE20 (Fig. 2). The Mochras data show higher 187Os/188Osi values just before the T-OAE CIE (~0.4), which increase to an acme of 0.8 during the T-OAE, and decrease to ~0.3 after the event23 (Fig. 2). While the absolute 187Os/188Osi values differ between the sites, the magnitude of the excursions at East Tributary and Mochras are similar at 0.4, and are almost half the magnitude observed at Yorkshire (0.7).
The differences observed between the 187Os/188Osi records at East Tributary, Mochras, and Yorkshire suggest there were regional differences in 187Os/188Ossw during the studied interval. These differences likely represent local processes such as differing degrees of hydrographic restriction from the open ocean and the amounts of local runoff and its 187Os/188Os composition. However, the similarity in the magnitude of the excursions recorded at East Tributary and Mochras suggest this likely represents the global record of change during the T-OAE. This observation, coupled with the more extreme 187Os/188Osi excursion record at Yorkshire, supports the suggestion that the Yorkshire 187Os/188Ossw record was influenced by a local riverine input of radiogenic osmium during the T-OAE21, and the East Tributary and Mochras records are more representative of global osmium seawater chemistry
With these observations in mind, we advocate, when possible, analyzing osmium isotope records from coeval stratigraphic successions deposited in different sedimentary and ocean basins18, 24,25,26 before attempting to interpret them as a global signal. This methodology is especially important regarding palaeoceanographic studies on intervals older than the Cretaceous since the preserved records are predominantly from continental margin and epicontinental successions, where geochemical signatures have a greater potential to be modified by local processes.
Quantifying the Early Jurassic marine osmium cycle
To gain a more quantitative measure of the changes in the marine osmium cycle during the Toarcian we employed a numerical box model that simulates the osmium inventory of the ocean and its isotopic composition (see Supplemental Information). Specifically, we test whether the osmium isotope excursion associated with the T-OAE (~300–500 kyr in duration)31, 32 can be reproduced by a transient increase in the weathering input of radiogenic osmium to the ocean. We also explored other situations that may have potentially driven the observed T-OAE osmium isotope record, but are likely implausible, such as decreasing the input flux of mantle-derived osmium to zero (see Table 1 for values explored and Supplemental Information for a discussion of these cases). Overall, the numerical model results show that the osmium isotope excursion can be reproduced by a transient three- to six-fold increase in the input of continental-derived osmium to the oceans over 100 to 200 kyr31, 32 (Fig. 4; more details of the modelling results including sensitivity tests can be found in the Supplemental Information).

Examples of the modelled osmium isotopic composition of the ocean over the T-OAE. (A) For this model run, the osmium isotopic composition of the continental input was increased to 2.0 and the flux of osmium from continents was increased two-fold (475.3 mol/yr) during the Toarcian OAE. This resulted in the seawater osmium isotope values to increase to 0.44, which does not reproduce the observed osmium isotope excursion observed at East Tributary. (B) Model run where the osmium isotopic composition and flux of the continental input of osmium was increased to 2.0 by ~3.4x respectively. This model run reproduced the osmium isotope excursion. (C) The osmium isotope composition of the continental input of osmium was kept at 1.4 during the Toarcian OAE, but the flux of osmium from continents was increased by ~6.3x to reproduce the osmium isotope excursion.
Changes in the 187Os/188Oscont to more radiogenic values through the differential weathering of lithologies such as shales and cratonic rocks33,34,35 could have played a role in the T-OAE osmium isotope record. We investigated the potential effect this change would have on the osmium budget during the event by running simulations where we elevated 187Os/188Oscont from 1.4 to 2 (see Supplemental Information for a discussion of the choice of the maximum 187Os/188Oscont value). In these simulations, a nearly three-fold increase of the input of continental-derived osmium to the oceans was still necessary to reproduce the excursion (Fig. 4), regardless of timescale used, and solely increasing 187Os/188Oscont to reasonable values cannot reproduce the observed excursion (see Supplemental Information). Given the plausible proposition of the changing composition of the continental weathering flux, we conservatively suggest that T-OAE weathering rates increased by as much as three-fold.
A potential source of radiogenic, continentally derived osmium was the remnants of the Central Pangaean Mountains, a Himalayan-scale mountain belt in eastern North America and northwestern Africa. This mountain belt was positioned at tropical and subtropical latitudes in the Early Jurassic (Fig. 1). The rifting of Pangaea during the Late Triassic and Early Jurassic would have exposed the core of the mountain range leaving this material open to weathering or erosion. General circulation models predict large increases in the air temperature and runoff during the T-OAE in the geographic region that contained these mountains36. These regional climatic changes would have facilitated enhanced chemical weathering, and makes this mountain belt a plausible source of the enhanced input of osmium to the oceans advocated here.
The weathering of organic-rich rocks and sediments would be another plausible way to raise the isotopic composition of the continental weathering flux, but also results in a net release of CO2 to the atmosphere37. However, enhanced continental runoff would also have increased nutrient delivery and stimulated primary productivity in aquatic environments leading to increased hypoxia, anoxia, and potentially euxinia5. Elevated burial of organic matter in these environments would have sequestered much more atmospheric CO2 than that associated with any black shale weathering, which we suggest represent only a fraction of the continental materials that were predominantly weathered during the event.
Differences in the osmium isotope response between OAE events
A striking feature of the 187Os/188Os records during the Mesozoic OAEs is the directionality of their excursions. The T-OAE records show a positive 187Os/188Os excursion, whereas the onset of the Cretaceous OAE 1a and OAE 2 both display negative excursions. The difference in the 187Os/188Os response to these events most likely lies in the environment where the LIPs were emplaced. The Cretaceous events are associated with subaqueous emplacements of the Ontong Java Plateau (OAE 1a) and the Caribbean and High Arctic LIPs (OAE 2). Emplacement of these LIPs would have supplied large amounts of unradiogenic, mantle-derived osmium directly into the oceans from weathering of basalts on the seafloor, resulting in osmium isotope excursions to nonradiogenic values25, 38,39,40.
The T-OAE, on the other hand, is associated with a subaerial emplacement of the Karoo-Ferrar LIP at high latitudes (Fig. 1), where the semi-arid climate would have made the relative weathering potential of this material low. In contrast to the younger OAEs, the Toarcian 187Os/188Osi records reflect enhancement of the weathering of continental materials facilitated by the injection of greenhouse gases into the atmosphere and subsequent climate changes. Notably, delivery of osmium from the Karoo-Ferrar LIP would have also been delayed, as compared to the Cretaceous LIPs. However, if weathering of the Karoo-Ferrar LIP was a significant source of osmium to the oceans during the T-OAE, then its lower 187Os/188Os compositions41,42,43,44,45 would necessitate an even greater contribution of continental material to generate the observed 187Os/188Osi excursion.
Implications and Conclusions
Based on the osmium isotope records and our modelling results, the transient increase in continental weathering rates during the T-OAE may be one of the largest observed during the Phanerozoic. Chemical weathering rates are also suggested to have significantly increased across the Permian-Triassic boundary46, Triassic-Jurassic boundary47, 48, and the Paleocene-Eocene Thermal Maximum49, all of which are associated with intervals of global warming, environmental deterioration, and extinction events50. The rapid response of the osmium isotope system during the T-OAE, as well as during other OAEs38,39,40, indicates that chemical weathering feedbacks may respond to episodes of rapid climatic warming on short timescales (103–106 years) and lead to a net drawdown of atmospheric CO2 5. Enhanced continental runoff would also have increased nutrient delivery and stimulated primary productivity in nearshore environments, leading to increased marine hypoxia, anoxia, and potentially euxinia5. CO2 would also have been sequestered through the deposition of organic-rich sediments in marine and lacustrine settings5, 6, 51.
In the case of the Toarcian OAE, increased weathering likely played a critical role in reversing the enhanced greenhouse state induced by Karoo-Ferrar magmatism. As atmospheric CO2 was consumed through these mechanisms, global temperatures would have declined5, 20. As modern atmospheric CO2 levels continue to increase at rates much higher than any point during the Cenozoic52, increased weathering, through the chemical and physical weathering feedbacks and stimulation of primary production and subsequent organic matter burial, may eventually act as a negative feedback to global warming, although on timescales much longer than what is necessary to mitigate the immediate environmental and ecological deterioration due to this warming53.
Methods
δ13C and total organic carbon analysis
δ13C and total organic carbon (TOC) were measured from each sample for rhenium, osmium, and trace metals (see below). The samples were prepared and analysed using the same methods from ref 15.
Rhenium and osmium isotopic analysis
In order to isolate primarily the hydrogenous rhenium and osmium from our samples, and minimize the removal of detrital rhenium and osmium, we followed the procedures of ref. 54. Between ~0.25 and 1 g of sample powder (dependent upon previously measured rhenium abundances via inductively-coupled plasma mass spectrometry) were digested with a known amount of 185Re and 190Os tracer (spike) solutions in 8 mL of a CrO3-H2SO4 solution; this reaction occurred in sealed Carius tubes, which were heated incrementally to 220 °C for 48 hours. The tubes were allowed to cool before opening. The osmium was immediately isolated and purified from the acid medium by solvent extraction using chloroform. This step was followed by the back reduction of Os from the chloroform into HBr. The Os fraction was further purified by micro-distillation. Rhenium was purified from the remaining CrO3-H2SO4 solution by a NaOH-Acetone solvent extraction55 and further purified using anion exchange chromatography. The purified Re and Os fractions were then loaded onto Ni and Pt filaments, respectively, and analysed for their isotopic composition using negative thermal-ionization mass spectrometry (NTIMS)56, 57 using a Thermo Scientific TRITON mass spectrometer with static Faraday collection for Re and ion-counting using a secondary electron multiplier in peak-hopping mode for Os. In-house Re and Os solutions were continuously analysed during the course of this study to ensure and monitor long-term mass spectrometry reproducibility. A 125 pg aliquot of the Re std solution and a 50 pg aliquot of DROsS yield 185Re/187Re values of 0.5983 ± 0.002 (1 SD, n = 6) and 187Os/188Os values of 0.16089 ± 0.0005 (1 SD, n = 8), respectively; both are identical to previously reported values57. The measured difference in 185Re/187Re values for the Re std solution and the accepted 185Re/187Re value (0.5974)58 is used for mass fractionation correction of the Re sample data. All Re and Os data are oxide and blank corrected. Procedural blanks for Re and Os in this study were 12 ± 3 pg/g and 0.07 ± 0.05 pg/g, respectively, with an 187Os/188Os value of 0.25 ± 0.15 (n = 4). The 187Re/188Os and 187Os/188Os uncertainties are determined through full propagation of uncertainties, including those in weighing, mass spectrometer measurements, spike calibrations, blank abundances and reproducibility of standard values.
Trace metal analysis
In order to compare the changes in [Re] and [Os] to sedimentation patterns across the T-OAE, we also analysed the concentrations of aluminum and titanium in each sample, which are used to estimate the contribution of terrigenous input to a sedimentary basin59, 60 (see Fig. 3 and SI dataset). Approximately 0.05 g of powder was added to a teflon beaker, followed by the addition of 4 mL of a 50:50 mixture of concentrated HCl and concentrated HNO3. This solution was placed inside a (CEM MARS 5) microwave assisted digestion system and run until all organic material had broken down at a temperature of 150 °C. The samples were then dried down and the silicates were dissolved using 4:1 HNO3 to HF, dried down, and re-dissolved in 5% HNO3 solution. A 100 μL solution split was spiked with an internal standard to measure elemental abundances using an Agilent 7500cs inductively-coupled plasma mass spectrometer in He and H mode. Internal standard was used to correct the samples for machine drift. International standards USGS SCO-1 and SDO-1 were also measured and had a reproducibility of ± 5%.
U-Pb analysis of zircons
CA-TIMS procedures described here are modified from refs 61,62,63. After rock samples have undergone standard mineral separation procedures zircons are handpicked in alcohol. The clearest, crack- and inclusion-free grains are selected, photographed, and then annealed in quartz glass crucibles at 900 °C for 60 hours. Annealed grains are transferred into 3.5 mL PFA screwtop beakers, ultrapure HF (up to 50% strength, 500 μL) and HNO3 (up to 14 N, 50 μL) are added and caps are closed finger tight. The beakers are placed in 125 mL PTFE liners (up to four per liner) and about 2 mL HF and 0.2 mL HNO3 of the same strength as acid within beakers containing samples are added to the liners. The liners are then slid into stainless steel Parr™ high pressure dissolution devices, which are sealed and brought up to a maximum of 200 °C for 8–16 hours (typically 175 °C for 12 hours). Beakers are removed from liners and zircon is separated from leachate. Zircons are rinsed with > 18 MΩ.cm water and subboiled acetone. Then 2 mL of subboiled 6 N HCl is added and beakers are set on a hotplate at 80°–130 °C for 30 minutes and again rinsed with water and acetone. Masses are estimated from the dimensions (volumes) of grains. Single grains are transferred into clean 300 μL PFA microcapsules (crucibles), and 50 μL 50% HF and 5 μL 14 N HNO3 are added. Each is spiked with a 233–235U-205Pb tracer solution (EARTHTIME ET535), capped, and again placed in a Parr liner (8–15 microcapsules per liner). HF and nitric acids in a 10:1 ratio, respectively, are added to the liner, which is then placed in a Parr high pressure device and dissolution is achieved at 220 °C for 40 hours. The resulting solutions are dried on a hotplate at 130 °C, 50 μL 6 N HCl is added to microcapsules and fluorides are dissolved in high-pressure Parr devices for 12 hours at 180 °C. HCl solutions are transferred into clean 7 mL PFA beakers and dried with 2 μL of 0.5 N H3PO4. Samples are loaded onto degassed, zone-refined Re filaments in 2 μL of silicic acid emitter64.
Isotopic ratios are measured with a modified single collector 354S (with Sector 54 electronics) thermal ionization mass spectrometer equipped with analogue Daly photomultipliers. Analytical blanks are 0.2 pg for U and up to 1.9 pg for Pb. U fractionation was determined directly on individual runs using the EARTHTIME ET535 mixed 233–235U-205Pb isotopic tracer and Pb isotopic ratios were corrected for fractionation of 0.25 ± 0.03%/amu, based on replicate analyses of NBS-982 reference material and the values recommended by ref. 65. Data reduction employed the excel-based program of ref. 66. Standard concordia diagrams were constructed and regression intercepts, weighted averages calculated with Isoplot67. Unless otherwise noted all errors are quoted at the 2-sigma or 95% level of confidence. Isotopic dates are calculated with the decay constants λ238 = 1.55125E-10 and λ235 = 9.8485E-10 (ref. 68) and a 238U/235U ratio of 137.88. EARTHTIME U-Pb synthetic solutions are analysed on an on-going basis to monitor the accuracy of results.
Five single zircon grains from the bentonite at −1.9 meters in the East Tributary section (see Fig. 3) were analysed by the uranium-lead chemical abrasion isotope dilution thermal ionization mass spectrometry technique (U-Pb CA-ID-TIMS). A weighted mean 206Pb/238U age of 188.58 ± 0.17 (0.25) [0.32] Ma, (MSWD = 0.89) is based on concordant and overlapping results for three of the analysed grains (see SI Dataset 2). Older results for the other two grains suggest that they are xenocrysts and/or contain inherited cores. It is important to note that this bentonite has a previously published multigrain U-Pb ID-TIMS age of 188.3 + 1.5/−1 Ma69.
Five single zircon grains from the bentonite at 2.35 meters in the East Tributary section (see Fig. 3) were analysed by the U-Pb CA-ID-TIMS technique. A weighted mean 206Pb/238U age of 185.49 ± 0.16 (0.25) [0.32] Ma, (MSWD = 1.17) is based on concordant and overlapping results for three of the analysed grains (see SI Dataset 2). Older results for the other two grains, one of which is discordant, suggest that they are xenocrysts and/or contain inherited cores.
Age model and calculation of 187Os/188Osi
The age model (see below) is constructed using a single grain U-Pb CA-ID-TIMS age of 188.58 ± 0.17 (0.25) [0.32] Ma from approximately two meters below the lowest interval with carbon isotope data in the East Tributary section15 and a single grain U-Pb CA-ID-TIMS age of 185.49 ± 0.16 (0.25) [0.32] Ma (see above) located at 2.35 meters in the section (see Fig. 3). Linear interpolation was used to calculate ages between the bentonites layers and between the age assigned for the Toarcian CIE. The onset of the CIE is placed at 183.1 Ma, with a total duration of 300 kyr31. Sedimentation rates are also assumed to remain constant after the Toarcian CIE. The initial osmium isotopic composition of the oceans (187Os/188Osi) was calculated using the following equation and the 187Re decay constant from ref. 70:
This equation accounts for the 187Os produced after deposition by the decay of 187Re. As stated above, the age component was derived from U-Pb ages from this succession (this study) and previously published dates for the age and estimated duration of the Toarcian CIE31. Furthermore, if a longer 500-kyr duration32 is assigned to the T-OAE CIE, the calculated 187Os/188Osi values do not change significantly and our interpretations do not change (see Supplemental Information).
References
Walker, J. C. G., Hays, P. B. & Kasting, J. F. A negative feedback mechanism for the long-term stabilization of Earth’s surface temperature. J. Geophys. Res. 86, 9776–9782 (1981).
Berner, R. A., Lasaga, A. C. & Garrels, R. M. The carbonate-silicate geochemical cycle and its effect on atmospheric carbon dioxide over the past 100 million years. Am. J. Sci. 283, 641–683 (1983).
Kump, L. R., Brantley, S. L. & Arthur, M. A. Chemical Weathering, Atmospheric CO2, and Climate. Ann. Rev. Earth Plan. Sci. 28, 611–667 (2000).
Foster, G. L. & Vance, D. Negligible glacial-interglacial variation in continental weathering rates. Nature 444, 918–921 (2006).
Jenkyns, H. C. Geochemistry of oceanic anoxic events. G3 11, Q03004 (2010).
Jenkyns, H. C. The Early Toarcian (Jurassic) Anoxic Event: Stratigraphic, Sedimentary, and Geochemical Evidence. Am. J. Sci. 288, 101–151 (1988).
Harries, P. J. & Little, C. T. S. The early Toarcian (Early Jurassic) and the Cenomanian-Turonian (Late Cretaceous) mass extinctions: similarities and contrasts. Palaeogeo. Palaeoclim. Palaeoecol 154, 39–66 (1999).
Pálfy, J. & Smith, P. L. Synchrony between Early Jurassic extinction, oceanic anoxic event, and the Karoo-Ferrar flood basalt volcanism. Geology 28, 747–750 (2000).
McElwain, J. C., Wade-Murphy, J. & Hesselbo, S. P. Changes in carbon dioxide during an oceanic anoxic event linked to intrusion into Gondwana coals. Nature 435, 479–482 (2005).
Beerling, D. J. & Brentnall, S. J. Numerical evaluation of mechanisms driving Early Jurassic changes in global carbon cycling. Geology 35, 247–250 (2007).
Svensen, H. et al. Hydrothermal venting of greenhouse gases triggering Early Jurassic global warming. Earth Plan. Sci. Lett 256, 554–566 (2007).
Hesselbo, S. P. et al. Massive dissociation of gas hydrate during a Jurassic oceanic anoxic event. Nature 406, 392–395 (2000).
Kemp, D. B., Coe, A. L., Cohen, A. S. & Schwark, L. Astronomical pacing of methane release in the Early Jurassic period. Nature 437, 396–399 (2005).
Pieńkowski, G., Hodbod, M. & Ullmann, C. V. Fungal decomposition of terrestrial organic matter accelerated Early Jurassic climate warming. Scientific Rep 6, 31030 (2016).
Them, T. R. II et al. High-resolution carbon isotope records of the Toarcian Oceanic Anoxic Event (Early Jurassic) from North America and implications for the global drivers of the Toarcian carbon cycle. Earth Plan. Sci. Lett 459, 118–126 (2017).
Peucker-Ehrenbrink, B. & Ravizza, G. The marine osmium isotope record. Terra Nova 12, 205–219 (2000).
Peucker-Ehrenbrink, B. Accretion of extraterrestrial matter during the last 80 million years and its effect on the marine osmium isotope record. Geochim. Cosmochim. Acta 60, 3187–3196 (1996).
Rooney, A. D. et al. Tracking millennial-scale Holocene glacial advance and retreat using osmium isotopes: Insights from the Greenland ice sheet. Quat. Sci. Rev. 138, 49–61 (2016).
Cohen, A. S., Coe, A. L., Bartlett, J. M. & Hawkesworth, C. J. Precise Re—Os ages of organic-rich mudrocks and the Os isotope composition of Jurassic seawater. Earth Plan. Sci. Lett 167, 159–173 (1999).
Cohen, A. S., Coe, A. L., Harding, S. M. & Schwark, L. Osmium isotope evidence for the regulation of atmospheric CO2 by continental weathering. Geology 32, 157–160 (2004).
McArthur, J. M., Algeo, T. J., van de Schootbrugge, B., Li, Q. & Howarth, R. J. Basinal restriction, black shales, Re-Os dating, and the Early Toarcian (Jurassic) oceanic anoxic event. Paleoceanography 23, PA4217 (2008).
Waltham, D. & Gröcke, D. R. Non-uniqueness and interpretation of the seawater 87Sr/86Sr curve. Geochim. Cosmochim. Acta 70, 384–394 (2006).
Percival, L. M. E. et al. Osmium isotope evidence for two pulses of increased continental weathering linked to Early Jurassic volcanism and climate change. Geology 44, 759–762 (2016).
Paquay, F. S. & Ravizza, G. Hetergeneous seawater 187Os/188Os during the Late Pleistocene glaciations. Earth Plan. Sci. Lett 349-350, 126–138 (2012).
Du Vivier, A. D. C. et al. Marine 187Os/188Os isotope stratigraphy reveals the interaction of volcanism and ocean circulation during Oceanic Anoxic Event 2. Earth Plan. Sci. Lett 389, 23–33 (2014).
Hall, R. L. Lithostratigraphy and biostratigraphy of the Fernie Formation (Jurassic) in the southern Canadian Rocky Mountains, in Stott, D. F. & Glass, D. J., eds, The Mesozoic of Middle North America. Can. Soc. Petr. Geol. Mem. 9, 233–247 (1984).
Hall, R. L. New Lower Jurassic ammonite faunas from the Fernie Formation, southern Canadian Rocky Mountains. Can. J. Earth Sci. 24, 1688–1704 (1987).
Asgar–Deen, M., Hall, R., Craig, J. & Riediger, C. New biostratigraphic data from the Lower Jurassic Fernie Formation in the subsurface of west-central Alberta and their stratigraphic implications. Can. J. Earth Sci. 40, 45–63 (2003).
Porter, S. J., Selby, D., Suzuki, K. & Gröcke, D. Opening of a trans-Pangaean marine corridor during the Early Jurassic: Insights from osmium isotopes across the Sinemurian-Pliensbachian GSSP, Robin Hood’s Bay, UK. Palaeogeo. Palaeoclim. Palaeoecol. 375, 50–58 (2013).
Cohen, A. S. & Coe, A. L. New geochemical evidence for the onset of volcanism in the Central Atlantic magmatic province and environmental change at the Triassic-Jurassic boundary. Geology 30, 267–270 (2002).
Sell, B. et al. Evaluating the temporal link between the Karoo LIP and climatic—biologic events of the Toarcian Stage with high-precision U-Pb geochronology. Earth Plan. Sci. Lett 408, 48–56 (2014).
Boulila, S. et al. Astronomical calibration of the Toarcian State: Implications for sequence stratigraphy and duration of the early Toarcian OAE. Earth Plan. Sci. Lett 386, 98–111 (2014).
Peucker-Ehrenbrink, B. & Hannigan, R. E. Effect of black shale weathering on the mobility of rhenium and platinum group elements. Geology 28, 475–478 (2000).
Jaffe, L. A., Peucker-Ehrenbrink, B. & Petsch, S. T. Mobility of rhenium, platinum group elements and organic carbon during black shale weathering. Earth Plan. Sci. Lett 198, 339–353 (2002).
Pierson-Wickmann, A.-C., Reisberg, L. & France-Lanord, C. Behavior of Re and Os during low-temperature alteration: Results from Himalayan soils and altered black shales. Geochim. Cosmochim. Acta 66, 1539–1548 (2002).
Dera, G. & Donnadieu, Y. Modeling evidences for global warming, Arctic seawater freshening, and sluggish ocean circulation during the Early Toarcian anoxic event. Paleoceanography 27, PA002283 (2012).
Georg, R. B., West, A. J., Vance, D., Newman, K. & Halliday, A. N. Is the marine osmium isotope record a probe for CO2 release from sedimentary rocks? Earth Plan. Sci. Lett 367, 28–38 (2013).
Tejada, M. L. G. et al. Ontong Java Plateau eruption as a trigger for the early Aptian oceanic anoxic event. Geology 37, 855–858 (2009).
Bottini, C., Cohen, A. S., Erba, E., Jenkyns, H. C. & Coe, A. L. Osmium-isotope evidence for volcanism, weathering, and ocean mixing during the early Aptian OAE 1a. Geology 40, 583–586 (2012).
Turgeon, S. C. & Creaser, R. A. Cretaceous oceanic anoxic event 2 triggered by a massive magmatic episode. Nature 454, 323–326 (2008).
Ellam, R. M., Carlson, R. W. & Shirey, S. B. Evidence from Re-Os isotopes for plume-lithosphere mixing in Karoo flood basalts genesis. Nature 359, 718–721 (1992).
Molzahn, M., Reisberg, L. & Wörner, G. Os, Sr, Nd, Pb, O isotope and trace element data from the Ferrar flood basalts, Antarctica: evidence for an enriched subcontinental lithospheric source. Earth Plan. Sci. Lett 144, 529–546 (1996).
Riley, T. R., Leat, P. T., Storey, B. C., Parkinson, I. J. & Millar, I. L. Ultramafic lamprophyres of the Ferrar large igneous province: evidence for a HIMU mantle component. Lithos 66, 63–76 (2003).
Heinonen, J. S., Carlson, R. W. & Luttinen, A. V. Isotopic (Sr, Nd, Pb, and Os) composition of highly magnesian dikes os Vestfjella, western Dronning Maud Land, Antarctica: A key to the origins of the Jurassic large igneous province? Chem. Geol. 277, 227–244 (2010).
Heinonen, J. S., Carlson, R. W., Riley, T. R., Luttinen, A. V. & Horan, M. F. Subduction-modified oceanic crust mixed with a depleted mantle reservoir in the sources of the Karoo continental flood basalt province. Earth Plan. Sci. Lett 394, 229–241 (2014).
Sheldon, N. D. Abrupt chemical weathering increase across the Permian-Triassic boundary. Palaeogeo. Palaeoclim. Palaeoecol 231, 315–321 (2006).
Beerling, D. J. & Berner, R. A. Biogeochemical constraints on the Triassic-Jurassic boundary carbon cycle event. Glob. Biogeo. Cycles 16, GB001637 (2002).
Kuroda, J., Hori, R. S., Suzuki, K., Gröcke, D. R. & Ohkouchi, N. Marine osmium isotope record across the Triassic-Jurassic boundary from a Pacific pelagic site. Geology 38, 1095–9098 (2010).
Ravizza, G., Norris, R. N. & Blusztajn, J. An osmium isotope excursion associated with the late Paleocene thermal maximum: Evidence of intensified chemical weathering. Paleoceanography 16, 155–163 (2001).
Hönisch, B. et al. The Geological Record of Ocean Acidification. Science 335, 1058–1063 (2012).
Xu, W. et al. Carbon sequestration in an expanded lake system during the Toarcian oceanic anoxic event. Nature Geosci 10, 129–134 (2017).
Zeebe, R. E., Ridgwell, A. & Zachos, J. C. Anthropogenic carbon release rate unprecedented during the past 66 million years. Nature 9, 325–329 (2016).
Parmesan, C. Ecological and Evolutionary Responses to Recent Climate Change. Annu. Rev. Ecol. Evol. Syst. 37, 637–669 (2006).
Selby, D. & Creaser, R. A. Re-Os geochronology of organic rich sediments: an evaluation of organic matter analysis methods. Chem. Geol. 200, 225–240 (2003).
Cumming, V. M., Poulton, S. W., Rooney, A. D. & Selby, D. Anoxia in the terrestrial environment during the late Mesoproterozoic. Geology 41, 583–586 (2013).
Creaser, R. A., Papanastassiou, D. A. & Wasserburg, G. J. Negative thermal ion mass spectrometry of osmium, rhenium and iridium. Geochim. Cosmochim. Acta 55, 397–401 (1991).
Völkening, J., Walczyk, T. & Heumann, K. G. Osmium isotope ratio determination by negative thermal ion mass spectrometry. Int. J. Mass Spectrom. Ion Process. 105, 147–159 (1991).
Gramlich, J. W., Murphy, T. J., Garner, E. L. & Shields, W. R. Absolute isotopic abundance ratio and atomic weight of a reference sample of rhenium. J. Res. Nat. Bur. Stds 77A, 691–698 (1973).
Peterson, L. C., Haug, G. H., Hughen, K. A. & Röhl, U. Rapid Changes in the Hydrologic Cycle of the Tropical Atlantic During the Last Glacial. Science 290, 1947–1951 (2000).
Latimer, J. C. & Filippelli, G. M. Terrigenous input and paleoproductivity in the Southern Ocean. Paleoceanography 16, 627–643 (2001).
Mundil, R., Ludwig, K. R., Metcalfe, I. & Renne, P. R. Age and timing of the Permian Mass Extinctions: U/Pb Dating of Closed-System Zircons. Science 305, 1760–1763 (2004).
Mattinson, J. M. Zircon U-Pb chemical abrasion (“CA-TIMS”) method: Combined annealing and multi-step partial dissolution analysis for improved precision and accuracy of zircon ages. Chem. Geol. 220, 47–66 (2005).
Scoates, J. S. & Friedman, R. M. Precise age of the platiniferous Merensky Reef, Bushveld Complex, South Africa, by the U-Pb zircon chemical abrasion ID-TIMS technique. Econ. Geol. 103, 465–471 (2008).
Gerstenberger, H. & Haase, G. A. Highly effective emitter substance for mass spectrometric Pb isotopic ratio determinations. Chem. Geol. 136, 309–312 (1997).
Thirlwall, M. F. Inter-laboratory and other errors in Pb isotope analyses investigated using a 207Pb-204Pb double spike. Chem. Geol. 163, 299–322 (2000).
Schmitz, M. D. & Schoene, B. Derivation of isotope ratios, errors, and error correlations for U-Pb geochronology using 205Pb-235U-(233U)-spiked isotope dilution thermal ionization mass spectrometric data. Geochem. Geophys. Geosyst. 8, Q08006 (2007).
Ludwig, K. R. Isoplot 3.00, A Geochronological Toolkit for Microsoft Excel. University of California at Berkeley, kludwig@bgc.org.
Jaffey, A. H., Flynn, K. F., Glendenin, L. E., Bentley, W. C. & Essling, A. M. Precision measurement of half-lives and specific activities of 235U and 238U. Phys. Rev. C4, 1889–1906 (1971).
Hall, R., McNicoll, V., Gröcke, D., Craig, J. & Johnston, K. Integrated stratigraphy of the lower and middle Fernie Formation in Alberta and British Columbia, western Canada. Riv. Ital. Paleon. Strat 110, 61–68 (2004).
Smoliar, M. I., Walker, R. J. & Morgan, J. W. Re-Os ages of Group IIA, IIIA, IVA and IVB iron meteorites. Science 271, 1099–1102 (1996).
Scotese, C. R. Atlas of Earth History. PALEOMAP Project, Arlington. Texas (2001).
Acknowledgements
TRT would like to thank the Virginia Tech College of Science Roundtable grant committee for the Make-a-Difference Scholarship and the ExxonMobil/Geological Society of America, American Association of Petroleum Geologists, and International Association of Sedimentologists graduate student grant programs for funding (IAS grant funded the pilot study). A grant to BCG (EAR-1324752) and JDO (OCE-1624895) from the National Science Foundation also funded this work. DS acknowledges the Total Endowment Fund. Thanks to Dr. Joanna Hesselink, H. Lin, C. Wall, N. Moerhius, and T. Ockerman for laboratory assistance, and to Angela Gerhardt, Emma Tulsky, and Selva Marroquín for their help in collecting samples. Sample collections were authorized by the following permits: Parks Canada, Permit No: YHTR-2014-16156; RTMP, Permit No: 13-058, 14-009, 15-019. Finally, we would like to thank two anonymous reviewers whose comments greatly improved the manuscript.
Author information
Authors and Affiliations
Contributions
T.R.T., B.C.G., D.S., and D.R.G. designed the study. T.R.T. and B.C.G. collected samples. T.R.T. and D.S. conducted the Re-Os geochemical analyses. J.D.O. conducted the elemental analyses. R.M.F. conducted the U-Pb CA-ID-TIMS analyses. T.R.T. and B.C.G. conducted the numerical modelling. All authors analysed the data. T.R.T. and B.C.G. wrote the paper with contributions from all the authors. T.R.T. prepared the figures.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Them, T.R., Gill, B.C., Selby, D. et al. Evidence for rapid weathering response to climatic warming during the Toarcian Oceanic Anoxic Event. Sci Rep 7, 5003 (2017). https://doi.org/10.1038/s41598-017-05307-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05307-y
This article is cited by
-
Disparity between Toarcian Oceanic Anoxic Event and Toarcian carbon isotope excursion
International Journal of Earth Sciences (2024)
-
Latest Pliensbachian to Early Toarcian depositional environment and organo-facies evolution in the North-German Basin (Hondelage Section)
International Journal of Earth Sciences (2024)
-
Lower Jurassic (Pliensbachian–Toarcian) marine paleoenvironment in Western Europe: sedimentology, geochemistry and organic petrology of the wells Mainzholzen and Wickensen, Hils Syncline, Lower Saxony Basin
International Journal of Earth Sciences (2024)
-
Global ocean redox changes before and during the Toarcian Oceanic Anoxic Event
Nature Communications (2023)
-
New age constraints on the Lower Jurassic Pliensbachian–Toarcian Boundary at Chacay Melehue (Neuquén Basin, Argentina)
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
