
Abstract
Identification of driver genes contributes to the understanding of cancer etiology and is imperative for the development of individualized therapies. Gene amplification is a major event in oncogenesis. Driver genes with tumor-specific amplification-dependent overexpression can be therapeutic targets. In this study, we aimed to identify amplification-dependent driver genes in 1,454 solid tumors, across more than 15 cancer types, by integrative analysis of gene expression and copy number. Amplification-dependent overexpression of 64 known driver oncogenes were found in 587 tumors (40%); genes frequently observed were MYC (25%) and MET (18%) in colorectal cancer; SKP2 (21%) in lung squamous cell carcinoma; HIST1H3B (19%) and MYCN (13%) in liver cancer; KIT (57%) in gastrointestinal stromal tumors; and FOXL2 (12%) in squamous cell carcinoma across tissues. Genomic aberrations in 138 known cancer driver genes and 491 established fusion genes were found in 1,127 tumors (78%). Further analyses of 820 cancer-related genes revealed 16 as potential driver genes, with amplification-dependent overexpression restricted to the remaining 22% of samples (327 tumors) initially undetermined genetic drivers. Among them, AXL, which encodes a receptor tyrosine kinase, was recurrently overexpressed and amplified in sarcomas. Our studies of amplification-dependent overexpression identified potential drug targets in individual tumors.
Similar content being viewed by others

Introduction
Driver genes involved in oncogenesis are generated by genomic alterations, including point mutations, insertions, deletions, translocations, and gene amplifications1. Driver genes are considered “druggable” targets, using molecular targeted therapies, in which molecules selectively bind protein products translated from genes with mutations, or expressed from amplified genes or fusion genes, to inhibit their oncogenic activities. Examples of such therapies include EGFR inhibitors, which target point mutations in EGFR 2, anti-HER2 antibodies recognizing the product of the ERBB2 gene amplification3, and ALK inhibitors, which target the EML4-ALK fusion gene4. Identification of novel driver genes has been accelerated by recent developments in DNA sequencing technologies, including next-generation sequencing (NGS), particularly in the context of large-scale cancer genomic studies, such as The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/). To date, the number of known driver genes varies from 1381 to 602 genes, which are listed in the database of the Cancer Gene Census and has been updated from the initial 291 genes5. The majority of known driver genes are based on mutations.
Gene amplification is a relatively frequent event in cancer genomes; however, genomic amplification is not always accompanied by elevated gene expression6. Conversely, overexpression is requisite for amplified genes to function as driver alterations. Since they are overexpressed, genes with amplification-dependent overexpression are ideal targets for molecular targeting therapies using antibodies. Thus, integration of copy number variations (CNVs) and mRNA expression levels to identify candidate driver genes have been reported7, 8, and the development of TCGA datasets has accelerated the search for driver genes using in silico methods7.
With the aim of introducing individualized medicine for cancer patients in the future, Shizuoka Cancer Center launched Project HOPE in 2014, which is based on the multi-omics analyses including whole exome sequencing (WES) and gene expression profiling (GEP)9. In this study, we describe gene expression and copy number analysis to determine the involvement of amplification-based driver genes in 1,454 tumors. Coupled with the analysis of 138 mutation-based and 491 fusion-based driver genes, we further explored candidate amplification-based driver genes by examination of a wider candidate cancer-related gene dataset in those tumors with driver origins undetermined by our initial analyses, resulting in the identification of 16 additional potential amplification-based driver genes.
Results
GEP of 64 driver oncogenes in 1,454 solid tumors
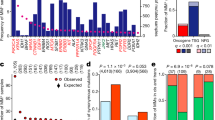
Gene expression analysis is requisite to confirm the status of tumor-specific genomic alterations, including mutations and amplifications, as actionable cancer driver genes. Thus, to determine potential driver carcinogenic genetic changes in 1,454 solid tumors (Supplementary Fig. 1), we first investigated the mRNA expression levels of 64 known driver oncogenes (Supplementary Table 1)1. Among the 64 oncogenes, 10 genes (CCND1, LMO1, MDM2, MDM4, MYC, MYCL, MYCN, NCOA3, NKX2-1, and SKP2) were categorized as amplification-based oncogenes1. The remaining 54 genes were mutation-based oncogenes, primary affected by base substitutions, intragenic insertions, and deletions. We assessed the fold change (FC) in expression levels between tumors and corresponding matched normal tissues by microarray analysis; genes with expression levels increased ≥5-fold in tumor tissues were defined as overexpressed. Among the 10 amplification-based oncogenes, expression levels of MYC family genes, including MYC, MYCL, and MYCN, varied among tumor tissue samples (Fig. 1 and Supplementary Fig. 2). A relatively high frequency of overexpression of MYC was observed in kidney (48%) and colorectal (37%) cancers; of MYCL (variants 1 and 2) in uterine (58%) and breast (40%) cancers; and of MYCN in uterine (58%), liver (44%), and ovarian (43%) cancers. In addition, CCND1 was frequently overexpressed in sarcoma (53%), 79% of which (42% of total sarcoma samples) showed a ≥10-fold increase in expression.

Elevated expression levels of 64 oncogenes in 1,454 solid tumors across 15 cancer types. Cased with strong (fold change ≥10) and moderate (5≤ fold change <10) overexpression are indicated by dark red and dark blue bars, respectively. The two groups of oncogenes, 10 amplification-based (Amp-based), and 54 mutation-based, oncogenes, are indicated on the left. Since some probes recognize multiple splice variants, the number of microarray probes to identify the 64 oncogenes was 71, and there were 13 probes among these corresponding to 12 genes which showed no elevated expression in any tumors, which are not shown. The numbers on the right indicate the number of samples with strong (fold change ≥10) and moderate (5≤ fold change <10) overexpression, as well as the sum of these samples (fold change ≥5). As indicated at the top, samples are arranged by tumor type, including colorectal, lung, stomach, head and neck (HN), liver, breast, pancreas, sarcoma, uterus, kidney, esophagogastric junction (EGJ), esophagus, gastrointestinal stromal tumor (GIST), ovary, melanoma, and other types of tumors. The number of samples of each type of tumor is indicated in parentheses.
Among the 54 mutation-based oncogenes, HIST1H3B and EZH2, which are involved in chromatin structure and remodeling, were overexpressed in 32% and 23%, respectively, of various types of tumors (Fig. 1 and Supplementary Fig. 2). These genes, along with FGFR3, appear to be co-overexpressed in colorectal cancer. Genes frequently overexpressed in specific tumors were as follows: ALK in melanoma (50%) as previously reported10; RET [reviewed in ref. 11] (37% for variant 2 and 35% for variant 4), FLT3 (44%) and ERBB2 12 (34%) in breast cancer; CSF1R (47%), DNMT1 (42%), and MYD88 (31%) in sarcoma; KIT 13, 14 (100%), AR (71%), SMO (50%), PDGFRA 14 (43%), and BCL2 15 (variant alpha) (42%) in gastrointestinal stromal tumors (GISTs). Overall, these data revealed different levels of expression of driver oncogenes across various cancer types.
Oncogenic driver gene amplification of 64 known oncogenes assessed by integrative analysis of GEP and CNVs
Overexpressed oncogenes with amplified respective chromosomal loci are candidate driver genes. In contrast, genomically amplified oncogenes without corresponding overexpression appear to have no, or less, involvement in oncogenesis. Thus, we next integrated the gene expression data for the 64 known oncogenes with the corresponding genomic copy number results, to predict oncogenic driver gene amplifications. As a primary analysis, we selected 12 genes from the 64 oncogenes. These include nine genes frequently overexpressed with copy number gain, including six amplification-based oncogenes (MYC, MYCL, MYCN, MDM2, NKX2-1, and SKP2) and three mutation-based oncogenes (HIST1H3B, EZH2, and CARD11) (Supplementary Fig. 3), and three mutation-based oncogenes, including ERBB2, EGFR, and MET, exhibiting amplifications associated with cancer6. As shown in Fig. 2A, the numbers of copies of these 12 oncogenes were diverse. Thus, we defined the degree of amplification by copy number as follows: genes with copy number ≥6 were defined as being highly amplified, while those with copy numbers of 3, 4, and 5 were defined as being moderately amplified. Next, the 12 oncogenes were divided into two groups based on their copy numbers in samples with ≥5-fold overexpression (Fig. 2A,B). One group contained genes where overexpression was frequent among those with high genomic amplification (copy number ≥6), and included EGFR (variant 1), ERBB2, and MDM2. The other group, contained the remaining nine genes, whose overexpression was frequent among samples with moderate genomic amplification (copy number 3–5), including MYC, MYCL (variants 1 and 2), MYCN, SKP2, NKX2-1, MET, HIST1H3B, EZH2, and CARD11. In addition to EGFR variant 1, ERBB2, and MDM2, samples overexpressing FGFR2, KRAS, and EGFR variant 3 were abundant among those high-level genomic amplification of these genes, specifically in stomach cancer (FGFR2), colorectal and stomach cancer (KRAS), and lung, and head and neck cancer (EGFR variant 3) samples (Fig. 2B and Supplementary Fig. 3). When samples overexpressing EGFR variants 1 and 3 included samples with both high and moderate-levels of genomic amplification, samples in which both EGFR variants were overexpressed with genomic amplification were abundant in lung, and head and neck cancer samples (Fig. 2B and Supplementary Fig. 4). Interestingly, samples in which only EGFR variant 3 was overexpressed with genomic amplification were abundant in colorectal cancer samples. EGFR variant 3 mRNA translates a soluble EGFR protein, p60 (isoform C), lacking transmembrane and tyrosine kinase domains, whereas EGFR variant 1 mRNA translates the full-length p170 EGFR (isoform A)16, 17. Because the formation of inactive heterodimers between different isoforms competitively prevents the formation of functional holoreceptors18, this result suggested that oncogenesis involving the EGFR pathway differs in samples with or without overexpressing EGFR variant 3 exhibiting genomic amplification.

Gene expression and copy number of 64 oncogenes. (A) Gene expression levels of 12 oncogenes, including EGFR variant 1, ERBB2, MDM2, MYC, MYCL variants 1 and 2, MYCN, SKP2, NKX2-1, MET, HIST1H3B, EZH2, and CARD11, were linked to the genomic copy number. Pearson’s correlation coefficient (r with P-value) between copy number and mRNA expression is indicated at the bottom right. Red horizontal bars indicate 5-fold on the log2 scale. (B) The relationship between gene overexpression and degree of genomic copy number gain. Tumors in which oncogenes were overexpressed by ≥5-fold were divided into three groups: those with high-level genomic amplification (copy number ≥6; dark blue bars), those with moderate genomic amplification (copy number 3–5; orange bars), and those without genomic amplification (copy number 1–2; light green bars). There were no cases with a copy number of zero among the indicated genes. Genes highlighted on the bottom correspond to the 12 oncogenes exhibited in panel A.
Using Pearson correlation coefficient, ERBB2 and MDM2 showed moderate relationships between copy number and FC (r = 0.53 and 0.49, respectively), whereas SKP2 (r = 0.35), EGFR variant 1 (r = 0.28), MYCL variants 1 and 2 (r = 0.26), and EZH2 (r = 0.20) were in the range between 0.2 and 0.4, indicating weak relationships. In particular, the majority of samples with high-level amplification of MYC were not overexpressed (r = 0.13), suggesting that these amplifications were passenger-like. Thus, we investigated the frequency of amplification in samples demonstrating overexpression (Table 1). Strong to moderate correlations between overexpression and amplification (defined as frequencies of ≥50%) were observed for EGFR (88% and 70% for variants 1 and 3, respectively), SKP2 (76%), MET (65%), MYC (64%), MDM2 (62%), DNMT3A (52%), and ERBB2 (50%). For these eight genes, Pearson correlation coefficient between copy number and FC among the samples overexpressed with FC ≥5 demonstrated moderate and weak relationships in EGFR variant 1 (r = 0.52) and MYC (r = 0.33), respectively (Supplementary Fig. 5), which increased from those obtained from all 1,454 samples (r = 0.28 and 0.13, respectively). This result indicates that overexpression with FC ≥5 of EGFR variant 1 and MYC is frequently involved in copy number gain. In contrast, weak correlations (≤10% frequency) were observed for IDH2 (10%), RET (10% and 8% for variants 2 and 4, respectively), DNMT1 (8%), MYCL variants 1 and 2 (8%), FGFR3 (6%), LMO1 (4%), FLT3 (3%), JAK3 (1%), and AR (0%).
The frequency of amplification-dependent overexpression, in which overexpression was accompanied by either high or moderate levels of genomic amplification, was calculated for individual tumor types (Table 2). For seven types of tumors, where ≥50 samples were available, genes identified as amplified and overexpressed with frequency of ≥10% were MYC (25%) and MET (18%) in colorectal cancer; SKP2 (21%), FOXL2 (18%), EGFR (16% and 14% for variants 3 and 1, respectively) in lung squamous cell carcinoma; HIST1H3B (19%) and MYCN (13%) in liver cancer; and HIST1H3B (12%) in breast cancer. Among the highly recurrent oncogenes amplified with overexpression in specific tumors, including MYC in colorectal cancer, MYCN in colorectal and liver cancers, and HIST1H3B and EZH2 in colorectal, lung, liver, and breast cancers, there was no significance between their status of amplification with overexpression and cancer stage (Supplementary Table 3) when significance was defined as P < 0.05 (Fisher’s exact test). However, MYCN amplification with overexpression was observed in colorectal cancer samples more frequently with stage III and IV than with stage I and II (P = 0.0672). In GISTs, 57% of samples exhibited KIT amplification-dependent overexpression. Another notable finding was that 21 of 25 samples (84%) with FOXL2 amplification-dependent overexpression were derived from squamous cell carcinoma of the lung (10 of 56), head and neck (9 of 101), and esophagus (2 of 14), and its frequency was 12% of a total of 176 squamous cell carcinoma samples (Supplementary Fig. 3).
Additionally, we classified 138 driver genes into the 12 core signaling pathways (Supplementary Table 1). There were five pathways, including cell cycle/apoptosis (CC/A), chromatin modification (CM), receptor tyrosine kinases (RTK), TGF-β, and transcriptional regulation (TR), related to the amplified and overexpressed oncogenes shown in Table 2. As shown in Fig. 3, the activation of receptor tyrosine kinases pathway was involved in all types of tumors, whereas the CM pathway was activated in the samples from liver and ovarian cancers, and sarcoma. Furthermore, the activation patterns were similar for samples from lung squamous cell carcinoma, head and neck, and esophageal cancers. Since amplification-dependent overexpression is predicted to be involved in oncogenesis, the genes amplified with overexpression are potential target molecules for anti-cancer agents.

Radar charts comparing the 12 core signaling pathways involved in amplified and overexpressed oncogenes in individual cancer types. Each axis of the diagrams represents a percentage of frequencies of pathway activation derived from amplified and overexpressed oncogenes, as listed in Table 2. As listed in Supplementary Table 1, 138 driver genes were assigned to the 12 signaling pathways, including APC, cell cycle/apoptosis (CC/A), chromatin modification (CM), DNA damage control (DDC), Hedgehog (HH), NOTCH, PI3K, RAS, receptor tyrosine kinase (RTK), STAT, TGF-β, and transcriptional regulation (TR).
Cancer-related genes with potential oncogenic driver gene amplification
Besides CNVs, we analyzed sequence-based alterations, including substitutions, insertions, and deletions in the 138 driver genes, along with 491 fusion genes. Integration of sequence- and amplification-based alteration data revealed that genomic driver aberrations were present in 1,127 of 1,454 tumors (78%) (Fig. 4, Supplementary Fig. 4), while the driver origins of the remaining 327 tumors (22%) were undetermined. Next, we expanded the number of target genes to 820 to clarify whether any other oncogenic driver gene amplifications were involved in the 327 tumors undetermined driver alterations by the initial analyses. Referring to various databases and publications19,20,21,22,23,24,25,26,27 (https://www.bcm.edu/research/medical-genetics-labs/test_detail.cfm?testcode=9705), we selected 820 cancer-related genes, referred to as SCC-820 (Supplementary Table 2). The SCC-820 set of genes includes the 138 cancer driver genes1. Initially, the levels of expression and copy numbers of SCC-820 genes were characterized in 1,454 tumors, and 589 of 879 microarray probes corresponding to SCC-820 genes exhibited amplification-dependent overexpression in 1,251 tumors (86%). Among them, INHBA and RECQL4 were frequently overexpressed and amplified in various tumor types (Fig. 5). INHBA overexpression promotes cell proliferation in esophageal adenocarcinoma28, while RECQL4 is associated with breast cancer tumor aggressiveness, due to both amplification and overexpression29. Thus, these genes have been potentially associated with oncogenesis in previous reports. In addition, SOX2 and TP63, map to chromosome 3q26.32–q29, were frequently overexpressed and amplified specifically in lung squamous cell carcinoma tumors (Fig. 6 and Supplementary Fig. 5A). Overexpression of CCNE1 with high-level genomic amplification was identified in lung, stomach, sarcoma, and esophagogastric junction tumors. Genes co-localizing with CCNE1 on chromosome 19q12–13.12, including TSHZ3, CEBPA, PDCD2L, ALKBH6, and KMT2B, were also co-overexpressed and co-amplified in some tumors.

Genomic aberrations of cancer driver genes in 1,454 solid tumors. (A) Bar graph depicting the number of samples having somatic structural alterations in individual tumor types. Genomic alterations were grouped by cases of overexpression with gene amplification in 64 oncogenes (Exp/Amp), cases of mutations, insertions, and deletions in 138 driver genes, along with the presence of fusion genes (Seq), and cases without any alterations (Unknown). (B) Pie chart depicting the frequency of somatic structural alterations in all tumor samples.

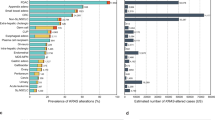
Frequency of overexpression with gene amplification of 820 cancer-related genes in individual tumor types. Genes overexpressed with ≥5-fold and a genomic copy number of ≥3 are ordered by type of tumor, as shaded. In colorectal (CRC), squamous cell carcinoma of the lung (Lg-sq), stomach (St), head and neck (HN), liver (Liv), and breast (Bre) cancer, genes with ≥10% frequency are shown. For the remaining tumor types, including tumors from lung adenocarcinoma (Lg-ad), pancreas (Pan), sarcoma (Sar), uterus (Ute), kidney (Kid), esophagogastric junction (EGJ), esophagus (Eso), gastrointestinal stromal tumor (GIST), ovary (Ov), and melanoma (Mel), genes with frequencies approximately ≥20% are listed. Frequencies observed in all 1,454 tumor samples are indicated as “All”. The frequency is indicated as a heat map (range 0–100%).

Tumor-specific overexpression with gene amplification among 820 cancer-related genes in 1,454 solid tumors. Genes overexpressed ≥5-fold with high (copy number ≥6) and moderate (copy number 3–5) genomic amplification are indicated by dark blue and orange bars, respectively. The tumor specificity of each gene is indicated on the right. As indicated at the top, samples are arranged by tumor type, including tumors from colorectal, lung, stomach, head and neck (HN), liver, breast, pancreas, sarcoma, uterus, kidney, esophagogastric junction (EGJ), esophagus, gastrointestinal stromal tumor (GIST), ovary, melanoma, and other types of tumors. The number of samples of each type of tumor is indicated in parentheses.
SOX2 30 and CCNE1 31 have previously been reported to function as driver oncogenes. These analyses of SCC820 genes identified additional genes (relative to those identified by analysis of the data on 64 oncogenes) with oncogenic potential that exhibited amplification-dependent overexpression.
Potential oncogenic driver gene amplification in tumors in which driver genes were not initially identified
Among the 1,454 tumors, 327 were categorized as having undetermined driver origins following mutation, copy number, and expression analyses of 138 driver and 491 fusion genes (Fig. 4 and Supplementary Fig. 4). For these 327 tumors, overexpression and amplification analysis using the SCC820 genes identified 214 (65%) and 113 (35%) tumors with and without amplification-dependent overexpression, respectively. There were 16 genes identified only in the 327 tumors (Table 3). Since no genomic alterations, other than amplification-dependent overexpression, were identified, these genes are candidate driver genes. Recurrent amplification-dependent overexpression of AXL, which encodes a receptor tyrosine kinase, was identified in two sarcoma samples. Among the 16 genes, AXL 32 and DIS3 33 have been previously reported to be associated with oncogenesis.
Discussion
We conducted integrative GEP and CNV analyses of 1,454 tumors to identify potential driver genes with amplification-dependent overexpression. When the number of copies of a gene increases, its expression is predicted to be elevated; however, there has been a report of gene amplification and copy number gain in the absence of overexpression6. Therefore, for the identification of driver genes, it is essential to investigate gene expression levels alongside copy number analysis. In this study, we also observed discrepancies between genomic copy number increase and levels of gene expression, particularly in cases where copy numbers were moderately elevated (3–5 copies) (Fig. 2). Moreover, elevation of gene expression was not always accompanied by copy number gain (Fig. 2 and Table 1).
Although there are no clear criteria defining gene amplification according to the number of copies, arbitrary numbers have been provided for some genes. For ALK, the presence of ≥6 copies has been defined as amplification, while having 3–5 copies was defined as copy number gain34. For RET, amplification defined as the presence of ≥5 copies, with copy number gain set at 3–4 copies35. Similar to these examples, we defined our own criteria for the grade of amplification by copy number as follows; copy numbers of ≥6 were defined as high-level amplification, while copy numbers of 3–5 were considered as moderate amplification. Depending on the number of copies, particular amplified sequences have been linked to clinical features, including MYCN and poor prognosis, and ERBB2 and drug sensitivity6. In addition, SKP2 overexpression and amplification has been related to metastasis in lung squamous cell carcinoma36. It remains to be seen whether the overexpressed and amplified genes identified in this study demonstrate such clinical significance. In particular, it will be of interest to determine differences between the two groups classified as having high and moderate amplification of ERBB2 and EGFR (Fig. 2B).
Vogelstein et al. selected 138 driver genes consisting of 64 oncogenes and 74 tumor suppressor genes (TSGs), the former of which were classified into two groups, including 10 amplification-based and 54 mutation-based oncogenes. Among the 10 amplification-based oncogenes, we identified that six genes (MDM2, MYC, MYCL, MYCN, NKX2-1, and SKP2) were amplified and overexpressed in ≥10 tumor samples (Supplementary Fig. 3). As among the most frequently amplified oncogenes37,38,39, MYC was amplified and overexpressed in a wide range of tumor tissues. Amplification with overexpression of MYCL, NKX2-1, and SKP2 in lung cancer, and MDM2 in sarcoma was observed as previously reported6. Although MYCN amplification with overexpression was observed in neuroblastoma, sarcoma, and lung cancer, our study identified that MYCN was amplified and overexpressed in 13% of liver cancer samples. Among the remaining four genes (CCND1, LMO1, MDM4, and NCOA3), we observed no amplification or overexpression of MDM4, which is amplified and overexpressed in glioma and retinoblastoma6, in our analysis of 1,454 tumors. Amplification with overexpression of CCND1 and NCOA3, which was previously observed in breast cancer6, were detected in four samples from colorectal, head and neck, liver, and ovarian cancer for CCND1 and eight samples from three sarcoma, two esophageal cancer, and one sample each from colorectal, lung, and breast cancer for NCOA3. Although LMO1 duplication was associated with more advanced disease and survival in neuroblastoma40, our analysis identified four samples from lung adenocarcinoma exhibiting LMO1 amplification and overexpression, three of which were derived from stage I and no stage information provided for the remaining sample.
The analysis of 64 known driver oncogenes revealed amplification-dependent overexpression in 587 of 1,454 tumors (40%) (Fig. 4B). The subsequent extended analysis including 820 cancer-related genes narrowed down the candidate driver genes associated with amplification-dependent overexpression, particularly in tumors with unidentified driver origins after the analyses of the 138-driver and 491-fusion genes. Sixteen genes (AXIN1, AXL, CD70, CNKSR1, DIS3, DST, EZH1, FAM175A, FLCN, FOXO1, HAU3, MTR, PER1, PIK3CB, TSC2, and ZNF384) were identified only in the 327 tumors that had undetermined drivers after the initial analyses. Of these, AXL and DIS3 have previously been suggested to have oncogenic functions32, 33. Recurrent amplification-dependent overexpression of AXL was observed only in two sarcomas (myxofibrosarcoma and leiomyosarcoma). In addition, a liposarcoma tumor sample among the 1,127 tumors with identified driver origins after the initial analyses exhibited a fold change in AXL expression approaching the cutoff level (FC = 4.97) and had four genomic copies of this gene. Including this case, AXL amplification-dependent overexpression was observed in three of a total of 19 sarcoma samples (16%). AXL (previously known as UFO), is a member of the TMA (TYRO3, MER, and AXL) receptor tyrosine kinase family and has important roles in various cancer processes [reviewed in ref. 41]. AXL amplification has been identified in colorectal cancer42 Moreover, AXL overexpression has been observed in many solid and hematopoietic malignancies, including Ewing sarcoma tumor tissues43 and sarcoma cell lines44. Since the relationship between levels of amplification and overexpression is currently unclear, from a clinical perspective, it will be of interest to determine the prognostic significance of AXL amplification-dependent overexpression in sarcoma tumors in the future.
Searching for genes overexpressed as a result of genomic amplification is one method of driver gene identification. It will be necessary and appropriate to eliminate passenger genes from these candidate drivers, particularly those derived from the analysis of 820 cancer-related genes using sophisticated in vitro and in silico experiments7. Another future challenge is to identify driver genes in the 113 tumors where the driver origin remained unknown after analysis on the SCC-820 dataset. Since our analyses were conducted in-house, the data we obtained from the multi-omics project can readily be coupled with clinical data to provide therapeutic and prognostic information for individual patients, as a step towards the development of personalized medicine.
Methods
Study setting
Ethical approval for all experimental protocols and study was obtained from the institutional review board at the Shizuoka Cancer Center (Authorization Number: 25–33). Written informed consent was obtained from all patients enrolled in the study. All experiments using clinical samples were carried out in accordance with the approved guidelines.
Clinical samples
Tumor tissue samples with sizes corresponding to weights of ≥0.1 g were dissected from surgical specimens, along with samples of surrounding normal tissue. The areas from which tumor samples were dissected were visually assessed as containing ≥50% tumor content. For RNA analysis, tissue samples were submerged in RNAlater solution (Thermo Fisher Scientific), minced, and stored overnight at 4 °C before RNA extraction. For DNA analysis, tumor and normal tissues were immediately frozen in liquid nitrogen before DNA extraction. In addition, whole blood was collected as a control for whole exome sequencing.
RNA isolation
Total RNA was extracted from approximately 10 mg of minced tissue samples using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Initially, tissue samples were mixed with QIAzol reagent from the kit and then ground with a TissueLyzer II (Qiagen) using a 5-mm zirconia bead for 10 min at room temperature. RNA samples were quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific) and their quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies) with an RNA 6000 Nano total RNA Kit (Agilent Technologies).
GEP analysis
RNA samples with RNA integrity number ≥6.0 was used for microarray analysis. Total RNA (100 ng) was amplified and fluorescence-labeled using the One-Color Low Input Quick Amp Labeling Kit (Agilent Technologies) according to the manufacturer’s instructions. Labeled samples were hybridized to the SurePrint G3 Human Gene Expression 8 × 60 K v2 Microarray (Agilent Technologies), which has 50,599 probes capable of detecting 29,833 genes registered in the Entrez Gene Database, published by the National Center for Biotechnology Information. Expression levels were calculated using previously described methods45, and data derived from tumor tissue samples collated with that from corresponding adjacent normal tissue specimens. Hybridization signals were detected using a DNA Microarray Scanner (Agilent Technologies) and scanned images were analyzed using Agilent Feature Extraction software. Microarray analysis was performed in accordance with the MIAME guidelines46.
Data analysis was performed using GeneSpring GX software (Agilent Technologies) and Microsoft Excel. Probes to be analyzed were selected according to the reference genome sequence, hg19, obtained from the UCSC Genome Browser47. Raw signal intensity values were log transformed and normalized to the 75th percentile. The fold change between tumor and normal tissues from the same patient was calculated using the normalized intensity values. Probes expressed at raw signal values < 10 in both tumor and normal tissues were excluded from further analysis. GEP data of SCC-820 genes are included in Supplementary Dataset 1 as MIAME-compliant data.
DNA isolation and WES analysis
DNA was extracted from tissue and blood samples using a QIAamp Kit (Qiagen) according to the manufacturer’s instructions, and subjected to WES on the Ion Proton System (Thermo Fisher Scientific) as reported previously48, 49. For data analysis, single-nucleotide variants with quality scores <30 or depth of coverage <20 were discarded. Torrent Suite software (ver. 4.4) was used to convert raw binary data into sequence reads that were mapped to the reference human genome (UCSC, hg19). Somatic mutations were identified by comparing data from tumor and corresponding blood samples. Driver mutations in 138 known driver genes1 were defined as those identified as pathogenic in the ClinVar database50, or those contained in the Database of Curated Mutations (DoCM, http://docm.genome.wustl.edu) or the UMD TP53 mutation database51. In addition, nonsense, frameshift, and splice site mutations in TSGs among 138 driver genes were also classified as driver mutations.
CNVs were detected using Ion Reporter Software Copy Number Variation Analysis (Thermo Fisher Scientific). The CNV detection algorithm was based on a hidden Markov model. CNVs with confidence scores ≥10 were included in the analysis. Using this system, the maximum copy number calculated was 10. CNV data of SCC-820 genes are included in Supplementary Dataset 2. The WES data was applied to estimate tumor purity using an in silico method52, which is included in Supplementary Dataset 2.
Detection of fusion genes
Fusion gene analysis was performed using the Ion Proton System, as previously reported53. In brief, total RNA (10 ng) was used as a template to prepare cDNA using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). The Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific) and the Ion Proton Sequencing 200 Kit (Thermo Fisher Scientific) were used to construct an Ion Torrent adapter-ligated library and perform nucleotide sequencing, respectively, according to the manufacturer’s protocols. All data were analyzed using the Ion Reporter server. The Ion AmpliSeq RNA Fusion workflow (Thermo Fisher Scientific) was used to detect fusion transcripts from a panel of 491 fusion genes.
References
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004).
Carter, P. et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl, Acad. Sci. USA 89, 4285–4289 (1992).
Soda, M. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007).
Futreal, P. A. et al. A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004).
Santarius, T., Shipley, J., Brewer, D., Stratton, M. R. & Cooper, C. S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 10, 59–64 (2010).
Chen, Y. et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One 9, e98293, doi:10.1371/journal.pone.0098293 (2014).
Caburet, S. et al. Combined comparative genomic hybridization and transcriptomic analyses of ovarian granulosa cell tumors point to novel candidate driver genes. BMC Cancer 15, 251, doi:10.1186/s12885-015-1283-0 (2015).
Yamaguchi, K. et al. Implementation of individualized medicine for cancer patients by multiomics-based analyses—the Project HOPE—. Biomed. Res. 35, 407–412 (2014).
Busam, K. J. et al. Primary and metastatic cutaneous melanomas express ALK through alternative transcriptional initiation. Am. J. Surg. Pathol. 40, 786–795 (2016).
Morandi, A., Plaza-Menacho, I. & Isacke, C. M. RET in breast cancer: functional and therapeutic implications. Trends Mol. Med. 17, 149–157 (2011).
Kraus, M. H., Popescu, N. C., Amsbaugh, S. C. & King, C. R. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 6, 605–610 (1987).
Went, P. T. et al. Prevalence of KIT expression in human tumors. J. Clin. Oncol. 22, 4514–4522 (2004).
Jung, S. H. et al. Expression of DOG1, PDGFRA, and p16 in gastrointestinal stromal tumors. Gut Liver 5, 171–180 (2011).
Antonescu, C. R. et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin. Cancer Res. 10, 3282–3290 (2004).
Reiter, J. L. & Maihle, N. J. A 1.8 kb alternative transcript from the human epidermal growth factor receptor gene encodes a truncated form of the receptor. Nucleic Acids Res. 24, 4050–4056 (1996).
Reiter, J. & Maihle, N. J. Characterization and expression of novel 60-kDa and 110-kDa EGFR isoforms in human placenta. Ann. N. Y. Acad. Sci. 995, 39–47 (2003).
Basu, A., Raghunath, M., Bishayee, S. & Das, M. Inhibition of tyrosine kinase activity of the epidermal growth factor (EGF) receptor by a truncated receptor form that binds to EGF: role for interreceptor interaction in kinase regulation. Mol. Cell Biol. 9, 671–677 (1989).
Meric-Bernstam, F. et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J. Clin. Oncol. 33, 2753–2762 (2015).
Meador, C. B. et al. Beyond histology: translating tumor genotypes into clinically effective targeted therapies. Clin. Cancer Res. 20, 2264–2275 (2014).
Jones, S. et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 7, 283ra53, doi:10.1126/scitranslmed.aaa7161 (2015).
Wagle, N. et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2, 82–93 (2012).
Ananda, G. et al. Development and validation of the JAX Cancer Treatment Profile™ for detection of clinically actionable mutations in solid tumors. Exp. Mol. Pathol. 98, 106–112 (2015).
Hovelson, D. H. et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 17, 385–399 (2015).
Cheng, D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Frampton, G. M. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031 (2013).
Seder, C. W. et al. INHBA overexpression promotes cell proliferation and may be epigenetically regulated in esophageal adenocarcinoma. J. Thorac. Oncol. 4, 455–462 (2009).
Arora, A. et al. RECQL4 helicase has oncogenic potential in sporadic breast cancers. J. Pathol. 238, 495–501 (2016).
Hussenet, T. et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One 5, e8960, doi:10.1371/journal.pone.0008960 (2010).
Etemadmoghadam, D. et al. Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS One 5, e15498, doi:10.1371/journal.pone.0015498 (2010).
Li, Y. et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 28, 3442–3455 (2009).
de Groen, F. L. et al. Gene-dosage dependent overexpression at the 13q amplicon identifies DIS3 as candidate oncogene in colorectal cancer progression. Genes Chromosomes Cancer 53, 339–348 (2014).
Yang, H. S. & Horten, B. Gain of copy number and amplification of the RET gene in lung cancer. Exp. Mol. Pathol. 97, 465–469 (2014).
Salido, M. et al. Increased ALK gene copy number and amplification are frequent in non-small cell lung cancer. J. Thorac. Oncol. 6, 21–27 (2011).
Yokoi, S. et al. Amplification and overexpression of SKP2 are associated with metastasis of non-small-cell lung cancers to lymph nodes. Am. J. Pathol. 165, 175–180 (2004).
Nesbit, C. E., Tersak, J. M. & Prochownik, E. V. MYC oncogenes and human neoplastic disease. Oncogene 18, 3004–3016 (1999).
Beroukhim, R. et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010).
Lin, C. Y. et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67 (2012).
Wang, K. et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 469, 216–220 (2011).
Brown, M., Black, J. R., Sharma, R., Stebbing, J. & Pinato, D. J. Gene of the month: Axl. J. Clin. Pathol. 69, 391–397 (2016).
Martinelli, E. et al. AXL is an oncotarget in human colorectal cancer. Oncotarget 6, 23281–23296 (2015).
Fleuren, E. D. et al. The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget 5, 12753–12768 (2014).
Bai, Y. et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 72, 2501–2511 (2012).
Hatakeyama, K. et al. Identification of a novel protein isoform derived from cancer-related splicing variants using combined analysis of transcriptome and proteome. Proteomics 11, 2275–2282 (2011).
Brazma, A. et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 29, 365–371 (2001).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Serizawa, M. et al. Novel tumor-specific mutations in receptor tyrosine kinase subdomain IX significantly reduce extracellular signal-regulated kinase activity. Anticancer Res. 36, 2733–2744 (2016).
Akiyama, Y. et al. Immune response-associated gene analysis of 1,000 cancer patients using whole-exome sequencing and gene expression profiling-Project HOPE. Biomed. Res. 37, 233–242 (2016).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Edlund, K. et al. Data-driven unbiased curation of the TP53 gene mutation database and validation by ultradeep sequencing of human tumors. Proc. Natl. Acad. Sci. USA 109, 9551–9556 (2012).
Larson, N. B. & Fridley, B. L. PurBayes: estimating tumor cellularity and subclonality in next-generation sequencing data. Bioinformatics 29, 1888–1889 (2013).
Urakami, K. et al. Next generation sequencing approach for detecting 491 fusion genes from human cancer. Biomed. Res. 37, 51–62 (2016).
Acknowledgements
We thank the members of the Shizuoka Cancer Center Hospital and Research Institute for their support and suggestions. This work was supported by the Shizuoka Prefectural Government, Japan.
Author information
Authors and Affiliations
Contributions
K.Y., M.K. and K.U. developed the concept. K.O. isolated RNA, analyzed all of the data and wrote the manuscript. K.H., Y.W., K.K., Y.D. and T.I. performed microarray experiments and collected data. T.N. developed the analytical pipeline software and performed the WES and fusion gene bioinformatics analysis. Y.S., T.T., Su.O., Sh.O., M.S. and K.U. isolated DNA, performed WES and fusion gene experiments and collected data. K.Y., M.K., K.U., T.M., Y.A. and K.M. contributed to data interpretation. K.Y., M.K. and K.U. obtained funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ohshima, K., Hatakeyama, K., Nagashima, T. et al. Integrated analysis of gene expression and copy number identified potential cancer driver genes with amplification-dependent overexpression in 1,454 solid tumors. Sci Rep 7, 641 (2017). https://doi.org/10.1038/s41598-017-00219-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00219-3
This article is cited by
-
Integrative multi-omic sequencing reveals the MMTV-Myc mouse model mimics human breast cancer heterogeneity
Breast Cancer Research (2023)
-
NCBP2 and TFRC are novel prognostic biomarkers in oral squamous cell carcinoma
Cancer Gene Therapy (2023)
-
Amplified therapeutic targets in high-grade serous ovarian carcinoma – a review of the literature with quantitative appraisal
Cancer Gene Therapy (2023)
-
Incidence and clinical significance of 491 known fusion genes in a large cohort of Japanese patients with colorectal cancer
International Journal of Clinical Oncology (2023)
-
Overview and clinical significance of multiple mutations in individual genes in hepatocellular carcinoma
BMC Cancer (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
