
Abstract
In rheumatoid arthritis (RA), chronic inflammation is thought to drive increased cardiovascular risk through accelerated atherosclerosis. It may also lead to a more high-risk plaque phenotype. We sought to investigate carotid plaque phenotype in RA patients using Dynamic Contrast-Enhanced MRI (DCE-MRI) and Fludeoxyglucose Positron Emission Tomography(FDG-PET). In this pilot study, RA patients and age/sex-matched controls were evaluated for cardiovascular risk factors and carotid plaque on ultrasound. Subjects with plaque >2 mm thick underwent DCE-MRI, and a subgroup of patients had FDG-PET. Comparison of MRI findings between groups and correlation between clinical, serological markers and imaging findings was undertaken. 130 patients and 62 controls were recruited. Plaque was more prevalent in the RA group (53.1% vs 37.0%, p = 0.038) and was independently associated with IL6 levels (HR[95%CI]: 2.03 [1.26, 3.26] per quartile). DCE-MRI data were available in 15 patients and 5 controls. Higher prevalence of plaque calcification was noted in RA, despite similar plaque size (73.3% vs 20%, p = 0.04). FDG-PET detected plaque inflammation in 12/13 patients scanned and degree of inflammation correlated with hs-CRP (r = 0.58, p = 0.04). This study confirms increased prevalence of atherosclerosis in RA and provides data to support the hypothesis that patients have a high-risk plaque phenotype.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease which affects 1% of the adult population1. It is characterised by synovial joint inflammation which causes progressive disabling arthritis. Patients also suffer with extra-articular disease and a high systemic inflammatory burden. RA is associated with premature death, the leading cause being cardiovascular disease (CVD)2. Studies have demonstrated that the excess burden of CVD in RA is equivalent to that seen in type two diabetes3. Evidence suggests a more aggressive clinical phenotype of CVD in RA, with patients having fewer warning symptoms prior to major events, and higher case-fatality rates4, 5.
Although an increased prevalence of some traditional risk factors is seen in RA, they do not fully explain the excessive risk. Chronic inflammation is thought to be a major driver of CVD in this population6. Although mechanisms are unclear, inflammation may not only influence traditional cardiovascular risk factors such as dyslipidaemia, but also have direct detrimental effects on the vessel wall7, 8. Recent therapeutic advances in RA have led to significant improvements in outcomes of joint disease. However, the mortality gap between those with and without RA persists, and excess cardiovascular risk is still evident9, 10. A better understanding of the underlying pathological processes driving increased risk, and the potential effects of RA treatment on vascular pathology, would improve our ability to target cardiovascular risk reduction more effectively in this population.
Atherosclerosis, the pathological mechanism underlying most CVD in RA, is also a chronic inflammatory condition with immune cells playing a key role in all stages of plaque development11. In addition, plaque rupture (the main trigger for clinical events) is influenced by plaque composition and presence of inflammation rather than simply by plaque size12, 13. High risk plaque features include the presence of calcification, lipid rich necrotic core (LRNC), neovascularisation and inflammatory cell infiltration. In the general population there is evidence linking pro-inflammatory cytokines with plaque progression and destabilisation14. IL6 and TNF levels have been shown to be independent predictors of future cardiovascular mortality in large meta-analyses15, 16. In RA, an increased prevalence and faster progression of atherosclerosis is associated with inflammation17, 18 and there is emerging evidence that RA patients may have a more rupture-prone plaque phenotype19. In a post mortem study, Aubrey et al. demonstrated less stenosed, more inflammatory coronary plaques in RA patients who had died from acute myocardial infarction (MI)19. Furthermore, Karpouzas et al., found that RA patients had a higher prevalence of high risk plaques on coronary CT20. The clinical phenotype of CVD with more sudden severe events may partly be explained by the hypothesis that patients have a more rupture prone phenotype5, 21.
Recent advances in non-invasive carotid artery imaging allow more detailed characterisation of plaque phenotype. Carotid plaque rupture is an important cause of stroke, however presence and composition of carotid plaque are also predictors of coronary disease22. Carotid magnetic resonance imaging (MRI) can be used to measure plaque burden and compositional features including LRNC, calcification and fibrous cap integrity23. A strong correlation has been established between MRI and histological findings, and MRI has been used in clinical studies to evaluate predictors of stroke, and response to statin therapy24, 25. More recently, carotid artery dynamic contrast enhanced MRI (DCE-MRI) has been developed to measure plaque neovascularisation and inflammation in stroke patients26. Serial images are taken before, during and after contrast injection, then contrast kinetics are modelled to estimate the transfer constant (K trans) of contrast and the fractional volume of plasma within the plaque (v P). Both K trans and v P have been shown to correlate with microvessel density, while K trans also correlated with macrophage infiltration of the plaque on histology27.
18F-fludeoxyglucose positron emission tomography (FDG-PET) is a nuclear imaging technique which can also be used to quantify carotid plaque inflammation. FDG is taken up preferentially in metabolically active tissues and when combined with MRI or CT, activity can be mapped to an anatomical location. In stroke patients, FDG is taken up preferentially in ‘culprit’ lesions and uptake associates strongly with macrophage density on histology28.
Maki-Petaja demonstrated an improvement in aortic inflammation on FDG-PET-CT in RA patients treated with anti-TNF therapy8. Haavisto et al. also recently demonstrated that carotid artery inflammation improved following treatment with anti-inflammatory therapy29. Although FDG-PET has been used to characterise vascular inflammation in RA, neither MRI nor PET have been utilised to investigate atherosclerotic plaque phenotype in RA. However the combination of imaging techniques could provide a comprehensive method with which to interrogate the relationship between chronic inflammation and atherosclerosis in RA, thus allowing a more stratified approach to CVD risk management.
The aim of the current study was test the hypothesis that RA patients have a high-risk carotid plaque phenotype which can be characterised non-invasively using DCE-MRI and PET-CT. We also aimed to evaluate the association between clinical and serological markers of RA disease activity and plaque presence and characteristics. Finally we aimed to test the feasibility of utilising DCE-MRI and FDG-PET to study carotid atherosclerosis in an RA population.
Patients and Methods
Study population
A cross-sectional pilot study of patients with established active RA (defined as a disease activity 28 score (DAS28) >3.2, diagnosed for more than 1 year) who fulfilled the 1987 ACR Criteria for RA30, together with age and sex matched controls, was undertaken. Patients were recruited from outpatient clinics in the North West of England between September 2012 and October 2014. Controls were recruited through the “best friend” method, whereby participants invite a friend to consider participation. Additionally, controls were recruited via advertisements within The University of Manchester and mailshots through The Greater Manchester Primary Care Research Network. Participants aged between 18 and 70 were included. Major exclusion criteria included recent statin use (within 2 months of inclusion), significant renal impairment (estimated glomerular filtration rate <50 ml/min/1.73 m2), any contra-indication to MRI, and a history of vasculitis. The study was approved by the NHS Health Research Authority (North West Research Ethics Committee, reference: 12/NW/0117). All participants provided written informed consent and all methods were performed in accordance with the ethically approved protocol.
Data collection
Clinical and laboratory assessments
Participants underwent assessment of traditional cardiovascular risk factors and, in patients, RA disease characteristics were assessed. This included assessment of joint disease activity using the DAS28 score: a composite measure of tender and swollen joints, patient reported disease activity and serological measure of inflammation which has been widely validated and is used in routine clinical practice. Disability was also evaluated using The Stanford Health Assessment Questionnaire (HAQ) in addition to history of extra-articular disease and treatment history. Fasting bloods were drawn for lipids, glucose, renal function, ESR measurement. High-sensitivity C-reactive protein (hs-CRP) was measured by an in-house ELISA method using anti-human CRP antibodies, calibrators and controls from Abcam (Cambridge, UK). Serum IL6, ICAM, VCAM1, E-selectin and P-selectin levels were all measured by ELISA using DuoSet development kits from R&D Systems (Abingdon, UK).
Carotid artery ultrasound
An ultrasound (US) was performed to screen for carotid plaque in the carotid bulb, common carotid artery and internal carotid artery within 2 cm above and below the bifurcation by one of three trained sonographers. All scans were performed on a Philips iU22 machine using a 9–3 MHz probe and standardised vascular protocol settings. Both arteries were scanned in transverse and longitudinal planes with the participants in a supine position. Plaque was defined in accordance with published recommendations (2 out of 3: Intimal medial thickening >1.5 mm, increased echogenicity, luminal protrusion31). For carotid plaque assessment on MRI, a minimum plaque thickness of >2 mm is required. Therefore only participants with plaques >2 mm thick were invited for MRI.
MRI assessment
Suitable participants attended for MRI within 2 weeks of initial assessment. Scans were performed on a 3T Philips Achieva MRI scanner (Philips Healthcare, The Netherlands) with a specially designed 8-channel carotid artery coil (SHCG, China). A number of sequences were acquired including T1-weighted, T2-weighted, proton density, magnetisation prepared rapid acquisition gradient echo (MP-RAGE) and time of flight angiography sequences (protocol details in supplemental data).
A DCE sequence was then performed. Briefly, 4 slices were positioned around the plaque and 20 frames were acquired (TR:126 ms, TE:4.61 ms, flip angle 50°, slice thickness 3 mm, matrix 260 × 260). 2 frames were acquired prior to injection of gadopentate dimeglumine (Magnevist, Bayer HealthCare Pharmaceuticals), concentration 0.05 mmol/kg, injection rate: 1cc/sec then 18 frames acquired during and after injection (17.5 s/frame).
Scans were anonymised and transferred to the Vascular Imaging Laboratory, University of Washington then analysed with CASCADE software using previously published methods (CASCADE, Seattle, WA)32, 33. Scans were analysed by 2 blinded readers. Plaque dimensions were measured and a normalised wall index (a measure of plaque burden) was calculated by dividing the measured wall area by the sum of the wall and luminal area in each slice. Then compositional features were measured including presence and size of LRNC, calcification, intra-plaque haemorrhage and fibrous cap integrity. Contrast kinetics within the plaque were evaluated using a 2-compartmental Patlak model from which K trans and v P were estimated.
FDG-PET sub-study
Consecutive RA patients undergoing MRI who had no history of cancer, uncontrolled diabetes or infection were invited to have a carotid FDG-PET-CT scan. Scans were performed on a Siemens Biograph mCT·64·S (Siemens Healthcare, Germany). Patients fasted for 6 hours prior to intravenous injection of 200MBq 18F-FDG. Following a 2 hour resting/uptake period the PET-CT scan was performed, localised around the carotid bifurcation of the index artery (detailed methods in supplemental data).
PET-CT images were reconstructed using Siemens UHD (see supplemental data) then co-registered with MRI T1-weighted images. Volumes of interest (VOIs) were drawn around the affected artery on MRI, then plaque FDG uptake measured as the maximum standardised uptake values (SUVmax). A second VOI was used to derive SUVmax from non-atheromatous wall. An example of VOIs on PET-MRI can be seen in Fig. 1. The physicist undertaking the analysis was blinded to clinical, serological and MRI measurements.

Identification of VOIs on PET-MRI. ROIs were drawn around the borders of plaque on each slice where plaque was seen (an example is seen in the axial image (A), yellow arrow points to VOI border). An equivalent number of ROIs are drawn around vessel wall on slices where no plaques are seen (an example is seen in (B)). An example of the ROIs on a sagittal section can be seen in (C) where each arrow points to the two defined ROIs, the superior one includes the atheromatous wall while the inferior ROI is of non-atheromatous wall.
Statistical analysis
This was a pilot study therefore no formal sample size was undertaken. Data were non-normally distributed thus cohort characteristics were summarized using median and interquartile range(IQR). Descriptive statistics were undertaken to compare clinical, serological and imaging findings in patient and control groups using non-parametric statistics. Associations of CV risk factors and RA related factors with presence of plaque were evaluated. Discrete variables were split into quartiles for further analysis. Hs-CRP and IL6 levels were undetectable in some subjects, therefore results were split into quartiles for analysis of association with plaque presence. Univariable logistic regression was used to evaluate associations of plaque with traditional and RA related risk factors. The relationship of RA related factors with plaque was interrogated with logistic regression models with age adjustment and then with adjustment for traditional risk factors. Finally a backwards stepwise multivariable regression was used to identify factors independently associated with plaque in RA patients. Association of disease activity and risk markers with localised plaque inflammation measured on MRI and PET were evaluated using the Spearman rank test.
Results
130 patients and 62 controls were recruited. Figure 2 demonstrates the flow of participants through the study. Baseline characteristics are summarised in Table 1. There were no significant differences in age or gender between the groups. However there was a higher prevalence of hypertension in the patient group (17.7% vs 3.9%, p = 0.01), and a higher systolic blood pressure was also noted compared with controls (136 vs 127 mmHg, p = 0.009). Patients also had lower total cholesterol levels and lower high density lipoprotein cholesterol (p = 0.013 and p = 0.045, respectively). There was an increased prevalence of carotid plaque in patients compared to controls (53.1% vs 37.0%, p = 0.038). When logistic regression was used to adjust for traditional cardiovascular risk factors and history of clinical CVD, the association between RA diagnosis and presence of plaque remained significant (odds ratio [95%CI]: 2.50 [1.08, 5.79]). Factors associated with presence of carotid plaque in patients are summarised in Table 2.

Schematic flow of data collection in patients and controls recruited into the study.
On univariable analysis age and smoking status were the only traditional risk factors associated with plaque. However there were also significant associations with RA related factors: DAS28 score, disability, ESR, IL6 and hs-CRP levels (as shown in Table 2). On adjustment for traditional risk factors, the association of plaque with DAS28 score and IL6 remained significant (detailed in Table 2). On multivariable analysis IL6 levels, age and smoking remained in the model (OR [95%CI]: 2.03 [1.26, 3.26], 1.10 [1.04, 1.18] and 8.523 [0.872, 88.13], respectively). While IL6 and hs-CRP were significantly correlated (spearman rho = 0.38, p < 0.001), re-testing of the stepwise regression without hs-CRP did not lead to any change in the point estimate for IL6. Additionally removal of IL6 from the regression did not lead to inclusion of hs-CRP in the final model, suggesting that co-linearity was not a major issue in the analysis.
Circulating markers of endothelial activation: ICAM and e-selectin were increased in patients compared with controls (p = 0.04, p < 0.001, respectively) and were associated with presence of plaque on univariable analysis (OR [95%]: 1.007 [1.002, 1.012], 1.020 [1.0, 1.041], respectively). While VCAM-1 levels were not significantly higher in patients, they were associated with presence of plaque (OR [CI95%]: 1.004 [1.001, 1.007]).
Plaque phenotype and inflammation on MRI
As shown in Fig. 2, of the plaques identified on US, 35/69 in patients and 8/20 in controls did not meet suitability criteria for MR scanning. Additionally, although the scans were generally well tolerated by subjects, a number of MR scans were aborted in both groups (n = 4) and 16/36 scans were not suitable for analysis. In most cases despite satisfactory imaging quality plaques were not large enough for DCE analysis. Analysis was completed in 15 patients and 5 controls (DCE analysis was not possible in one control due to slice positioning). A representative MR image of plaque is shown in Fig. 3.

Representative images acquired during MRI. The small yellow arrows correspond to the outer border of plaque, the green arrows highlight an area of calcification within plaque and *highlights the lumen in each sequence. (A) A 2D time of flight with the yellow arrow pointing towards the bifurcation. (B) A black blood sagittal oblique section through the bifurcation. (C) A corresponding cross sectional black blood imaging sequence through the common carotid artery bifurcation where plaque can be seen at the posterior aspect of the vessel wall. (D) A corresponding bright blood image at the same level. (E) A DCE parameter map of the cross sectional image. Ktrans is estimated using a Patlak model and a Ktrans map is generated. The Ktrans signal is shown in green. The boundaries of the plaque defined on the T1 weighted sequence are applied to this map to estimate the Ktrans measurement within the plaque. The white arrow highlights the Ktrans signal within the plaque area.
The RA group undergoing MRI were older than the whole RA cohort (59.6[56.2, 64.5] vs 54.2[48.0, 60.9] years, p = 0.004) and had higher median ESR (29.5[17, 46] mm/hr vs 13[7, 27] mm/hr, p = 0.002). There were no significant differences in any other traditional cardiovascular risk factors or disease related factors among the RA group with MRI data and the whole RA cohort.
The prevalence of traditional risk factors in the patients and controls in whom MRI data were analysed, is described in Table 3. There was no difference in age (median [IQR]: 59.6[56.2, 64.9] and 59.0[52.5, 65.5] years, p = 0.57). Two patients but no controls were smokers. There was a trend towards higher LDL levels in the control group (median [IQR]: 2.96[2.13, 2.28] and 3.87[3.28, 4.33] mmol/l in patients and controls respectively, p = 0.081) and higher systolic blood pressure in the patient group (median [IQR]: 147[129, 161] vs 127[117, 140] mmHg in patients and controls respectively, p = 0.079). One subject in the RA group had a history of stable angina. No control subjects had a history of clinical CVD.
There was no significant difference in plaque size between the groups (seen in Table 3). However, there was a significantly higher prevalence of calcification in patients compared to controls (73.3% vs 20%, p = 0.038). There were no significant differences in any other compositional features. While a degree of contrast enhancement was demonstrated in all patients, there was no significant difference in DCE parameters between the two groups. There was a trend towards an inverse association between calcium content of plaque and K trans measurement in patients (r-0.448, p = 0.093). There was no significant association between K trans measurements in plaque and hs-CRP levels (r = 0.002, p = 0.99), IL6 levels (r = 0.26, p = 0.36) or DAS28 score (r = −0.125, p = 0.61).
Plaque inflammation on 18FFDG-PET-MRI
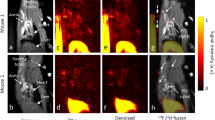
13 RA patients underwent carotid artery PET-CT. In all cases, registration was possible with MRI T1- weighted images and representative example images are presented in Fig. 4. FDG uptake was detected in all 13 cases and 12 out of 13 cases had an SUVmax > 1.85 (the proposed threshold for carotid wall inflammation34). The median (IQR) SUVmax of plaque was 2.18 (2.00, 2.65) and SUVmax correlated significantly with hs-CRP (r = 0.58, p = 0.04). There was no association with plaque volume, K trans, IL6 levels or DAS28 score (r = 0.20[p = 0.609]; r = 0.14[p = 0.74]; r = 0.13[p = 0.697]; r = −0.15[p = 0.617], respectively).

Provides an example of a carotid FDG-PET-MRI in an RA patient. (A) A cross sectional image at the level of the plaque. (B) Provides a more magnified image of the plaque on this slice which demonstrates increased uptake around the vessel wall where plaque has been identified (highlighted by the red arrow). (C) A coronal section in the same patient with the red arrow highlighting the area of increased uptake within the plaque in this view.
Median (IQR) SUVmax in non-atheromatous artery was 2.23(1.62, 2.56). There was a trend towards a correlation between SUVmax in atheromatous and non-atheromatous wall (r = 0.42, p = 0.078). 5/13 subjects had more than 30% higher SUVmax values in plaque than in non-atheromatous wall, suggesting preferential plaque inflammation in these cases.
Discussion
This study adds to the literature demonstrating higher prevalence of atherosclerosis in RA and an association between plaque and both traditional risk factors and inflammation. The association of hs-CRP with plaque presence and inflammation and the independent association of IL6 with plaque accords with the theory of the IL6/CRP axis being a significant driver of cardiovascular risk in the general population and the hypothesis that excessive inflammation contributes to increased risk in RA15.
MRI findings
Despite there being no significant difference in plaque size, an increased prevalence of carotid plaque calcification was noted in patients. Previous published data suggests that 14 subjects are required per group to compare plaque composition on MRI35. Although the control group was small, the presence of calcification in the RA group (n = 15) was also higher than described in studies of asymptomatic individuals in the literature. Underhill et al. evaluated carotid plaque composition in 191 patients with asymptomatic carotid stenosis, comparing findings in those with and without significant coronary artery disease. The mean age of participants included in this study ranged from 57.8 and 60.5 years (in male without coronary disease and female with coronary disease respectively), similar to the age of patients in the current study. Underhill et al. demonstrated a prevalence of calcification of 40.2% and 16% in patients with and without coronary disease respectively36. We found plaque calcification in 70% of patients with RA, only one of whom had a history of stable angina. Carotid plaque calcification is a predictor of subsequent stroke and more severe coronary disease37, 38. Although calcification is a sign of advanced plaque, calcium deposition can occur early, often in response to tissue necrosis or cholesterol deposition. Pro-inflammatory cytokines, in particular TNF, are known to upregulate signalling pathways promoting vascular calcification, and higher levels of pro-inflammatory cytokines have been found preferentially in areas of calcified plaque39, 40. In RA increased vascular calcification has been noted and found to be associated with CRP levels41. However to date, measurement of vascular calcification has been thought to reflect an overall increase in atherosclerosis burden in RA. Although the size of the control group is a major limitation in the current study, when compared with the literature, our data provides preliminary evidence to support early plaque calcification in RA and may suggest an altered natural history of plaque formation and progression. This highlights the need for further investigation into the role of chronic inflammation on vascular calcification in RA.
We found no difference in DCE measurements of plaque inflammation and neovascularisation between groups. There are a number of factors that may contribute to the negative finding. The first is the small sample size. At the time of study set-up, carotid DCE-MRI was a novel technique and little was known about the sample size required to power a study comparing DCE parameters. However, a study published in 2014 suggested that 50 MRI datasets per group were required to achieve statistical power42. The increased prevalence of calcification in the RA group may also have confounded measurements of K trans. Calcium nodules are avascular, hence no contrast would enter this area of plaque which in turn could lead to a lower measured K trans in plaques with calcium. We noted a trend towards an inverse correlation between calcium content and K trans measurement, which accords with previous studies27, 43. Thus, higher calcium content of plaque is likely to have contributed to lower overall K trans values in the RA cohort in this study. Development of methods to exclude areas of calcification from the region of interest on DCE-MRI would go some way to address this issue and may be an area for further research.
A number of MRI scans (16/36) were not suitable for analysis mainly because the plaque thickness was <2 mm on MRI, despite reaching this threshold on US. US is operator-dependent even with expert sonographers. We also noted that in some cases, what appeared as plaque on US, was actually an area of minimal thickening at the origin of a small collateral branch off the carotid bulb on MRI. This may have led to an overestimation of plaque thickness on US, a phenomenon not well described in the literature. No false positive or negative rates are available for US and MRI. However, in a study where MRI scans were performed in patients with plaque causing >15% stenosis on US, only 31/123 cases had plaque >2 mm thick44. Imaging small plaques can be technically challenging and evidence suggests the inclusion of plaques causing more than 30% stenosis or measuring plaque thickness on MRI at inclusion, may be a more reliable way of identifying plaque suitable for DCE-MRI analysis in future studies42, 45. While DCE-MRI appears to be an effective method of evaluating plaque inflammation in stroke patients, the large numbers that would need to be screened in order to obtain a sample size of 50 subjects with asymptomatic plaques may be a limiting factor when considering implementation of carotid DCE-MRI in larger RA studies in the future. However, only 14 subjects per group are required to power a study for compositional analysis of plaque, thus MRI is still a valuable and feasible option for the study of other high risk features of plaque in future studies34. We note the technical challenges of carotid MRI and that in any future study using these methods in an RA population, these feasibility factors should be considered.
PET findings
PET-MRI analysis was performed in 13 patients and a degree of FDG uptake was detected in all cases. Although normal values for FDG uptake within the carotid artery are not well established, a recent paper by Van Der Valk et al. proposed a threshold of SUVmax > 1.85 for significant carotid artery inflammation34. This value was proposed as it was above the 90th percentile in healthy volunteers who had no clinical cardiovascular disease, nor any significant risk factors. Other studies in Takayasu's arteritis have also suggested that an SUV max >1.3 signifies vascular inflammation46. If we apply the more conservative thresholds to our data 12/13 patients would be deemed to have significant inflammation. However we acknowledge that there were differences in acquisition and reconstruction methods between Van der Valk’s study and our own, so these may not be completely comparable data. Although no control group was included in the current study, in the published literature the prevalence of plaque inflammation in asymptomatic subjects is estimated to be 29% thus the current data would support the hypothesis that patients with RA may have more inflammatory plaques47.
Maki-Petaja et al. also demonstrated that patients with active RA had aortic inflammation on FDG-PET and that vascular inflammation improved following anti-TNF therapy8. Our data showing FDG uptake in non-atheromatous walls in RA patients, supports the hypothesis that RA patients have generalised arterial inflammation. However, in 5 cases we found a >30% higher SUVmax in plaque than in non-atheromatous wall, suggesting that plaque inflammation, is not simply a reflection of generalised wall inflammation, but a biomarker of an active inflammatory process that may result in plaque vulnerability and an enhanced risk of a future CVE. The significant correlation between CRP and FDG uptake supports the hypothesis that systemic inflammation not only influences plaque development but also plaque stability. Other studies have also reported an association between CRP and atherosclerosis, as well as future cardiovascular events in RA (48, 49). Our study adds to this literature by demonstrating a link between systemic and local plaque inflammation in RA and may explain why atherosclerotic lesions in RA are more vulnerable to early rupture.
PET-CT was well tolerated by patients and co-registration was possible with MRI sequences in all cases, suggesting PET may be an effective method of quantifying plaque inflammation in future RA studies.
Limitations
We acknowledge several limitations to the study, including the limited power to detect differences in MRI measurements. We do note however that it provides valuable feasibility data with which to power future studies in this population. Additionally, subjects on statins were excluded from the study, as these agents influence MRI and PET measurements. This likely led to exclusion of patients at particularly high risk of CVD which whilst limiting generalisability would, if anything, bias us towards observing fewer differences between patients and controls.
Although the groups were age- and sex-matched, there was a higher prevalence of some traditional risk factors in the patient group, which meant that traditional risk factors may have contributed to the differences in plaque prevalence and phenotype observed on MRI. However the association between a number of measures of inflammation including joint disease activity and IL6 levels remained significant on adjustment for traditional risk factors. This emphasises the importance of disease specific factors in the development of atherosclerosis in RA. Additionally the independent association between IL6 and plaque in patients strongly suggests that inflammation is playing an important role in cardiovascular risk in RA.
We did not recruit a control group for the PET sub-study as the radiation exposure could not be justified in the context of a pilot study. PET and MRI were performed on different scanners and images were co-registered. While images were co-registered satisfactorily in all cases, small anatomical discrepancies due to differences in neck flexion could not be completely excluded and therefore, potentially may have contributed to a measurement error of SUVmax. This has been an inherent problem in the past, with PET-MRI studies being performed on separate scanners, and not restricted to the current study.
In conclusion, we have shown an increased prevalence of atherosclerosis which was associated with classical cardiovascular risk factors and inflammation. On MRI we noted plaque calcification in RA patients, providing preliminary evidence to support earlier calcification and a more high risk plaque phenotype in this patient group. MRI and FDG-PET are well tolerated, feasible techniques to investigate plaque morphology and inflammation. However careful power and sample calculations are needed due to the technical demands involved. MRI and PET may provide the opportunity to interrogate the natural history of plaque and elucidate the influence of disease activity and anti-inflammatory therapies on atherosclerosis in RA. Additionally the development of combined PET-MRI scanners may allow a comprehensive assessment of morphology and inflammation of plaque in one imaging session which may be more feasible in patients with inflammatory joint diseases.
References
Alamanos, Y., Voulgari, P. V. & Drosos, A. A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic review. Semin Arthritis Rheum. 36, 182–188, doi:10.1016/j.semarthrit.2006.08.006 (2006).
Gabriel, S. E. et al. Survival in rheumatoid arthritis: a population-based analysis of trends over 40 years. Arthritis Rheum. 48, 54–58, doi:10.1002/art.10705 (2003).
Peters, M. J. et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 61(11), 1571–9, doi:10.1002/art.v61:11 (2009).
Goodson, N. et al. Cardiovascular admissions and mortality in an inception cohort of patients with rheumatoid arthritis with onset in the 1980s and 1990s. Ann. Rheum. Dis. 64, 1595–1601, doi:10.1136/ard.2004.034777 (2005).
Maradit-Kremers, H. H. et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 52(2), 402–11, doi:10.1002/(ISSN)1529-0131 (2005).
Skeoch, S. & Bruce, I. N. Atherosclerosis in rheumatoid arthritis is it all about inflammation. Nat Rev Rheumatol. 11(7), 390–400, doi:10.1038/nrrheum.2015.40 (2015).
Myasoedova, E. et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann. Rheum. 70(3), 482–7, doi:10.1136/ard.2010.135871 (2011).
Maki-Petaja, K. M. et al. Anti-tumor necrosis factor-alpha therapy reduces aortic inflammation and stiffness in patients with rheumatoid arthritis. Circulation. 126(21), 2473–80, doi:10.1161/CIRCULATIONAHA.112.120410 (2012).
Humphreys, J. H. et al. Mortality trends in patients with early rheumatoid arthritis over 20 years: results from the Norfolk Arthritis Register. Arthritis Care Res (Hoboken). 66(9), 1296–301, doi:10.1002/acr.v66.9 (2014).
Ljung, L. et al. The risk of acute coronary syndrome in rheumatoid arthritis in relation to tumour necrosis factor inhibitors and the risk in the general population: a national cohort study. Arthritis Research & Therapy. 416, 127, doi:10.1186/ar4584 (2016).
Libby, P. Inflammation in atherosclerosis. Nature. 420(6917), 868–74, doi:10.1038/nature01323 (2002).
Rothwell, P. M., Gutnikov, S. A. & Warlow, C. P. Reanalysis of the final results of the European Carotid Surgery Trial. Stroke. 34(2), 514–23, doi:10.1161/01.STR.0000054671.71777.C7 (2003).
Redgrave, J. N. et al. Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation. 113(19), 2320–8, doi:10.1161/CIRCULATIONAHA.105.589044 (2006).
Legein, B. et al. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. 70(20), 3847–69, doi:10.1007/s00018-013-1289-1 (2013).
Sarwar, N. et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 379(9822), 1205–13, doi:10.1016/S0140-6736(11)61931-4 (2012).
Kaptoge, S. et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur. Heart J. 35, 578–589, doi:10.1093/eurheartj/eht367 (2014).
Sandoo, A. et al. Vascular function and morphology in rheumatoid arthritis: a systematic review. Rheumatology (Oxford) 50, 2125–2139, doi:10.1093/rheumatology/ker275 (2011).
Giles, J. T. et al. Longitudinal predictors of progression of carotid atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 63, 3216–3225, doi:10.1002/art.v63.11 (2011).
Aubry, M. C. et al. Differences in atherosclerotic coronary heart disease between subjects with and without rheumatoid arthritis. J. Rheumatol. 34, 937–942 (2007).
Karpouzas, G. A. et al. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann. Rheum. Dis. 73, 1797–1804, doi:10.1136/annrheumdis-2013-203617 (2014).
Sodergren, A. et al. Increased incidence of stroke and impaired prognosis after stroke among patients with seropositive rheumatoid arthritis. Clin Exp Rheumatol. 27(4), 641–4 (2009).
Evans, M. R. et al. Carotid atherosclerosis predicts incident acute coronary syndromes in rheumatoid arthritis. Arthritis Rheum. 63(5), 1211–20, doi:10.1002/art.30265 (2011).
Saam, T. et al. Prevalence of American Heart Association type VI carotid atherosclerotic lesions identified by magnetic resonance imaging for different levels of stenosis as measured by duplex ultrasound. J Am Coll Cardio. 51(10), 1014–21, doi:10.1016/j.jacc.2007.10.054 (2008).
Gupta, A. et al. Carotid plaque MRI and stroke risk: a systematic review and meta-analysis. Stroke. 44(11), 3071–7, doi:10.1161/STROKEAHA.113.002551 (2013).
Zhao, X. Q. et al. MR imaging of carotid plaque composition during lipid-lowering therapy a prospective assessment of effect and time course. JACC Cardiovasc Imaging. 4(9), 977–86, doi:10.1016/j.jcmg.2011.06.013 (2011).
Kerwin, W. et al. Quantitative magnetic resonance imaging analysis of neovasculature volume in carotid atherosclerotic plaque. Circulation. 107(6), 851–6, doi:10.1161/01.CIR.0000048145.52309.31 (2003).
Kerwin, W. S. et al. Inflammation in carotid atherosclerotic plaque: a dynamic contrast-enhanced MR imaging study. Radiology. 241(2), 459–68, doi:10.1148/radiol.2412051336 (2006).
Davies, J. R. et al. Identification of culprit lesions after transient ischemic attack by combined 18F fluorodeoxyglucose positron-emission tomography and high-resolution magnetic resonance imaging. Stroke. 36(12), 2642–7, doi:10.1161/01.STR.0000190896.67743.b1 (2005).
Haavisto, M. et al. Influence of triple disease modifying anti-rheumatic therapy on carotid artery inflammation in drug-naïve patients with recent onset of rheumatoid arthritis. Rheumatology (Oxford). 55(10), 1777–85, doi:10.1093/rheumatology/kew240 (2016).
Arnett, F. C. et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 31(3), 315–24, doi:10.1002/(ISSN)1529-0131 (1988).
Li, R. et al. B-mode-detected carotid artery plaque in a general population. Atherosclerosis Risk in Communities (ARIC) Study Investigators. Stroke. 25(12), 2377–83, doi:10.1161/01.STR.25.12.2377 (1994).
Liu, F. et al. Automated in vivo segmentation of carotid plaque MRI with Morphology-Enhanced probability maps. Magn Reson Med. 55(3), 659–68, doi:10.1002/mrm.20814 (2006).
Kerwin, W. S. et al. MR imaging of adventitial vasa vasorum in carotid atherosclerosis. Magn Reson Med. 59(3), 507–14, doi:10.1002/mrm.21532 (2008).
der Valk, Van et al. Thresholds for arterial wall inflammation quantified by 18F-FDG PET Imaging. JACC Cardiovasc Imaging. 9(10), 1198–1207, doi:10.1016/j.jcmg.2016.04.007 (2016).
Saam, T. et al. Sample size calculation for clinical trials using magnetic resonance imaging for the quantitative assessment of carotid atherosclerosis. J Cardiovasc Magn Reson. 7(5), 799–808, doi:10.1080/10976640500287703 (2005).
Underhill, H. R. et al. Differences in carotid arterial morphology and composition between individuals with and without obstructive coronary artery disease: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 12, 10–31 (2008).
Saba, L. et al. Imaging of the carotid artery. Atherosclerosis. 220(2), 294–309, doi:10.1016/j.atherosclerosis.2011.08.048 (2012).
Zhao, X. et al. Prevalence of compositional features in subclinical carotid atherosclerosis determined by high resolution magnetic resonance imaging in Chinese patients with coronary artery disease. Stroke. 41(6), 1157–62, doi:10.1161/STROKEAHA.110.580027 (2010).
Dhore, C. R. et al. Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 21(12), 1998–2003, doi:10.1161/hq1201.100229 (2001).
Fuller, K. et al. TNF-alpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 143(3), 1108–18, doi:10.1210/endo.143.3.8701 (2002).
Wang, S. et al. Prevalence and extent of calcification over aorta, coronary and carotid arteries in patients with rheumatoid arthritis. J Intern Med. 266(5), 445–52, doi:10.1111/jim.2009.266.issue-5 (2009).
Chen, H. et al. Scan-rescan reproducibility of quantitative assessment of inflammatory carotid atherosclerotic plaque using dynamic contrast-enhanced 3T CMR in a multi-center study. J Cardiovasc Magn Reson. 16, 51, doi:10.1186/s12968-014-0051-7 (2014).
Kerwin, W. S. et al. MR imaging of adventitial vasa vasorum in carotid atherosclerosis. Magn Reson Med. 59(3), 507–14, doi:10.1002/mrm.21532 (2008).
Dong, L. et al. Carotid artery atherosclerosis: effect of intensive lipid therapy on the vasa vasorum-evaluation by using dynamic contrast-enhanced MR imaging. Radiology. 260(1), 224–31, doi:10.1148/radiol.11101264 (2011).
Gaens, M. E. et al. Dynamic contrast-enhanced MR imaging of carotid atherosclerotic plaque: model selection, reproducibility, and validation. Radiology. 266(1), 271–9, doi:10.1148/radiol.12120499 (2013).
Kobayashi, Y. et al. Aortic wall inflammation due to takayasu arteritis imaged with 18F-FDG PET coregistered with enhanced CT. J Nucl Med. 46(6), 917–22 (2005).
Tahara, N. et al. The prevalence of inflammation in carotid atherosclerosis: analysis with fluorodeoxyglucose-positron emission tomography. Eur Heart J. 28(18), 2243–8, doi:10.1093/eurheartj/ehm245 (2007).
Acknowledgements
Sarah Skeoch received funding from The North West England Medical Research Council Fellowship Scheme in Clinical Pharmacology and Therapeutics, which is funded by the Medical Research Council (grant number G1000417/94909), ICON, GlaxoSmithKline, AstraZeneca and the Medical Evaluation Unit (SS). Funding was also provided by the Astra-Zeneca University of Manchester Strategic Alliance Fund. Professor Bruce is a National Institute for Health Research (NIHR) Senior Investigator and is funded by the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Unit and The NIHR Manchester Wellcome Trust Clinical Research Facility. He also acknowledges support from Arthritis Research UK and The Manchester Academic Health Science Centre. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. We thank the Arthritis Research UK for their support: Arthritis Research UK grant number 20380. The project is supported by the Manchester Academic Health Sciences Centre (MAHSC), by the NIHR Manchester Wellcome Trust Clinical Research Facility and the Greater Manchester Clinical Research Network. Philips Healthcare also provided support for imaging acquisition methods.
Author information
Authors and Affiliations
Contributions
Study conception and design: S. Skeoch, I. Bruce, P. Hubbard Cristinacce, H. Williams, M. Alexander, J. Waterton, P. Hockings, T. Hatsukami, C. Yuan; data collection, study management and data analysis including laboratory and imaging analysis: S. Skeoch, P. Hubbard Cristinacce, H. Williams, P. Pemberton, D. Xu, S. Jie, M., J. James; statistical analysis; S. Skeoch, I. Bruce; interpretation of results: S. Skeoch, I. Bruce, Y. Alexander, J. Waterton; compilation of manuscript: all authors.
Corresponding author
Ethics declarations
Competing Interests
Professor Yuan has received grants from Philips Healthcare and is a board member of Philips Radiology Advisory Board. Professor Hockings reports receiving a salary from Astra Zeneca and Arantos Medical, during the conduct of this study. Professor Waterton has received a salary from Astra Zeneca during the conduct of the study.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skeoch, S., Cristinacce, P.L.H., Williams, H. et al. Imaging atherosclerosis in rheumatoid arthritis: evidence for increased prevalence, altered phenotype and a link between systemic and localised plaque inflammation. Sci Rep 7, 827 (2017). https://doi.org/10.1038/s41598-017-00989-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00989-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
