
Abstract
The differential diagnosis of immune (ITP) and hereditary macrothrombocytopenia (HM) is key to patient management. The immature platelet fraction (IPF) represents the subset of circulating platelets with higher RNA content, and has been shown to distinguish hypo- from hyperproliferative thrombocytopenias. Here we evaluated the diagnostic accuracy of IPF in the differential diagnosis between HM and other thrombocytopenias in a population of patients with post-chemotherapy thrombocytopenia (n = 56), bone marrow failure (n = 22), ITP (n = 105) and HM (n = 27). TPO levels were also measured in HM and ITP matched for platelet counts. Platelet counts were similar in all patient groups. Higher IPF values were observed in both ITP (12.3%; 2.4–65.6%) and HM (29.8%; 4.6–65.9%) compared to hypoproliferative thrombocytopenias. IPF values were also higher in HM compared to ITP, yielding a diagnostic accuracy of 0.80 (95%CI 0.70–0.90; P < 0.0001) to distinguish these two conditions. Intra- and inter-assays reproducibility of IPF in HM patients revealed that this is a stable parameter. In conclusion, IPF is increased in HM compared to both ITP and other thrombocytopenias and contributes to the differentiation between ITP and HM. Further studies are warranted to understand the biological rationale of these findings and to its incorporation in diagnostic algorithms of HM.
Similar content being viewed by others

Introduction
The differential diagnosis of thrombocytopenias includes a variety of conditions such as hematologic malignancies, bone marrow failure (BMF), hypersplenism, immune thrombocytopenia (ITP), microangiopathic hemolytic anemias and hereditary macrothrombocytopenia (HM). Among these, the differential diagnosis between ITP and HM can be challenging due to the absence of specific tests, particularly in patients with mild bleeding symptoms1,2,3. Recently, the feasibility of using parameters of the complete blood count (CBC) to support this differential diagnosis was illustrated by a series of studies which demonstrated and validated that the mean platelet volume (MPV) can help the segregation of patients with ITP and HM4, 5.
In recent years, new parameters were incorporated to the CBC, including the immature platelet fraction (IPF), which represents a population of newly formed platelets containing a greater amount of residual RNA6. Initially, the IPF was measured by flow cytometry, and described as reticulated platelets7. Recently, studies have reported the clinical utility of measuring immature platelets in clinical settings using automated hematology analyzers8,9,10. The correlation between flow cytometry and hematology analyzers in quantifying this population has been previously demonstrated11.
Although the utility of the IPF in the differential diagnosis between hypo- and hyperproliferative thrombocytopenias has been already reported8, 12, less information is available about its use in the differential diagnosis between ITP and HM. In 2000, Fabris et al. evaluated reticulated platelets by flow cytometry in a population of 29 patients with HM, observing reduced IPF values when compared to ITP13. More recently, Miyazaki et al.14, in a study with 15 patients with HM in which the IPF was measured in an automated hematology analyzer observed significantly higher IPF values in HM compared to ITP. To further elucidate the value of IPF determination in the diagnosis of thrombocytopenias, with focus on the differential diagnosis between ITP and HM, we investigated the diagnostic accuracy and the precision of IPF measurements in a population of patients with different causes of thrombocytopenia, including a well-characterized cohort of patients with HM. In addition, we also evaluated whether thrombopoietin (TPO) levels could further facilitate this diagnosis.
Methods
Study design and patient population
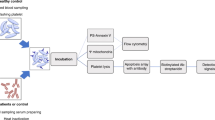
This was a cross-sectional diagnostic accuracy study, designed according to STARD (Standards for the Reporting of Diagnostic accuracy studies) guidelines15. The study population consisted of patients with thrombocytopenia confirmed in two independent samples and microscopic analysis, in regular clinical follow-up in the Hemostasis outpatient clinic of University of Campinas, or admitted to the hematology ward of the same institution. The inclusion criteria was a confirmed diagnosis of any of the following: (i) ITP (based on previously established guidelines)16; (ii) bone marrow failure (including aplastic anemia and myelodisplatic syndromes with a platelet count below 150 × 109/L)17, 18; (ii) post-chemotherapy (Ctx) thrombocytopenia (in admitted patients with hematological malignancies) with a platelet count below 150 × 109/L; and (iv) HM, with platelet counts below 150 × 109/L. Diagnosis of HM was established by clinical and laboratory criteria as determined by international guidelines19,20,21. These included the exclusion of all other causes of thrombocytopenia, objective confirmation of thrombocytopenia in first-degree relatives, platelet aggregation studies, molecular analyses and specific tests such as platelet glycoprotein studies and electron microscopy in selected cases. Exclusion criteria included: a platelet count above 150 × 109/L in ITP patients at the day of enrollment; (ii) the presence of conditions known to influence IPF values such as sepsis and other inflammatory diseases22, or (iii) the use of antiplatelet agents. The study was approved by the Institutional Review Board of University of Campinas (certificate number 411.620/2013) and performed in accordance with the Declaration of Helsinki. All patients provided written informed consent prior to enrollment.
Recruitment occurred between July 2013 and February 2015. ITP, HM and BMF patients were included consecutively, aiming for an enrollment of 100 patients with ITP, 20 with BMF and all patients with HM. Post-Ctx patients were included using a convenience sampling strategy, which consisted in weekly visits to the hematology ward, with a target of 50 patients.
Sample collection and processing
Samples were collected by venipuncture by the same nursing team responsible for the collection of routine clinical samples, and using the same standard operating procedures in both clinical sites. Blood was drawn in Vacutainer® EDTA K2 tubes (Becton Dickinson-BD, Franklin Lakes, Nova Jersey, EUA). IPF measurements were performed within four hours from sample collection, which is the threshold suggested in the equipment operational procedures. After IPF analysis, tubes were centrifuged (2,500 g for 10 min at room temperature), and plasma aliquots were frozen at −80 °C until analysis.
IPF measurement
Whole blood samples were analyzed in a Sysmex XE 5000 hematology analyzer (Sysmex, Kobe, Japan). Briefly, the IPF fraction was identified in the reticulocyte channel, employing a fluorescent dyes containing polymethine and oxazine. These dyes penetrate the cell membrane, staining residual RNA of red blood cells (reticulocytes) and platelets. In addition, these two populations are separated by cell size. A computer algorithm then discriminates mature platelets and the IPF, which was expressed as a percentage of total platelets (IPF%). In addition, we also calculated the absolute count of the IPF (A-IPF), which has been previously reported as superior to the IPF% in some clinical settings9, 10. Finally, we also evaluated the intra- and inter-assay reproducibility of IPF measurements in a subgroup of patients with HM. Platelet counts and mean platelet volume (MPV) were obtained from the same samples, by the same hematological analyzer, which measures the MPV by impedance. In the laboratory where IPF measurements were performed, commercial internal quality control samples (Sysmex e-Check, XE) are run before the beginning of each 8 h-routine, as well as after any corrective or preventive maintenance of the hematological analyzer. In addition, the laboratory participates in a regular national external quality control program and is within the scope of the University laboratory quality management system.
TPO measurement
Circulating TPO levels were measured in plasma samples using a commercial immunoassay (Quantikine, “Human thrombopoietin Immunoassay”, R&D Systems, Minneapolis, USA), based on a sandwich ELISA technique, according to the manufacturer’s protocol, in all HM patients and in seventy-four ITP patients. Of note, none of these patients were using TPO receptor agonists. A group of eight patients with a history of ITP diagnosis that were not excluded from the study due to platelet counts above 150 × 109/L were used as a control group for the measurement of TPO levels. All of these patients were off-ITP treatment for at least one year and had platelet counts above 250 × 109/L at the time of testing.
Statistical Analysis
Results are expressed as median and range, or means ± SD, unless mentioned otherwise. Differences between quantitative variables were evaluated by Mann-Whitney or Kruskal-Wallis with Dunn’s post-test for comparison with 2 or more than 2 variables respectively. Associations involving categorical clinical variables were analyzed using the Fisher exact test. Correlation analyses were performed using the Pearson or Spearman correlation (rho) test, according to data distribution. The diagnostic accuracy of IPF for the segregation of patients by each diagnosis was estimated by the ROC (receiver operating characteristic) procedure, which allows the simultaneous analysis of sensitivity and specificity of each test in relation to selected clinical outcomes. Results are reported with confidence intervals and level of significance. A P value < 0.05 was considered statistically significant.
Results
Two-hundred forty-eight patients were enrolled in this study, of which 38 were excluded due to platelet counts above 150 × 109/L (all of them from the ITP group). In total, 210 patients were analyzed, divided in the following subgroups: Post-Ctx (n = 56), BMF (n = 22), ITP (n = 105) and HM (n = 27). Demographic and clinical characteristics are shown in Table 1. For patients with HM, these data are shown individually (Table 2). In agreement with previous reports, the MPV was not reported by the analyzer used in this study in most patients with HM (Table 1) owing to abnormalities in platelet distribution curves4, 5. In all other samples, IPF was normally reported. Of note, no differences were found in platelet counts between ITP and HM patients.
Similar IPF values were observed between patients with Post-Ctx thrombocytopenia and BMF. All other comparisons yielded significant differences in IPF between different patient groups (Fig. 1), with both ITP and HM patients with higher IPF levels than Post-Ctx and BMF. In addition, HM patients presented a significantly higher median IPF level (29.8%; 4.6–65.9%) than ITP patients (12.3%; 2.4–65.6%; P < 0.0001). Similar results were obtained when the IPF was analyzed in absolute values (A-IPF) (data not shown).

Immature platelet fraction (%) of patients with different causes of thrombocytopenia; Post-Ctx: post chemotherapy; BMF: bone marrow failure; ITP: immune thrombocytopenia; HM: hereditary macrothrombocytopenia. Kruskal-Wallis test with Dunn’s post test (**P < 0.001, *P < 0.05).
An inverse correlation between IPF and platelet counts was observed in BMF, HM, and ITP, but not in Pos-Ctx patients (Fig. 2). IPF values correlated with MPV only in Post-Ctx (rho = 0.57; P < 0.0001).

Scatter plots showing correlation between immature platelet fraction (IPF%) and platelet counts (X 109/L) of patients with different causes of thrombocytopenia. (a) Post-chemotherapy, (b) bone marrow failure, (c) immune thrombocytopenia, (d) hereditary macrothrombocytopenia. Spearman correlation coefficient is shown.
The diagnostic accuracy of IPF% and A-IPF measurements for the differential diagnosis of thrombocytopenia was estimated using ROC analysis. The AUC for the differential diagnosis between ITP and HM was 0.80 (95%CI 0.70–0.90; P < 0.0001). Using an arbitrary IPF% cut-off value of 17.4%, which presented the best combination of sensitivity and specificity, ITP could be distinguished from HM with a sensitivity of 70% and a specificity of 90%. The positive and negative predictive values were 81.48% and 71.43 respectively. A similar result was obtained using the A-IPF, which yielded and AUC of 0.77 (95%CI 0.67–0.87; P < 0.0001). Therefore, all additional analyses were performed using the IPF%.
As previously described, ITP patients had significantly lower TPO levels compared to control subjects23 despite the former group having much lower platelet counts than the latter. Even when compared to HM patients with similar levels of thrombocytopenia, ITP patients presented significantly lower TPO levels (Fig. 3). No significant correlation could be observed with platelet counts or IPF% in HM. Interestingly, despite lower TPO levels in ITP significant correlations were observed between TPO levels and platelet count (rho = −0.50; P < 0.001) and with IPF% (rho = 0.44; P < 0.001). The estimated diagnostic accuracy of TPO measurement for the differential diagnosis between ITP and HM was 0.66 (95%Cl 0.56–0.77; P = 0.007).

Thrombopoietin (TPO) levels were measured by ELISA in patients with immune thrombocytopenia (ITP; n = 74) and hereditary macrothrombocytopenia (HM; n = 27). Eight ITP patients in complete remission for at least one year and off any treatment were evaluated as a control population. Horizontal bars represent: medians and interquartile ranges. Kruskal-Wallis test with Dunn’s post test; *P = 0.04 and **P = 0.03.
The intra- and inter-assay reproducibility of IPF% measurements were evaluated in six HM patients, all of them with IPF% values in the upper range of the IPF% distribution (Table 3). The coefficient of variation (CV%) of three sequential IPF% measurements (intra-assay reproducibility) was 5.4 ± 3.1, similar to the CV% of platelet counts from the same samples. In addition, we repeated the IPF% measurements of these patients after 12 months from the first measurement (Table 3). While platelet counts fluctuations were observed between these two measurements in some of these patients (CV% = 30.2 ± 29.0), IPF% remained fairly constant (CV% = 7.6 ± 3.9), suggesting that this is a stable parameter in HM, irrespective of fluctuations in platelet counts.
Despite the low number of patients with HM, we performed a subgroup analysis to investigate whether IPF values differed between patients with different types of HM (Table 2). While no difference was observed between patients with molecularly confirmed MYH9-related platelet disorders (IPF = 43.2%, range 22.4–62.7%) or Bernard-Soulier syndrome (IPF = 38.3%, range 28.3–65.9%), lower IPF values were observed in the remaining patients classified as non-specified hereditary macrothrombocytopenia (IPF = 17.5%, range 4.6–51.7%; P < 0.05).
Discussion
The differential diagnosis between ITP and HM represents a significant clinical problem since tests used to distinguish these two conditions are not available in most clinical laboratories, and even when available, cannot discern among all clinical etiologies. The development of hematological analyzers in the last decades has allowed the incorporation of new parameters in the diagnosis of hematological disorders; and the feasibility of this strategy for the diagnosis of HM has been shown by a series of studies evaluating platelet size in these patients. These studies demonstrated that MPV is significantly higher in patients with HM compared to ITP4, a finding that was later validated in a multicenter study using different instruments to measure this parameter5. Using a population of 210 patients with different causes of thrombocytopenia, the main result of our study was that another platelet parameter derived from automated an hematology analyzer reliably differentiates HM from ITP, with an estimated diagnostic accuracy similar to that reported in the literature for MPV4, 5.
In our population, IPF% values were significantly higher (almost three times) in the HM group compared to hypoproliferative thrombocytopenias such as BMF and Post-Ctx. More importantly, IPF% values were higher in HM than ITP patients with similar platelet counts. An inverse correlation between platelet counts and IPF% was observed not only in ITP patients but also in patients with BMF and HM. The absence of such correlation in patients with Post-Ctx thrombocytopenia is most likely due to the blunted thrombopoietic response secondary to chemotherapy. The ROC analysis estimated significant diagnostic accuracies of IPF% and A-IPF in the differential diagnosis of ITP and HM even in a group of patients with similar levels of thrombocytopenia (median 52 × 109/L for both groups). It should be noted that in contrast to other clinical settings, no relevant difference was observed in when IPF was reported as a percentage or in absolute values.
The increase of IPF in patients with hyperproliferative thrombocytopenias such as ITP has been recognized since the original demonstration that the RNA content of circulating platelets could be used as an indicator of thrombopoietic activity24. Formerly measured by flow cytometry, the evaluation of these recently produced platelets has been reported in other patient populations with different thrombocytopenic etiologies12. This was first assessed in 2000 in HM, in a study with 29 patients with chronic hereditary thrombocytopenia and 23 patients with ITP. Using a flow cytometry-based method, markedly lower reticulated platelet counts were observed in patients with HM compared to ITP13. More recently, the IPF% was evaluated in a group of 15 patients with congenital macrothrombocytopenia using an automated hematological analyzer as in our study. In contrast to Fabris et al., markedly higher IPF% levels were observed in HM patients compared to ITP patients14. Herein, we confirm and extend these results in an independent and larger population of 27 well-characterized patients with HM and 105 patients with ITP. The validation of IPF% measurement as a diagnostic tool in HM is important due to differences in the management of these patients compared to ITP and the current lack of accessible tests for this purpose. Moreover, our study found good intra- and inter-assay reproducibility of IPF% measurements in patients with HM. This is particularly important due to the observation of Myazaki et al., that small platelet aggregates could increase IPF% values14, and to the fact that mechanisms underlying the increase in IPF% values in these patients have not been elucidated. In this context, the low CV of IPF% measurements both in the same sample and in different samples from the same patient support that the IPF% in HM is a stable and robust indicator, paving the way for studies aimed to evaluate its incorporation in the clinical evaluation of these patients.
We also evaluated whether TPO measurements could contribute to the differential diagnosis between HM and ITP, or provide insights about the mechanisms underlying the variation of IPF values observed in these patients. Despite the fact that HM patients had higher TPO levels compared to ITP patients, this difference was much smaller than the one that we reported in IPF. In fact, the estimated diagnostic accuracy for the differential diagnosis between HM and ITP was higher for the IPF (both in % and in absolute count) compared to TPO. In regard to the fact that no correlation was found between TPO levels with either platelet counts or IPF in HM, this suggests that increased IPF is not associated with increased thrombopoietic activity in HM.
The mechanisms underlying the increase of IPF in HM remain to be elucidated. The previously reported increase in platelet volume of around 50 to 100%5, 25 is probably not sufficient to explain the 200–300% increase of IPF in these patients. In addition, formation of platelet aggregates was ruled out by the microscopic evaluation of blood smears, and by a review of the scatter plots of IPF measurements from all patients with HM. The strong correlation between IPF measurements performed one year apart suggests that the higher RNA content of these platelets could be a stable characteristic associated with the cellular mechanisms of thrombocytopenia in these patients. Cellular mechanisms responsible for thrombocytopenia in HM are complex and dependent on the molecular etiology of each disease entity. Increased counts of immature platelets were described in an animal model of HM due to RASA3 mutations in which platelet turnover is increased26. However, most of our patients presented with more classical forms of thrombocytopenia such as BSS and MYH9-related disorders in which thrombocytopenia is attributed to defects in proplatelet formation27,28,29. Interestingly, in MYH9 megakaryocytes, the physiological suppression of proplatelet extension exerted by the interaction with type I collagen is lost leading to premature or abnormal platelet release from the bone marrow29. We therefore speculate that the extremely high IPF values observed in most of our patients with HM are an expression of the premature release of platelets to the circulation. Interestingly, patients in whom the diagnosis of MYH9-related platelet disorders or BSS were confirmed by molecular or flow cytometry assays presented significantly higher IPF values than patients with a diagnosis of non-specified HMT. However, due to the limited number of patients in this subgroup analysis, these data should be considered preliminary. The confirmation of this hypothesis in future studies would provide a biological rationale to the use of IPF as a diagnostic tool in HM.
In accordance with the concept that ITP is a condition associated with suboptimal increases in TPO levels23, our analysis demonstrated significantly lower TPO levels in ITP patients compared to control subjects with normal platelet counts. Nevertheless, suboptimal TPO increase did not preclude the demonstration of a marked negative correlation between TPO levels and platelet counts in ITP (rho = −0.50), which was even stronger when only patients using steroid were evaluated (data not shown). This suggests that in ITP, increase in TPO levels is blunted by yet unknown mechanisms which are apparently improved by steroid therapy.
Our study presents limitations that need to be acknowledged, with lack of flow cytometry data being possibly the most relevant. As mentioned earlier divergent results were obtained when the IPF was measured in HM patients by flow cytometry in the study by Fabris et al.13 or using automated hematological analyzers, as in the study by Myiazaki14 and ours. Few studies have formally evaluated the correlation of IPF measurements by these two methods but in two of the most recent ones, this correlation was limited to ITP patients11, 30. Considering the challenges of standardizing platelet flow cytometry analyses compared to a CBC, we believe that it is fair to state that the former method should not be regarded as a gold-standard for IPF measurement. In fact, the vast majority of studies addressing other uses of IPF do not perform parallel flow cytometry analysis. Moreover, the concordance of our results with those from Myiazaki et al. in two independent populations supports the conclusion that IPF is increased in HM, possibly showing the premature release of platelets into the circulation. In addition, the analyzer used in our study measures MPV by impedance, which is a method that in patients with HM is known to report MPV results in less than half of the cases due to abnormalities in the platelet distribution curve4, 5. This technical limitation was also present in our study, and precluded some interesting analyses such as the comparison of the estimated diagnostic accuracy of IPF and MPV for the differential diagnosis between HM and ITP, and the evaluation of the combined performance of IPF and MPV in this context. On the other hand, it illustrates the challenges of using the MPV as a diagnostic test for HM, highlighting the importance of additional assays such as the IPF. Interestingly, when MPV values from our patients obtained from the same sample in a different hematological analyzer (Advia 2120) were used, the estimated diagnostic accuracy for the differential diagnosis of HM and ITP (AUC = 0.64; 95%CI 0.50–0.79; P = 0.02) was lower than that of the IPF.
In conclusion, our results validate previous demonstrations that IPF is increased in HM in an independent and larger population of patients. We also demonstrate the reproducibility of this method, thus supporting the feasibility of its incorporation in the diagnostic approach of HM. Given the easy accessibility to this parameter for routine clinical laboratories, and the importance of differentiating HM and ITP, further studies are warranted to the incorporation of the IPF in diagnostic algorithms of HM, and to further investigate new questions raised by our study such as the mechanisms of IPF elevation in HM.
References
Wong, E. Y. & Rose, M. G. Why does my patient have thrombocytopenia? Hematol Oncol Clin North Am 26, 231–252, vii, doi:10.1016/j.hoc.2012.02.006 (2012).
Rodeghiero, F. et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 113, 2386–2393, doi:10.1182/blood-2008-07-162503 (2009).
Drachman, J. G. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 103, 390–398, doi:10.1182/blood-2003-05-1742 (2004).
Noris, P. et al. Platelet size distinguishes between inherited macrothrombocytopenias and immune thrombocytopenia. J Thromb Haemost 7, 2131–2136, doi:10.1111/j.1538-7836.2009.03614.x (2009).
Noris, P. et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol 162, 112–119, doi:10.1111/bjh.12349 (2013).
Buttarello, M. & Plebani, M. Automated blood cell counts: state of the art. Am J Clin Pathol 130, 104–116, doi:10.1309/EK3C7CTDKNVPXVTN (2008).
Rinder, H. M., Munz, U. J., Ault, K. A., Bonan, J. L. & Smith, B. R. Reticulated platelets in the evaluation of thrombopoietic disorders. Arch Pathol Lab Med 117, 606–610 (1993).
Hoffmann, J. J. Reticulated platelets: analytical aspects and clinical utility. Clin Chem Lab Med 52, 1107–1117, doi:10.1515/cclm-2014-0165 (2014).
Hong, H., Xiao, W. & Maitta, R. W. Steady increment of immature platelet fraction is suppressed by irradiation in single-donor platelet components during storage. PLoS One 9, e85465, doi:10.1371/journal.pone.0085465 (2014).
Hong, H., Xiao, W., Stempak, L. M., Sandhaus, L. M. & Maitta, R. W. Absolute immature platelet count dynamics in diagnosing and monitoring the clinical course of thrombotic thrombocytopenic purpura. Transfusion 55, 756–765, doi:10.1111/trf.12912 (2015).
Pons, I. et al. Correlation between immature platelet fraction and reticulated platelets. Usefulness in the etiology diagnosis of thrombocytopenia. Eur J Haematol 85, 158–163, doi:10.1111/j.1600-0609.2010.01468.x (2010).
Briggs, C., Kunka, S., Hart, D., Oguni, S. & Machin, S. J. Assessment of an immature platelet fraction (IPF) in peripheral thrombocytopenia. Br J Haematol 126, 93–99, doi:10.1111/j.1365-2141.2004.04987.x (2004).
Fabris, F. et al. Indirect study of thrombopoiesis (TPO, reticulated platelets, glycocalicin) in patients with hereditary macrothrombocytopenia. Eur J Haematol 64, 151–156 (2000).
Miyazaki, K. et al. Immature platelet fraction measurement is influenced by platelet size and is a useful parameter for discrimination of macrothrombocytopenia. Hematology 20, 587–592, doi:10.1179/1607845415Y.0000000021 (2015).
Bossuyt, P. M. et al. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. Ann Intern Med 138, W1–12 (2003).
Neunert, C. et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 117, 4190–4207, doi:10.1182/blood-2010-08-302984 (2011).
Scheinberg, P. & Young, N. S. How I treat acquired aplastic anemia. Blood 120, 1185–1196, doi:10.1182/blood-2011-12-274019 (2012).
Cazzola, M. Introduction to a review series: the 2016 revision of the WHO classification of tumors of hematopoietic and lymphoid tissues. Blood 127, 2361–2364, doi:10.1182/blood-2016-03-657379 (2016).
Bolton-Maggs, P. H. et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol 135, 603–633, doi:10.1111/j.1365-2141.2006.06343.x (2006).
Nurden, A. T. & Nurden, P. Inherited thrombocytopenias. Haematologica 92, 1158–1164 (2007).
Nurden, A. T. & Nurden, P. Congenital platelet disorders and understanding of platelet function. Br J Haematol 165, 165–178, doi:10.1111/bjh.12662 (2014).
Enz Hubert, R. M. et al. Association of the immature platelet fraction with sepsis diagnosis and severity. Sci Rep 5, 8019, doi:10.1038/srep08019 (2015).
Nichol, J. L. Endogenous TPO (eTPO) levels in health and disease: possible clues for therapeutic intervention. Stem Cells 16(Suppl 2), 165–175, doi:10.1002/stem.5530160719 (1998).
Kienast, J. & Schmitz, G. Flow cytometric analysis of thiazole orange uptake by platelets: a diagnostic aid in the evaluation of thrombocytopenic disorders. Blood 75, 116–121 (1990).
Noris, P. et al. Platelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disorders. Blood 124, e4–e10, doi:10.1182/blood-2014-03-564328 (2014).
Stefanini, L. et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest 125, 1419–1432, doi:10.1172/JCI77993 (2015).
Strassel, C. et al. Intrinsic impaired proplatelet formation and microtubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica 94, 800–810, doi:10.3324/haematol.2008.001032 (2009).
Pecci, A. et al. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost 102, 90–96, doi:10.1160/TH09-01-0068 (2009).
Balduini, A. et al. Proplatelet formation in heterozygous Bernard-Soulier syndrome type Bolzano. J Thromb Haemost 7, 478–484, doi:10.1111/j.1538-7836.2008.03255.x (2009).
Ibrahim, H. et al. Detection and quantification of circulating immature platelets: agreement between flow cytometric and automated detection. J Thromb Thrombolysis 42, 77–83, doi:10.1007/s11239-016-1338-3 (2016).
Acknowledgements
This study was financially supported by Fapesp, grant 2013/09319-0, CNPq, Brazil and Faepex/UNICAMP. The Hematology and Hemotherapy Center - Hemocentro UNICAMP, forms part of the National Institute of Science and Technology of Blood, Brazil (INCT do Sangue CNPq/MCT/FAPESP).
Author information
Authors and Affiliations
Contributions
F.L.B. Ferreira collected collected and analyzed data and drafted the manuscript; M.P. Colella, S.S. Medina, C. Costa-Lima, F.A. Orsi and J.M. Annichino-Bizzacchi recruited patients; M.M.L. Fiusa and L.N.G. Costa performed TPO analysis; K.Y. Fertrin and M.F.P. Gilberti supervised and performed CBC and IPF analysis; M.C. Ozelo contributed to study design and provided access to HM patients; E.V. De Paula designed the study, analyzed the data and drafted the manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferreira, F.L.B., Colella, M.P., Medina, S.S. et al. Evaluation of the immature platelet fraction contribute to the differential diagnosis of hereditary, immune and other acquired thrombocytopenias. Sci Rep 7, 3355 (2017). https://doi.org/10.1038/s41598-017-03668-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03668-y
This article is cited by
-
Early prediction of platelet recovery with immature platelet fraction in patients receiving hematopoietic stem cell transplantation
Annals of Hematology (2024)
-
Reference guide for the diagnosis of adult primary immune thrombocytopenia, 2023 edition
International Journal of Hematology (2024)
-
Macrothrombocytopenia: Role of Automated Platelet Data in Diagnosis
Indian Journal of Hematology and Blood Transfusion (2023)
-
A familial case of MYH9 gene mutation associated with multiple functional and structural platelet abnormalities
Scientific Reports (2022)
-
Platelet features allow to differentiate immune thrombocytopenia from inherited thrombocytopenia
Annals of Hematology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
