
Abstract
Deafblindness is mostly due to Usher syndrome caused by recessive mutations in the known genes. Mutation-negative patients therefore either have distinct diseases, mutations in yet unknown Usher genes or in extra-exonic parts of the known genes – to date a largely unexplored possibility. In a consanguineous Saudi family segregating Usher syndrome type 1 (USH1), NGS of genes for Usher syndrome, deafness and retinal dystrophy and subsequent whole-exome sequencing each failed to identify a mutation. Genome-wide linkage analysis revealed two small candidate regions on chromosome 3, one containing the USH3A gene CLRN1, which has never been associated with Usher syndrome in Saudi Arabia. Whole-genome sequencing (WGS) identified a homozygous deep intronic mutation, c.254–649T > G, predicted to generate a novel donor splice site. CLRN1 minigene-based analysis confirmed the splicing of an aberrant exon due to usage of this novel motif, resulting in a frameshift and a premature termination codon. We identified this mutation in an additional two of seven unrelated mutation-negative Saudi USH1 patients. Locus-specific markers indicated that c.254–649T > G CLRN1 represents a founder allele that may significantly contribute to deafblindness in this population. Our finding underlines the potential of WGS to uncover atypically localized, hidden mutations in patients who lack exonic mutations in the known disease genes.
Similar content being viewed by others

Introduction
Usher syndrome is the most common cause of inherited deafblindness1. Type 1 (USH1) is characterized by congenital deafness and early (first decade) retinitis pigmentosa (RP), whereas type 2 (USH2) displays progressive hearing impairment and RP of later onset. USH3 is characterized by progressive hearing loss, RP, and variable peripheral vestibular dysfunction2. However, disease resulting from mutations in the USH3A gene, CLRN1, is variable, ranging from non-syndromic RP3 to USH14. The advent of next-generation sequencing (NGS) has enabled panel-sequencing of the 11 known Usher genes, and its application in a recent study on European deafblind patients identified the causative mutations in the majority5. In a Saudi Arabian family with four siblings affected by Usher syndrome type 1, escalating the genetic investigations from gene panel NGS over genome-wide linkage analysis to whole-exome sequencing (WES) and finally whole-genome sequencing (WGS) led up to the molecular diagnosis. Our study demonstrates the potential of WGS to unlock hidden mutations.
Results
NGS of gene panels for retinal dystrophy and for deafness
Apart from a heterozygous frameshift mutation in TUBGCP6, c.5001_5003delinsCA (p.Gln1667Hisfs*11), NGS of the known genes for Usher syndrome, for other syndromic and isolated hearing loss, and for retinal degeneration did not identify any mutations. Biallelic TUBGCP6 mutations cause microcephalic primordial dwarfism and additional congenital anomalies, including retinopathy6. Given the recessive inheritance and additional symptoms associated with mutations in TUBGCP6 (which are not present in the affected family members analyzed in our study), the apparently monoallelic variant most likely represents carriership for an unrelated disorder. Our results from NGS panel analysis thus largely excluded not only mutations in the coding sequences of the Usher syndrome genes and genes causing similar syndromes (e.g. USH3-like PHARC due to ABHD12 mutations7), but also simultaneous mutations in a deafness gene and an RP gene mimicking Usher syndrome. Quantitative analysis of NGS reads did not indicate large copy number variations (CNVs) such as deletions of one or several contiguous exons.
Whole-exome sequencing (WES)
WES data were filtered for rare homozygous variants (see Methods), which revealed 38 such variants in 37 genes. As could be expected after mutation-negative panel-NGS, none of these variants affected a gene implicated in Usher syndrome, RP, or recessive deafness. The family structure with distant parental consanguinity and four affected siblings was highly suitable for an efficient linkage analysis. Hence, to identify the causative mutation, we set out to apply this approach (Fig. 1A).

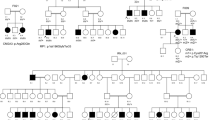
(A) Pedigree of the Saudi family with Usher syndrome type 1. Parents are distantly related. The sample of patient II:1 were subjected to NGS of an Usher gene panel and subsequently to WES – without finding a mutation. (B) Samples of the parents and the four affected children were subjected to genome-wide linkage analysis. The graphical view of LOD score calculation illustrates a combined maximum parametric LOD score of 3.01 for two neighboring regions on chromosome 3. (C) The candidate interval on chromosome 3q25.1 comprises the CLRN1 gene (the position of the polymorphic microsatellite marker D3S1315 is indicated; the other microsatellite markers that have been used for haplotyping flank this HBD region at the centromeric or telomeric side), whereas (D) the 3q25.2 region does not contain a known Usher gene. CLRN1-AS1, CLRN1 antisense RNA 1; MED12L, Mediator of RNA polymerase II transcription subunit 12-like protein; ARHGEF26, Rho guanine nucleotide exchange factor 26; ARHGEF26-AS1, ARHGEF26 antisense RNA 1; DHX36, DEAH-box helicase 36; GPR149, G protein-coupled receptor 149.
Genome-wide linkage analysis
Compatible with the distant consanguinity of the parents, we identified only two neighboring regions with homozygosity by descent (HBD) of very small size and a combined maximum parametric LOD score of 3.01 (Fig. 1B) on chromosome 3q25.1 (150,609,866–150,911,683; 301.817 kb; Fig. 1C) and 3q25.2 (153,396,096–154,676,122; 1.28 Mb; Fig. 1D). These regions contained two and three annotated genes, respectively. Of note, the USH3A gene, CLRN1, was contained in the 3q25.1 region. None of the 38 homozygous variants identified by WES located in the two neighboring candidate regions on chromosome 3.
Whole-genome sequencing (WGS)
Given the results of panel-NGS, WES and genome-wide linkage analysis, we hypothesized that the causative mutation was likely to reside in a non-coding region, and possibly within the CLRN1 gene. Of note, and in line with the results from genome-wide linkage analysis, filtering of WGS data for rare homozygous variants (see Methods) identified only one such variant, located within one of the two mapped adjacent HBD regions on chromosome 3: g.150660197A > C (c.254–649T > G) in CLRN1. Because the c.254–649T > G CLRN1 affects an intron (between “exon 0b” – an exon contained in transcript NM_001256819.1 – and exon 1) of a proven Usher syndrome gene, and because in silico analysis predicted aberrant splicing (see below), we focused on this alteration. Moreover, it has not been annotated in the 1000 Genomes Project. Compatible with its deep intronic location, the variant is absent from exonic sequence databases. It has not been reported previously and is therefore also absent from the HGMD.
Minigene splice assay
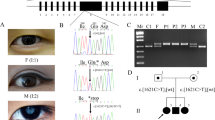
In silico analysis of the c.254–649T > G mutation using Spliceview and Maximum Entropy predicts that the mutation generates a novel donor splice site (score of 85 [Spliceview] and 8.76 [Maximum Entropy model]) compared to no predicted donor site in the wild-type sequence). Several potential acceptor sites are predicted in the wild-type sequence 5′ of the alteration. Because we could not detect CLRN1 in RT-PCR analysis from whole blood of the patients, we established a minigene-based assay suitable for analysis in commonly used human cell lines. Due to the genomic dimension of CLRN1 (>46 kb, Figs 2 and 3A), minigene-based analysis of CLRN1 mRNA splicing could not be examined with a construct comprising all five exons (exons 0, 0b, 1, 1b, 2) and introns. Hence, we designed a CLRN1 minigene of convenient size encompassing approx. 3.6 kb and harboring three annotated CLRN1 exons and the interjacent native introns (Fig. 3B). According to the established nomenclature of the major CLRN1 transcripts8, the respective exons were termed exon 0b, exon 1 and exon 1b (Fig. 3A,B). The minigenes containing the healthy (herein referred to as wild-type, WT) and the mutant variant were transiently transfected to HEK293 cells. In subsequent RT-PCR analysis for the WT CLRN1 minigene, we exclusively detected the correctly spliced transcript, validating the suitability of the minigene assay in this cell line. Splicing of the mutant construct with the c.254–649T > G mutation resulted in an additional band besides the correctly spliced band, indicating aberrant splicing (Fig. 3C,D). Subsequent semi-quantitative analysis of the band intensities for the c.254–649T > G mutation revealed that, compared to the correctly spliced variant, this aberrant splice product is predominant (87% versus 13%, Fig. 4). Sequencing of the band corresponding to the novel splice variant showed that the c.254–649T > G mutation generates an aberrant exon in intron 0b. This aberrant exon comprises 230 bp (Fig. 3B,E). If included into the major CLRN1 isoform (isoform a, NM_174878.2), the aberrant exon leads to a frameshift and a premature stop codon, predicting either a truncated protein (106 residues compared to 232 residues of the NM_174878.2-deduced wild-type protein, with the inclusion of 22 unrelated residues) or an unstable transcript subjected to nonsense-mediated decay (NMD). However, irrespective of the CLRN1 splice isoform, the insertion of the aberrant exon is expected to result in profound alteration or complete deficiency of CLRN1 protein. According to the ACMG guidelines, the c.254–649T > G variant can be claimed as pathogenic (Table 3 and Table 5 in Richardson et al.9), with the following classification criteria for pathogenic variants applying here: (i) 1 Very strong (PVS1 null variant: canonical splice site (being generated by the mutation) in a gene where loss-of-function is a known mechanism of disease) AND (a) ≥1 Strong (PS3: functional studies supportive of a damaging effect, PS4: observation of the variant in multiple unrelated patients with the same phenotype). In addition, the variant is absent from controls (Moderate, PM2).

Scheme of UCSC-annotated Refseq CLRN1 isoforms (hg38). (A) For our construct, we refer to a combination of isoforms d/3 (NM_001195794.1) and e (NM_001256819.1) that also includes the exons of the “major isoform”, isoform a (NM_174878.2)8: exons 0, 1 and 2. We designated additional exons, which are located between exons 0 and exon 1 and between exons 1 and exon 2, respectively, as exon 0b and exon 1b. (B) Protein sequences of the three above isoforms. In NM_001256819.1, the inclusion of exon 0b shifts the reading frame of exon 1, resulting in a different peptide sequence for exon 1 in this isoform. Note that, in contrast to the scheme in Fig. 3, the isoforms are in antisense orientation.

CLRN1 minigene splice assay. (A) Schematic to scale overview of the genomic CLRN1 structure including the 5′ and 3′ untranslated regions (5′ UTR and 3′ UTR, respectively). bp, basepairs. Exons (ex) are shown as colored boxes. (B) CLRN1 to scale minigene used for the splice experiments shown in (C,D). The position of the c.254–649 T > G mutation is indicated by an arrowhead and the novel exon generated by the mutation (referred to as “aberrant exon”) is displayed as a dashed purple box. The dashed lines represent a schematic magnification of the 3′ and 5′ splice sites flanking the aberrant exon. The consensus sequences for the putative branch point (BP), the polypyrimidine tract (PPT), the 3′ acceptor splice site (ASS), and the 5′ donor splice site (DSS) of the aberrant exon are underlined. The putative lariat-forming adenosine in the PPT is shown in upper case. The binding positions of the primers used for the RT-PCR shown in (C) are indicated by arrows. (C) Representative RT-PCR analysis from HEK293 cells transiently transfected with the single constructs as indicated. As control, RT-PCR from non-transfected HEK293 cells was performed. The length and exon composition of the two splice products for WT (432 bp) and the c.254–649T > G mutant (432 bp and 662 bp) are shown on the right panel. The position of the primer used for Sanger sequencing shown in (D) is indicated by an arrow. (D) Representative electropherograms of the exon-exon boundaries for both RT-PCR products of the c.254–649T > G mutant.

Quantification of the RT-PCR results shown in Fig. 3C. (A) Quantification of eight RT-PCR experiments resulting from four independent transfection experiments for both WT and c.254–649T > G mutant for the 432 bp and 662 bp bands. The single values are given as percentages of the total band intensities ± standard error of the mean (SEM). (B) Quantification of the single WT and the c.254–649T > G cDNAs used for the calculation of band intensities displayed in (A). Shown are the mean CT values (n = 3) ± SEM for aminolevulinic acid synthase (ALAS) as housekeeper gene. The single values are summarized in Table 1. For statistical analysis, one-way ANOVA followed by Tukeys test was used. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Targeted mutation analysis in NGS-panel-negative Saudi Arabian USH patients and haplotype analysis
We identified the c.254–649T > G CLRN1 mutation in homozygous state in two (here referred to as USH-KSA1 and USH-KSA2) out of seven additional patients with Usher syndrome type 1 from Saudi Arabia in whom targeted NGS covering all coding exons of the known Usher syndrome genes had not identified any mutation. Genotyping of locus-specific microsatellite markers (D3S1299, D3S1315, D3S3625, D3S1279 and D3S4531, spanning about 1 Mb) in all members of the index family and both additional patients, USH-KSA1 and USH-KSA2, revealed a disease-associated haplotype which was preserved on the paternal allele in the index family and in patients USH-KSA1 and USH-KSA2 over the whole range covered by the above markers, indicating that these patients have a common ancestor who carried the mutation (Fig. 5).

Genotyping of polymorphic microsatellite markers from the CLRN1 (USH3A) locus on chromosome 3q25.1 in (A) the index family and (B) the two additional Saudi patients (USH-KSA1 and USH-KSA2) who also carry the c.254–649T > G CLRN1 mutation in homozygous state. Grey numbers indicate the marker positions in Mb on chromosome 3 (according to hg38). Numbers indicate the PCR product size obtained with the primer pairs given in the UCSC Genome Browser. Only D3S1315 localizes within one of the two HBD regions that has been mapped in the index family. D3S1299 is located centromeric of that HBD region while D3S3625, D3S1279 and D3S4531 are telomeric of that region. The putative original c.254–649T > G-associated haplotype (red) is preserved on the paternal allele in the index family (paternal and maternal haplotypes are indicated by boxes of different colors), on one allele of patient USH-KSA2, and on both alleles of patient USH-KSA1.
Discussion
About 85% of the mutations underlying Mendelian traits localize in the protein-coding exons of these genes, or they affect the splice sites adjacent to the coding sequences10. For phenotypes like Usher syndrome, with mutations in the known genes explaining the vast majority of cases, targeted NGS of panels comprising these genes are highly effective in confirming the clinical diagnosis. The determination of the causative mutations is important for personalized management of patients because it enables clinical prognoses (differentiation of clinical subtypes), in particular in hearing-impaired children before onset of retinal degeneration. Although combined impairment of hearing and vision, commonly termed deafblindness, is due to Usher syndrome in most cases, other conditions should be excluded, amongst other reasons because they may be treatable (e.g. Refsum syndrome that may respond to phytanic acid-reduced diet11).
The phenotype in the family reported herein is clearly inherited (parental consanguinity, four affected siblings) and compatible with Usher syndrome. Lack of mutations in the exons of the genes known to cause Usher syndrome (and clinically overlapping conditions) may indicate rare atypically localized mutations, outside the protein-coding exons. Such mutations have been reported for different retinopathies including Usher syndrome: They may affect non-coding exons and possibly affect gene transcription, as we and others have shown for EYS-related RP12. Deep intronic mutations, as in case of OFD1-associated RP13, the prevalent c.2991 + 1655A > G CEP290 mutation in LCA14, or certain USH2A mutations in USH215, 16) have been shown to generate aberrant exons through missplicing. Moreover, mutations may reside outside genes. For example, structural variations and point mutations may disturb normal chromatin folding with consecutive gene misexpression, a disease mechanism known from developmental disorders and cancer17. Finally, the recent identification of mutations in CEP78 in patients with an Usher-like phenotype18 illustrates that novel disease genes have to be taken into account even in mutation-negative Usher syndrome patients.
It has been estimated that about one third of disease-causing mutations may cause aberrant splicing. While splice mutations affecting splice site consensus sequences (some 10% of disease-causing mutations19) are easy to recognize, those in less conserved sequence motifs are more difficult, but, if exonic, are at least captured by Sanger, NGS-panel, or whole-exome sequencing. Because deep intronic splice site mutations escape detection by standard sequencing approaches, they represent the most challenging mutation of this category. Our finding highlights the diagnostic potential of whole-genome sequencing (WGS) in finding mutations in the 99% of the genome that are not protein-coding. WGS has been shown to be superior to WES in identifying disease-causing mutations, amongst other things because of more uniform coverage and its ability to detect structural genomic mutations20, 21. However, the need for extensive data storage and the high costs of WGS have so far impeded its routine diagnostic application. Moreover, a minigene assay was necessary in our study to prove pathogenicity of c.254–649T > G CLRN1 . This would be impossible in a routine diagnostic setting and demonstrates that final interpretation of deep intronic variants suspected to cause aberrant splicing will remain challenging. Furthermore, in our minigene assay the investigated exons are not in their native genomic and cellular environment. We assume that c.254–649T > G-associated missplicing is very likely to occur in retinal and cochlear cells and in the way we describe here, but this cannot be finally proven by our data.
With only 30 supposedly pathogenic variants annotated in the Human Gene Mutation Database22, the CLRN1-associated subtype of Usher syndrome, USH3A, is very rare. However, due to founder mutations, it represents the predominant subtype in Finland23 and in some Jewish populations2. To our knowledge, our findings represent the first description of USH3A in the Saudi Arabian population. Its hidden localization has prevented its identification so far. The presence of c.254–649T > G CLRN1 in three Saudi USH1 patients and a mutation-associated haplotype (spanning at least 1 Mb) indicate a founder mutation that may significantly contribute to Usher syndrome in this population. We therefore recommend to consider this mutation in genetic analysis of patients with all clinical subtypes, explicitly including USH1 (all patients in our study were diagnosed as USH1). Because therapeutic strategies for USH3A are being developed24, pinpointing the molecular diagnosis may become crucial for USH3A patients’ medical care in the future. As a future treatment strategy to eliminate the abnormal splicing due to c.254–649T > G CLRN1 , CRISPR/Cas9-based genome editing may become a promising approach for patients with this mutation. Because modifications of deep intronic regions do not affect the coding sequence of the respective gene, this technology seems predestined for treatment of disease-associated mutations in these regions.
Methods
All methods were carried out in accordance with the approved guidelines.
Patients
The study was approved by the institutional review boards of the Ethics Committee of the University Hospital of Cologne and the King Khaled Eye Specialist Hospital, Riyadh. Informed consent for genetic investigations was obtained from the parents. Clinical and specimen investigations were conducted according to the Declaration of Helsinki.
NGS of gene panels for inherited retinal dystrophies and deafness
The coding exons of 11 Usher syndrome genes (MYO7A/USH1B, USH1C, CDH23/USH1D, PCDH15/USH1F, USH1G, USH2A, DFNB31/USH2D, GPR98/USH2C, CLRN1/USH3A, PDZD7/digenic/USH2A-modifier, CIB2; 398 exons) and 17 genes whose mutations underlie conditions clinically similar to Usher syndrome (CEP250, HARS, ABHD12, PEX1, PEX2, PEX3, PEX5, PEX6, PEX7, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26, PHYH) were enriched using Roche/NimbleGen sequence capture technology, sequenced on an Illumina HiSeq 1500 system and bioinformatically evaluated as described previously12. Another gene whose biallelic mutations have very recently been reported to cause Usher syndrome, CEP78 18, was not yet included in our panel. However, because patients with CEP78 mutations appear to have cone-rod dystrophy rather than RP25, 26, we would not consider CEP78 a bona fide Usher gene. Quantitative readout of NGS reads to exclude CNVs was carried out as described previously12. Besides the explicitely mentioned genes above, the used NGS panels contain virtually all genes known to be involved in non-syndromic and syndromic forms of hearing loss (n = 119; Suppl. Table 1) and retinal degeneration (n = 155; Suppl. Table 2), respectively, at the time of panel design (2015). These genes were enriched and sequenced in parallel (with very little redundancy: a few genes, like ABHD12, CLRN1 and USH1C are present on both panels because their mutations may cause either syndromic hearing loss or non-syndromic RP). The bioinformatic pipeline was consulted for putatively pathogenic variants not only in Usher syndrome genes, but also in these genes.
Genome-wide linkage analysis
DNA was extracted from peripheral blood samples using standard methods. DNA samples of the parents and the four affected siblings (family as displayed in Fig. 1A) were analyzed for genome wide linkage using the Infinium CoreExome-24 v1.1 BeadChip (Illumina Inc., San Diego, CA) according to the manufacturer’s protocol. Subsequent data handling was performed using the graphical user interface ALOHOMORA27. Relationship errors were identified by using the program Graphical Relationship Representation28. The program PedCheck was applied to find Mendelian errors29 and data for SNPs with such errors were removed from the data set. Non-Mendelian errors were identified by using the program MERLIN30 and unlikely genotypes for related samples were deleted. Linkage analysis was performed assuming autosomal-recessive inheritance, full penetrance, consanguinity, and a disease gene frequency of 0.0001. Multipoint LOD scores were calculated using the program Allegro31. Haplotypes were reconstructed with Allegro and presented graphically with HaploPainter32. Regions of homozygosity by descent (HBD) were annotated with their positions corresponding to NCBI Build 37.
Whole-exome sequencing
Genomic DNA of patient II:1 (Fig. 1A) was subjected to whole-exome sequencing, WES. Exome capture was performed using the Agilent SureSelectXT Human All Exon 50 Mb kit following manufacturer’s procedures (Agilent, Santa Clara, CA, USA) and sequenced with Illumina paired end sequencing (protocol v1.2). Briefly, DNA was sheared by fragmentation (Covaris, Woburn, MA, USA) and purified using Agencourt AMPure XP beads (Beckman Coulter, Fullerton, CA, USA). Resulting fragments were analysed using an Agilent 2100 Bioanalyzer. Fragment ends were repaired and adaptors were ligated to the fragments. The library was purified using Agencourt AMPure XP beads and amplified by PCR before hybridisation with biotinylated RNA baits. Bound genomic DNA was purified with streptavidin coated magnetic Dynabeads (Invitrogen, Carlsbad, CA, USA) and re-amplified to include barcoding tags before pooling for sequencing on an paired-end, 100 cycle run on an Illumina HiSeq 2000 according to manufacturer’s protocols. Briefly, primary data were filtered according to signal purity by the Illumina Realtime Analysis (RTA) software v1.8. Subsequently, reads were mapped to the human genome reference build hg19 using the bwa-aln33 alignment algorithm. GATK v1.634 was used to mark duplicated reads, for local realignment around short insertions and deletions, to recalibrate the base quality scores and to call SNPs (incorporating variant quality score recalibration) and short indels35. Scripts developed in-house at the Cologne Center for Genomics were used to detect protein changes, affected donor and acceptor splice sites, and overlaps with known variants. Analysis for acceptor and donor splice site mutations and for the activation of new aberrant splice sites was carried out with a Maximum Entropy model36 and filtered for effect changes. In particular, and because the patients came from a consanguineous background, we filtered for high-quality (coverage > 15; quality > 25) rare (MAF < 0.005) homozygous variants (dbSNP build 135, the database of the 1000 Genomes Project build 20110521, TGP37), and the Exome Variant Server, NHLBI Exome Sequencing Project, Seattle, build ESP650038). We also filtered against an in-house database containing all variants from 511 exomes from epilepsy patients to exclude pipeline-related artifacts/false positives (MAF < 0.004). In addition to the above large-scale sequencing databases consulted, a local pipeline35 and interface was used (Varbank v.2.3; https://varbank.ccg.uni-koeln.de) as described previously39, 40, and we searched the Exome Aggregation Consortium (ExAC) database41 (as of 05/2016), which aggregates numerous databases including the current versions of the ESP and the TGP, for homozygous candidate variants from the mapped regions.
Whole-genome sequencing
The library was prepared and size selected by using the Illumina® TruSeq® DNA Sample Preparation Kit and Agencourt AMPure XP beads, starting with 1,2 µg genomic DNA and followed by one cycle of PCR to complete adapter structure. The library was validated with the Agilent 2200 TapeStation and quantified by qPCR. Using an Illumina HiSeq X Ten Sequencer, we generated 423M 150-bp paired-end reads corresponding to 126,75 Gb of sequence data and an average coverage of 39-fold.
Bioinformatic analysis of WGS data
845,688,028 150 bp paired-end reads were generated from sequencing. They were mapped to the hg19 reference genome using BWA-ALN33 (version 0.6.2). After mapping, duplicates were marked using Picard (version 1.64; http://picard.sourceforge.net) and basecalling quality score recalibration and local indel realignment was performed using GATK34 (version 1.6.11). Enrichment statistics computed by Picard on the resulting BAM file showed a sufficient and rather uniform coverage of the 1.6 Mb target region (mean coverage 39×, 87.6% of target covered by at least 30×, 98.9% of target covered by at least 20×, 99.8% of target covered by at least 10×). Variants were called genome wide using samtools mpileup42 (version 0.1.18) and in the complete target region using GATK UnifiedGenotyper (version 1.6.11). The resulting variants were annotated with software developed at the CCG based on the ENSEMBL b68 gene models and filtered to exclude variants of low confidence (alternative allele frequency <10%, number of reads at variant position <5, variant quality score <10, number of reads supporting the variant <3). The remaining variants were annotated with their presence in public databases (dbSNP43, 1000 Genomes Project44, Exome Variant Server (EVS http://evs.gs.washington.edu/EVS/), dbVAR and DGVa45, GERP46, ENSEMBL47, and the commercial HGMD professional database48) as well as a CCG inhouse exome collection of 511 samples. Effects on splicing were predicted using the maximum entropy approach from Yeo and Burge36 and SIFT49, POLYPHEN50, and RVIS51 scores for all coding variants were taken into account. The GATK UnifiedGenotyper variant list was used to compute regions of homozygosity with Allegro31. The annotated variant lists were uploaded to the CCG’s varbank (https://varbank.ccg.uni-koeln.de) database for further evaluation.
Sanger sequencing
Validation of the CLRN1 candidate variant c.254–649T > G, segregation analysis and screening of so far NGS-panel-negative Saudi Arabian Usher syndrome patients for this mutation were carried out by Sanger sequencing. For this, we PCR-amplified a 571 bp fragment comprising the position of the mutation, using the forward primer CLRN1-mF: 5′-ggttataagctctgtgagacaac-3′ and the reverse primer CLRN1-mR: 5′-ccaagcctttaatgacctttctcg-3′. PCR amplification was carried out on a Biometra T3000 PCR cycler (Analytik Jena, Jena, Germany) as follows: 1× (95 °C, 15 min), 15× (95 °C, 1 min./68 °C (reduced by 0.5 °C in every subsequent cycle), 1 min./72 °C, 1 min.), 30× (95 °C, 1 min./60 °C, 1 min./72 °C, 1 min.), 1× 72 °C, 10 min.
Minigene splice assay
Attempts to investigate CLRN1 splicing via cDNA amplification and sequencing based on RNA isolated from whole blood of the patients were not successful. We thus chose a splicing minigene splice assay based on the CLRN1 genomic sequence (Figs 2 and 3A). Several CLRN1 isoforms have been annotated, and apart from the three protein-coding exons 0, 1 and 2 of isoform a (NM_174878.2), the major isoform8, there was no consistent numbering of exons available. Our investigation of splicing was based on two transcript isoforms which include additional exons between exon 0 and exon 1 (in isoform e, NM_001256819.1) and between exons 1 and 2 (isoform d, NM_001195794.1) (Fig. 3B). For maintaining compatibility with exon numbering of isoform a8, we designated these additional exons as exon 0b and 1b, respectively (Fig. 3A,B). The WT and mutant minigenes (3,552 bp each) were synthesized by BioCat (Heidelberg, Germany) and delivered in the pcDNA3.1 eGFP standard vector. For RT-PCR analysis, HEK293 cells were transiently transfected using the CaPO3 method. 24 h post transfection, cells were harvested and the RNA was isolated using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Subsequent cDNA synthesis was conducted with equal amounts of RNA (1 µg each) using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). For subsequent PCR, following primer were used: C-ex0b_F 5′- ctatcttgttgttgatgcaggc-3′ and C-ex2_R 5′-gtgtcaagagcaagaaagtacc-3′. The single PCR products representing the WT and the c.254–649T > G mutant splice isoforms were extracted, purified, and sequenced. Sequencing was conducted by Eurofins Genomics (Ebersberg, Germany) using the following primer: C-ex0b_seq_F 5′-ccttcatgggactcccaacag-3′. Semi-quantitative analysis of the band intensities from electrophoresis on an agarose gel was performed on eight technical replicates resulting from four different transfections. For this purpose, the single sets of two RT-PCR experiments for the WT and the c.254–649T > G mutant were conducted with a variable number of cycles ranging between 25–30. PCR was performed with the Herculase II Fusion DNA Polymerase (Agilent Genomics) using the following conditions: 1× (95 °C, 2 min), 25–30× (95 °C, 20 sec; 60 °C 20 sec; 72 °C, 2 min), 1× (72 °C, 5 min). The absolute intensities of the single PCR bands were calculated by the Image Lab software (BioRad, Hercules, CA, U.S.A.). The single cDNAs resulting from the four independent transfections for WT and for the c.254–649T > G mutant were quantified by a StepOnePlus Real-Time PCR System (Applied Biosystems) using SYBR Select Master Mix (Applied Biosystems). The following primers specific for human aminolevulinic acid synthase (ALAS) as a housekeeper gene were used: ALAS fwd: 5′-GATGTCAGCCACCTCAGAGAAC-3′ and ALAS rev: 5′-CATCCACGAAGGTGATTGCTCC-3′. For quantification, three technical replicates for each cDNA were conducted and the statistical comparison between the groups was done with one-way ANOVA, followed by the Tukey’s test for multiple comparisons. p < 0.05 was considered statistically significant.
Characterization of the haplotype associated with the c.254–649T > G mutation in CLRN1
Genotyping of locus-specific microsatellite markers (D3S1299, D3S1315, D3S3625, D3S1279 and D3S4531) was carried out in all members of the index family and in patients USH-KSA1 and USH-KSA2, using primers as given in the respective entries of the UCSC Genome Browser. For marker amplification, we applied the tailed primer method as described previously52. The forward primer of each marker was extended with a “tail” sequence 5′-TACGCATCCCAGTTTGAGACG-3′, and a FAM-labeled oligonucleotide complementary to this tail was added to the PCR reaction. The lengths of the PCR products (generated with Qiagen Hotstar Taq Polymerase) were determined by electrophoresis on an ABI-377 DNA sequencer. Genotypes were determined by GeneMapper (Applied Biosystems). PCR amplification of all markers was carried out as follows: 1× (95 °C, 15 min), 10× (95 °C, 30 sec./60 °C (reduced by 0.5 °C in every subsequent cycle), 40 sec./72 °C, 45 sec.), 25× (95 °C, 30 sec./57 °C, 40 sec./72 °C, 45 sec.), 1× 72 °C, 20 min.
References
Mathur, P. & Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta 1852, 406–420, doi:10.1016/j.bbadis.2014.11.020 (2015).
Ness, S. L. et al. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J Med Genet 40, 767–772, doi:10.1136/jmg.40.10.767 (2003).
Khan, M. I. et al. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology 118, 1444–1448, doi:10.1016/j.ophtha.2010.10.047 (2011).
Ebermann, I. et al. Deafblindness in French Canadians from Quebec: a predominant founder mutation in the USH1C gene provides the first genetic link with the Acadian population. Genome Biol 8, R47, doi:10.1186/gb-2007-8-4-r47 (2007).
Bonnet, C. et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur J Hum Genet 24, 1730–1738, doi:10.1038/ejhg.2016.99 (2016).
Martin, C. A. et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet 46, 1283–1292, doi:10.1038/ng.3122 (2014).
Eisenberger, T. et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J Rare Dis 7, 59, doi:10.1186/1750-1172-7-59 (2012).
Adato, A. et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet 10, 339–350, doi:10.1038/sj.ejhg.5200831 (2002).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, doi:10.1038/gim.2015.30 (2015).
Majewski, J., Schwartzentruber, J., Lalonde, E., Montpetit, A. & Jabado, N. What can exome sequencing do for you? J Med Genet 48, 580–589, doi:10.1136/jmedgenet-2011-100223 (2011).
Kohlschütter, A. et al. A child with night blindness: preventing serious symptoms of Refsum disease. J Child Neurol 27, 654–656, doi:10.1177/0883073811424799 (2012).
Eisenberger, T. et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One 8, e78496, doi:10.1371/journal.pone.0078496 (2013).
Webb, T. R. et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet 21, 3647–3654, doi:10.1093/hmg/dds194 (2012).
den Hollander, A. I. et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 79, 556–561, doi:10.1086/507318 (2006).
Liquori, A. et al. Whole USH2A Gene Sequencing Identifies Several New Deep Intronic Mutations. Hum Mutat 37, 184–193, doi:10.1002/humu.22926 (2016).
Vache, C. et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum Mutat 33, 104–108, doi:10.1002/humu.21634 (2012).
Spielmann, M. & Mundlos, S. Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet 25, R157–R165, doi:10.1093/hmg/ddw205 (2016).
Fu, Q. et al. CEP78 is mutated in a distinct type of Usher syndrome. J Med Genet 54, 190–195, doi:10.1136/jmedgenet-2016-104166 (2016).
Krawczak, M. et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat 28, 150–158, doi:10.1002/humu.20400 (2007).
Belkadi, A. et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA 112, 5473–5478, doi:10.1073/pnas.1418631112 (2015).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347, doi:10.1038/nature13394 (2014).
Stenson, P. D. et al. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinformatics Chapter 1, Unit1 13, doi:10.1002/0471250953.bi0113s39 (Chapter 1, 2012).
Joensuu, T. et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 69, 673–684, doi:10.1086/323610 (2001).
Alagramam, K. N. et al. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat Chem Biol 12, 444–451, doi:10.1038/nchembio.2069 (2016).
Namburi, P. et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am J Hum Genet 99, 777–784, doi:10.1016/j.ajhg.2016.07.010 (2016).
Nikopoulos, K. et al. Mutations in CEP78 Cause Cone-Rod Dystrophy and Hearing Loss Associated with Primary-Cilia Defects. Am J Hum Genet 99, 770–776, doi:10.1016/j.ajhg.2016.07.009 (2016).
Ruschendorf, F. & Nurnberg, P. ALOHOMORA: a tool for linkage analysis using 10 K SNP array data. Bioinformatics 21, 2123–2125, doi:10.1093/bioinformatics/bti264 (2005).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. C. & Cardon, L. R. GRR: graphical representation of relationship errors. Bioinformatics 17, 742–743, doi:10.1093/bioinformatics/17.8.742 (2001).
O’Connell, J. R. & Weeks, D. E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63, 259–266, doi:10.1086/301904 (1998).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30, 97–101, doi:10.1038/ng786 (2002).
Gudbjartsson, D. F., Jonasson, K., Frigge, M. L. & Kong, A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25, 12–13, doi:10.1038/75514 (2000).
Thiele, H. & Nurnberg, P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 21, 1730–1732, doi:10.1093/bioinformatics/bth488 (2005).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi:10.1093/bioinformatics/btp324 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, doi:10.1101/gr.107524.110 (2010).
Kawalia, A. et al. Leveraging the power of high performance computing for next generation sequencing data analysis: tricks and twists from a high throughput exome workflow. PLoS One 10, e0126321, doi:10.1371/journal.pone.0126321 (2015).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11, 377–394, doi:10.1089/1066527041410418 (2004).
Via, M., Gignoux, C. & Burchard, E. G. The 1000 Genomes Project: new opportunities for research and social challenges. Genome Med 2, 3, doi:10.1186/gm124 (2010).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220, doi:10.1038/nature11690 (2013).
Elsayed, S. M. et al. Non-manifesting AHI1 truncations indicate localized loss-of-function tolerance in a severe Mendelian disease gene. Hum Mol Genet 24, 2594–2603, doi:10.1093/hmg/ddv022 (2015).
Beck, B. B. et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum Mutat 35, 1153–1162, doi:10.1002/humu.22618 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, doi:10.1038/nature19057 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, doi:10.1093/bioinformatics/btp352 (2009).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311, doi:10.1093/nar/29.1.308 (2001).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65, doi:10.1038/nature11632 (2012).
Lappalainen, I. et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res 41, D936–941, doi:10.1093/nar/gks1213 (2013).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6, e1001025, doi:10.1371/journal.pcbi.1001025 (2010).
Flicek, P. et al. Ensembl 2014. Nucleic Acids Res 42, D749–755, doi:10.1093/nar/gkt1196 (2014).
Stenson, P. D. et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133, 1–9, doi:10.1007/s00439-013-1358-4 (2014).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081, doi:10.1038/nprot.2009.86 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi:10.1038/nmeth0410-248 (2010).
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S. & Goldstein, D. B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9, e1003709, doi:10.1371/journal.pgen.1003709 (2013).
Jagiello, P. et al. New genomic region for Wegener’s granulomatosis as revealed by an extended association screen with 202 apoptosis-related genes. Hum Genet 114, 468–477, doi:10.1007/s00439-004-1092-z (2004).
Acknowledgements
WGS for this study was conducted at the Garvan Institute of Medical Research. All computations underlying the analyses of WES and WGS data were performed on the CHEOPS high performance compute cluster of the Computing Center of the University of Cologne. NGS of gene panels and bioinformatic processing of data therefrom were conducted at the Center for Human Genetics at Bioscientia. We thank the family described herein for their support and cooperation, and Annika Heinemann-Dott and Michaela Thoenes for excellent technical assistance. HJB was supported by the GEERS-Stiftung and by Forschung contra Blindheit, Initiative Usher-Syndrom e.V. EB was supported by the Deutsche Forschungsgemeinschaft, grant no. BE 4830/1–1.
Author information
Authors and Affiliations
Contributions
A.O.K., E.B., C.B., C.N., S.M., G.N. and H.J.B. wrote the manuscript. A.O.K. carried out detailed clinical investigation of the patients. E.B. and L.M.R. carried out lab experiments for splicing analysis. J.A., S.M., P.N. and C.B. performed bioinformatic analyses of NGS data. G.N. analysed linkage data. E.B. was responsible for the minigene-based analysis of splicing. H.J.B. designed the study.
Corresponding authors
Ethics declarations
Competing Interests
C.B. and H.J.B. are employees of Bioscientia which is part of a publicly traded diagnostic company. The other authors have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, A.O., Becirovic, E., Betz, C. et al. A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci Rep 7, 1411 (2017). https://doi.org/10.1038/s41598-017-01577-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01577-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
