
Abstract
Cassava (Manihot esculenta Crantz) is a vital tropical root crop providing essential dietary energy to over 800 million people in tropical and subtropical regions. As a climate-resilient crop, its significance grows as the human population expands. However, yield improvement faces challenges from biotic and abiotic stress and limited breeding. Advanced sequencing and assembly techniques enabled the generation of a highly accurate, nearly complete, haplotype-resolved genome of the African cassava cultivar TMEB117. It is the most accurate cassava genome sequence to date with a base-level accuracy of QV > 64, N50 > 35 Mbp, and 98.9% BUSCO completeness. Over 60% of the genome comprises repetitive elements. We predicted over 45,000 gene models for both haplotypes. This achievement offers valuable insights into the heterozygosity genome organization of the cassava genome, with improved accuracy, completeness, and phased genomes. Due to its high susceptibility to African Cassava Mosaic Virus (ACMV) infections compared to other cassava varieties, TMEB117 provides an ideal reference for studying virus resistance mechanisms, including epigenetic variations and smallRNA expressions.
Similar content being viewed by others

Background & Summary
Plants exhibit remarkable genetic diversity, often as a mosaic of different variants within a single individual. Crops like cassava, mango, and rubber tree are often highly heterozygous because of either outcrossing or clonal propagation1,2,3. Plants propagated clonally through methods such as stem cutting retain genetic variation, making it challenging to create high-quality reference genomes. Despite this challenge, the recent advancements in sequencing technologies have made it possible for researchers to explore the complex genomic architecture of these crops4. By uncovering and analyzing the genomic diversity of plants, we can fully harness their potential and facilitate innovations in the fields like breeding, agronomy, and food security.
Cassava (Manihot esculenta Crantz) is a vital crop for subsistence farmers in tropical and subtropical regions across the globe, providing a source of food and industrial purposes. Cassava is utilized to produce various products such as starch, bioethanol, and other bio-based products such as feed, medicine, cosmetics, and biopolymers5. Subsistence farmers in Africa prefer cultivating cassava as it yields substantial harvests under diverse environmental conditions6. Additionally, cassava roots have an ideal harvesting age and can be harvested flexibly, offering the benefits of a longer in-ground storage7. However, the crop faces pests, diseases, drought, weeds, and environmental factors that limit its productivity. Developing more resilient and productive cassava varieties through conventional breeding is time-consuming. Therefore, having a complete haplotype-resolved genome with high accuracy can be a valuable resource in cassava breeding and genomics.
Cassava has a haploid genome of about 750 Mbp, a highly heterozygous and repetitive plant genome8. Despite the use of various sequencing technologies over time, there are unresolved gaps in the genome. The reference genome AM560-2, derived from a Colombian cassava line MCol505, has undergone steady improvement over a decade and has had five major releases, with the current version being AM560-2 version 84. While this reference genome benefits the cassava community, it does not capture the genetic diversity in African cassava cultivars grown by smallholder farmers due to its homozygous nature. Recently, attempts have been made to assemble genomes of African cassava lines such as TME3 and 60444, using a combination of Illumina short reads, PacBio long reads, bio-nano optical mapping, and chromatin conformation capture (Hi-C) sequence technologies producing assemblies of N50 of 98 and 117 Kbp. The assembled genomes had large contiguous assemblies but lacked haplotypic separation, containing multiple copies of duplicated sequences in the primary assembly9. The TME7 genome was assembled using a combination of Illumina, PacBio, and Hi-C sequencing technology to generate a contiguous genome assembly of N50 of approximately 320 Kbp. This genotype was successfully deduplicated and phased using Hi-C sequence data10. The most recent African cassava genotype to be assembled is TME204, which was phased using Hi-C technologies and PacBio high-fidelity (HiFi) sequencing reads, resulting in a highly contiguous assembly of N50 > 18 Mbp11. PacBio HiFi sequencing technology has proven effective in creating long and highly accurate reads for assembling complex genomes12. Recent studies demonstrate its potential in assembling high-quality plant genomes, including Populus tomentosa Carr, the 35.6 Gb California redwood genome and Bletilla striata13,14,15.
In this study, we have generated a haplotype-resolved diploid assembly of TMEB117, a farmer-preferred cassava cultivar, using PacBio HiFi reads. TMEB117 (also called TME117, TME 117, and ISUNIKANKIYAN) is a Nigerian cassava landrace highly susceptible to African cassava mosaic virus (ACMV)16. This genotype served as a reference for ACMV studies16 and a high-resolution genome will pave the way for future investigations in epigenetics and small RNA expression analysis to learn more about the mechanisms of ACMV resistance in cassava to support future breeding programs. The TMEB117 hap1 assembled genome had a total size of 694 Mbp and hap2 665 Mbp with a contig N50 length of 18 Mbp (hap1) and 17 Mbp (hap2) (Table 1). These assembled haplotigs were further ordered and scaffolded using the TME204 reference genome to produce a chromosome-scaled genome for TMEB117 and enhanced contiguity with an improved N50 length exceeding 35 Mbp in both haplotypes (Table 1). The haplotype-specific annotations for TMEB117 hap1 and hap2 genomes resulted in 47,138 and 49,163 gene models, respectively. Within the TMEB117 hap 1 genome, a total of 442 Mbp (65.34%) was occupied by repetitive elements, whereas hap2, 408 Mbp (60.32%), encompassed by transposable elements. Evaluation of the final genome exhibited a high completeness of 98.9%, according to BUSCO17. The two haplotype genomes attained a high base-level accuracy of QV > 64. Furthermore, in the raw data, we detected reads that closely matched the entire genome of the fungus Alternaria alternata, despite the plants being healthy and showing no symptoms. We eliminated these contaminant reads and excluded them from the final assembly. The phased and annotated homologous chromosomes provide a comprehensive perspective of cassava’s heterozygous genome organization with improved accuracy and completeness at a haplotype-resolved level (Fig. 1a). These chromosome pairs are anticipated to be a valuable resource for cassava breeders and essential for functional analysis to characterize molecular mechanisms important agronomical.

Overview of the cassava cultivar TMEB117 genome. (a) Circos plot displays repeat and gene densities for the two haplotypes visualized in 1 Mbp sliding windows. The tracks from the outer to inner show, (i) Repeat density for hap1 genome (ii) Gene density for hap1 genome (iii) Repeat density for hap2 genome (iv) Gene density for hap2 genome. (b) Cassava plant in a pot from the screen house. (c) BUSCO score of the TMEB117 genome.
Methods
Sampling, sequencing, sequence quality and contamination check
Cassava plants of the TMEB117 genotype, obtained from the International Institute of Tropical Agriculture (IITA) Genebank collection18, were grown in pots in a screen house (Fig. 1b). Third and fourth fully-expanded leaves of a potted plantlet hardened from in vitro culture were used for genomic DNA extraction using an optimized version of the CTAB (2% CTAB, 2% PVP-40, 20 mM Tris-HCl, pH 8.0, 1.4 M NaCl, 20 mM ethylenediaminetetraacetic acid) total nucleic acid extraction protocol as described by Carluccio, A. V. et al.19. After RNase treatment, the resulting DNA was cleaned using the Genomic DNA clean and concentrator kit (Zymo Research) according to the manufacturer’s instructions. We improved the gene annotation step by utilizing RNA data from another project, previously extracted from leaves of approximately two-month-old greenhouse plants at IITA Ibadan. The extraction process employed a combination of CTAB and spin column-based purification methods. The total RNA was then sequenced using the Illumina HiSeq. 2500 with a paired-end 2 × 100 cycle approach. The total DNA sample was sequenced with PacBio technology (PacBio Sequel II platform) using two SMRT cells. In the first cell, 875,686 reads and 1,163,062 reads in the second cell were generated. The raw sequence reads obtained from the two SMRT cells were combined. Fastp version 0.23.120 (parameters: --length_required 10000 --length_limit 30000) was used to filter the raw sequencing reads, with acceptable read lengths between 10 Kbp to 30 Kbp. Adapter-contaminated reads were removed using HiFiAdapterFilt21 with the default setting. Filtering and adapter removal resulted in 2,029,912 retained out of 2,038,748 raw reads (99.57%). The quality of the remaining reads was assessed with the default setting of FastQC version 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and the GC content graphs from the FastQC outputs were further investigated. GC content showed three peaks (Fig. S1a) (see figure deposited at Figshare)22. It is generally anticipated that the distribution of GC content follows a normal distribution close to the theoretical distribution. However, the peaks deviate from the theoretical normal distribution in this case. The first peak at ~35% and the second peak at 44.5% mean GC contents represent the GC content of nuclear and mitochondrial genomes, respectively23,24. Since the GC % of the chloroplast genome is similar to the nuclear genome, the third peak at ~51% mean GC content could be explained by contamination. Pbbm2 version 1.10.0 (https://github.com/PacificBiosciences/pbmm2), an SMRT minimap2 version 2.1525 (parameters:–unmapped–log-level INFO–log-file) wrapper was used to then map the raw reads to the cassava reference genome AM560-2 version 826. BLAST search was conducted on unmapped reads to detect the presence of contaminants. BLAST results indicated that the third peak of the GC content originates from reads of A. alternata27. We further mapped the filtered reads to the A. alternata genome. A total 58,703 reads (2.89%) mapped with a consensus of 23.7 Mbp, roughly 69% of the A. alternata genome of 34.38 Mbp. The GC content of the A. alternata genome is 51%, confirming the third peak of the GC content plot. We extracted all the reads mapping to the A. alternata genome, resulting in 1,971,209 sequence reads with GC content having two peaks in the GC content plot, which we process as clean reads for the assembly (Fig. S1b) (see figure deposited at Figshare)22. Extracting mapped and unmapped reads was done using samtools v1.15.1 (parameters: samtools fastq -F 4 & -f 4)28.
Genome assembly, scaffolding, and assembly quality check
Clean HiFi sequence reads of 45x estimated coverage with read length N50 of 17,513 bp (Table S1) (see table deposited in Figshare)29 were assembled de novo using hifiasm v0.16.1-r375 default settings, HiCanu v2.3 (parameters:-p out -pacbio-hifi genomeSize = 750 m -useGrid = false -merylThreads = 4 -merylMemory = 8 corOverlapper = ovl), and Flye v2.9.1-b1780 (parameters: --pacbio-hifi -o out --genome-size 750 m) assembly tools13,30,31. Utilizing outcomes from benchmarking analysis of these tools11 and outputs of these three assemblies, we opted to use the hifiasm assembly based on contiguity and achievement of haplotype resolved assemblies. Assembly statistics were compiled using QUAST32 default setting. The assembly comprised two haplotigs, hap1 and hap2. Before scaffolding, the two haplotigs assembly metrics showed hap1 and hap2 to consist of 362 contigs with a total length of 694 Mbp with a contig N50 length of 18 Mbp and 159 contigs with a total size of 665 Mbp with contig N50 length of 17 Mbp, respectively (Table 1). Scaffolding and ordering of the contigs were improved by RagTag33 (parameter: scaffold -o out -t 12) using TME204 reference, a chromosome-scaled genome11. The contiguity of the TMEB117 chromosome-scaled genome was improved, with hap1 N50 of 37 Mbp and hap2 N50 of 35 Mbp (Table 1). Eighteen pseudo-molecules representing the chromosomes were compiled for further annotation analysis. The unplaced contigs were separated from the sequences of chromosomes. These unplaced contigs of both haplotypes were then mapped to the chloroplast and mitochondrial genomes23,24. The alignment revealed a complete 100% coverage to the chloroplast genome and 90.39% and 62.63% coverage for hap1 and hap2, respectively, to the mitochondrial genome. These haplotigs provided representative sequences for the mitochondrial and chloroplast genome. The final scaffolded assembly of TMEB117 eighteen chromosomes was utilized for downstream analysis.
Repeat landscape and gene annotation
The annotation of transposable elements (TE) was accomplished by using the Extensive de novo TE Annotator (EDTA)34 (parameters:–genome–overwrite 1–sensitive 1–anno 1–evaluate 1), combining structure and homology-based detection to identify predominant TEs in the assembled genome. The pipeline applies various tools, such as HelotronScanner, LTR_FINDER, LTRHarvest, LTR_retriever, TIR-Learner, RepeatModeler2, and RepeatMasker35,36,37,38,39,40,41, to classify novel TE sequences. We screened the outputs of EDTA using R and tidyverse package, resulting in non-redundant TE annotations and visualizations for both haplotypes. The generated repeat-masked genome was subsequently used for gene prediction. Consistent with other African cassava genomes10,11, over 50% of the genome constitutes repetitive elements. Specifically, in this study, 65.34% and 60.32% of the genome in hap1 and hap2 are transposable elements. The long terminal repeats – retrotransposons (LTR-RTs) are the most abundant, covering 57.37% (hap1) and 54.42% (hap2) of the genome size (Fig. 2a,b). Gypsy was the most abundant retrotransposons superfamily, occupying 41.14% (hap1) and 38.35% (hap2) of the genome (Table S2a,b) (see tables deposited at Figshare)29. The annotations are classified as families and superfamilies. Between the two haplotype genomes, there is a minimal difference in transposable element annotation percentage (Fig. 2a,b). However, the distribution of the TEs across the chromosomes differs between the two haplotypes (Fig. 2c,d). We used the Funannotate v1.8.9 singularity pipeline (see the script in the code availability section) to annotate the TMEB117 genome. The annotation pipeline involves three primary steps: genome masking, gene prediction, and functional annotation. Prior to annotation, the genome assembly as an output of EDTA annotation underwent soft-masked using scripts provided by the EDTA tool make_masked.pl. PASA alignment tool42 was used to generate an initial set of gene models by integrating RNA-seq data and protein homology to improve the accuracy of gene models. We used a set of 568,002 reviewed and curated protein sequences from a diverse array of species found in the UniProtKB/Swiss-Prot database release 2022_03 for the gene prediction step. Gene prediction was conducted using ab initio gene prediction tools, Augustus v3.3, SNAP v2013-02-16, and GlimmerHMM v3.0.443,44 were employed for gene prediction. EVidenceModeler v1.1.145 integrated gene models from various gene predictors and generated a consensus of the gene models. These gene models were used to generate protein sequences, which underwent filtering to remove proteins with less than 50 amino acids and to check for homology to transposable elements. The predicted genes were functionally annotated using the EggNOG database, UniProtKB, MEROPS, and CAZYmes46,47,48,49, giving insights into the biological functions and pathways. The resulting annotations were manually curated to correct errors and adjust gene models as necessary using the Funnannotate interface. Non-overlapping tRNA genes were predicted using tRNAscan-SE v2.0.950. Transcript evidence was generated by Trinity v2.11.051 through de novo transcript assembly, which was used in correcting, enhancing, and updating the predicted gene models. The haplotype-specific annotations for hap1 and hap2 genomes resulted in 47,138 and 49,163 genes, 53,264 and 55,222 transcripts, 836 and 814 tRNA, and 52,428 and 54,408 proteins, respectively. BUSCO analysis reveals 90% protein sequence completeness for both haplotypes.

Illustrate the proportion and distribution of TEs across the chromosomes, as annotated by EDTA. (a) Shows the proportion of TEs identified in the hap1 genome, with the most abundant type LTR-RT (represented in the blue segment in the pie chart), covering 57.37% of the genome. (b) In hap2, LTR-RT remains the predominant TE family covering 55.07% of the genome. (c) Provides an overview of the distribution of TE families across all cassava chromosomes. (d) Slight difference in the distribution of TEs families annotated in the chromosomes of the hap2 genome compared to the hap1 genome.
Orthologue analysis
The predicted protein sequences from the study were further analyzed by comparing them to other phased African cassava genomes, namely TMEB7 and TME204, with the AM560-2 v8.1 genome. Each haplotype was analyzed separately with the AM560-2 v8.1 genome. The OrthoVenn2 online tool (https://orthovenn2.bioinfotoolkits.net/) was used to identify orthologous protein groups across the genomes. The analysis was conducted with default parameters, including an inflation value of 1.5 for the Markov clustering algorithm and a BLASTP e-value of 1e-2. The resulting orthologous groups were visualized using the OrthoVenn2 web interface, which displayed Venn diagrams that indicate the number of unique and shared groups across the genomes. The OrthoVenn2 analysis identified 37,384 and 37,518 orthologous clusters, with 18,770 and 19,588 core genome orthologs for hap1 and hap2, respectively, indicating the presence of conserved groups across the genomes. In the TMEB117 hap1 and hap2 genomes, we observe fewer unique protein sequences (931 and 1042) than other cassava genomes, as shown in Fig. 3a,b.

Venn diagram of the number of gene families shared among and unique to the haplotype genomes of three African cassava cultivars: TMEB117 (hap1 and hap2), TME204 (hap1 and hap2), TME7 (hap1 and hap2), in comparison to the reference genome AM560-2 v8. (a) 18,770 core gene families shared among the first haplotigs comparison with the reference genome AM560-2 v8. The second comparison (b)19,588 core genes on the second haplotig comparison with the reference genome AM560-2 v8. 931 gene families were unique in TMEB117 hap1 genome and 1042 in the hap2 genome.
Data Records
Raw PacBio Hifi reads utilized for the assembly can be accessed from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under BioProject PRJNA1002255, with accession numbers SRR2551717652 and SRR2551717553. The two chromosome-scaled haploid genomes have been submitted under distinct BioProject identifiers within NCBI, PRJNA1002865 for hap1 and PRJNA1002864 for hap2 with accession numbers JAWPHJ00000000054 and JAWPHK00000000055, respectively. The transcriptome data employed to annotate the genomes are available at NCBI with accession numbers SRR2553733956, SRR2553734057, and SRR2553733858. The genome annotation files are uploaded to Zenodo59.
Technical Validation
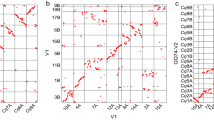
The genome was validated by ensuring that the constructed assembly conforms with the data used to generate it. Over 99% of the raw sequence reads mapped to both the haplotigs. To evaluate the completeness, we used BUSCO v5.3.2 (parameters: -m genome eudicots_odb10) with orthologs from the eudicots lineage datasets, which included 2326 reference sets of genes specific to plants. BUSCO completeness score for the hap1 and hap2 assemblies was 98.9% (Fig. 1c). We employed Blobtools60 using the default setting to ascertain the absence of contamination by blasting the nucleotide database against the assembly and mapping the coverage of the assembly using HiFi reads to generate blobs. Blobs in the blob plot (Fig. S2) (see figure deposited at Figshare)22 were plotted at expected GC content percentages consistent with the GC plot after contamination removal. BLAST hits from the blob plot showed Streptophyta, which is a clade of plants. Therefore, we conclude that the assembly was free from contaminants. Haplotypic separation and assembly quality were achieved by performing a k-mer-based analysis using Merqury61 (k-mer = 21). The assembly has a quality value (QV) score of 64.38 for hap1 and 67.99 for hap2. The k-mer completeness for each haplotype assembly and the combined set was 78.63%, 77.95%, and 98.79%, respectively. This was approximately 20% of k-mers being haplotype-specific. So far, this genome is of better quality assembly, based on the QV score, compared to already assembled African cassava cultivars10,11 (Table 2). Figure 4 illustrates that the assembled sequence resulted in a nearly completely haplotype-resolved genome, as indicated by the copy number and assembly k-mer plots. Most heterozygous haplotype-specific k-mers were observed once in the assembled sequence, and the majority of homozygous k-mers were shared by the two genome haplotypes (Fig. 4a). In the heterozygous peak, slightly fewer k-mers differed between the two haplotypes (Fig. 4b), confirming that the reconstruction of heterozygous variants was almost thorough. We assessed the completeness of the predicted protein sequences within the eudicots lineage using BUSCO v5.3.2 (parameters: -m proteins eudicots_odb10) to validate the gene annotations. BUSCO analysis reveals 90% protein sequence completeness for both haplotypes (Fig. 1c). Subsequently, we performed a conditional reciprocal BLAST62, extracting the predicted gene model sequences from both haplotypes and compared them to the gene sequences of AM560-2, the cassava reference genome. Out of the predicted 47,138 genes in hap1 and 49,163 genes in hap2, we identified 30,456 in hap1 and 30,370 in hap2 reference gene sequences. Particularly, the reference AM560-2 genome, with a total of 32,805 genes, exhibited similarity to most predicted genes of both haplotypes of the TMEB117 genome.

The completeness of resolved haplotypes assessed by Merqury copy number spectrum plots (a) and assembly plots (b). The x-axis represents the k-mer multiplicity, while the y-axis shows the abundance of k-mers. The grey region. represents the abundance of k-mers in the HiFi reads missing in the scaffold of the genome. (a) Copy number spectrum plot - the red peak observed at ~ 25x indicates heterozygous k-mers (1-copy k-mers), while the blue peak at ~ 50x represents the homozygous k-mers (2-copy k-mers). The other peaks show low levels of duplicated k-mers. (b) Assembly plot – k-mers coloured by their uniqueness, red peak (hap1), blue peak (hap2). At the heterozygous peak (25x), there is a slight difference in the k-mers indicating reconstruction of heterozygous variants was almost complete. Shared k-mers are shown in green which is at the 50x k-mer multiplicity.
Code availability
No custom programming or coding was used. Instead, the analysis utilized bash commands and the corresponding scripts stored within the GitHub repository accessible at: https://github.com/LandiMi2/GenomeAssemblyTMEB117.
References
Wang, P. et al. The genome evolution and domestication of tropical fruit mango. Genome Biol 21 (2020).
Tang, C. et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat Plants 2 (2016).
Bredeson, J. V. et al. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat Biotechnol 34, 562–570 (2016).
Lyons, J. B. et al. Current status and impending progress for cassava structural genomics. Plant Molecular Biology vol. 109, 177–191, https://doi.org/10.1007/s11103-020-01104-w (2022).
Li, S. et al. The industrial applications of cassava: current status, opportunities and prospects. Journal of the Science of Food and Agriculture 97, 2282–2290, https://doi.org/10.1002/jsfa.8287 (2017).
Ceballos, H. H., Iglesias, C. A., Pe´rezpe´rez, J. C. & Dixon, A. G. O. Cassava breeding: opportunities and challenges.
Uchechukwu-Agua, A. D., Caleb, O. J. & Opara, U. L. Postharvest Handling and Storage of Fresh Cassava Root and Products: a Review. Food and Bioprocess Technology 8, 729–748, https://doi.org/10.1007/s11947-015-1478-z (2015).
Prochnik, S. et al. The Cassava Genome: Current Progress, Future Directions. Tropical Plant Biology 5, 88–94, https://doi.org/10.1007/s12042-011-9088-z (2012).
Kuon, J. E. et al. Haplotype-resolved genomes of geminivirus-resistant and geminivirus-susceptible African cassava cultivars. BMC Biol 17, 1–15 (2019).
Mansfeld, B. N. et al. Large structural variations in the haplotype-resolved African cassava genome. Plant Journal 108, 1830–1848 (2021).
Qi, W. et al. The haplotype-resolved chromosome pairs of a heterozygous diploid African cassava cultivar reveal novel pan-genome and allele-specific transcriptome features. Gigascience 11 (2022).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data 7 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
An, X. et al. High quality haplotype-resolved genome assemblies of Populus tomentosa Carr., a stabilized interspecific hybrid species widespread in Asia. Mol Ecol Resour 22, 786–802 (2022).
Jiang, L. et al. Haplotype-resolved genome assembly of Bletilla striata (Thunb.) Reichb.f. to elucidate medicinal value. Plant Journal 111, 1340–1353 (2022).
Quantification of African cassava mosaic virus (ACMV) and East African cassava mosaic virus (EACMV-UG) in single and mixed infected Cassava (Manihot esculenta Crantz) using quantitative PCR - 1-s2.0-S0166093415003262-main.
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Paliwal, R., Adegboyega, T. T., Abberton, M., Faloye, B. & Oyatomi, O. Potential of genomics for the improvement of underutilized legumes in sub-Saharan Africa. Legume Science 3, https://doi.org/10.1002/leg3.69 (2021).
Carluccio, A. V. et al. Set up from the beginning: The origin and early development of cassava storage roots. Plant Cell Environ 45, 1779–1795 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. in Bioinformatics vol. 34 i884–i890 (Oxford University Press, 2018).
Sim, S. B., Corpuz, R. L., Simmonds, T. J. & Geib, S. M. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics 23 (2022).
Landi, M. Supplementary figures cassava TMEB117 genome. Figshare. https://doi.org/10.6084/m9.figshare.23792292.v2 (2023).
Tao, Q., Cao, J., Zhu, L. & Lin, H. The complete mitochondrial genome of an important root crop cassava (Manihot esculenta). Mitochondrial DNA B Resour 4, 1081–1082 (2019).
Daniell, H. et al. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theoretical and Applied Genetics 116, 723–737 (2008).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Bredeson, J. V. et al. ‘An improved reference assembly for cassava (Manihot esculenta Crantz)’. In preparation.
Gai, Y. et al. Chromosome-scale genome sequence of Alternaria alternata causing alternaria brown spot of citrus. Molecular Plant-Microbe Interactions 34 (2021).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Landi, M. Supplementary tables. Figshare. https://doi.org/10.6084/m9.figshare.23792298.v1 (2023).
Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37, 540–546 (2019).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol 23, (2022).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, (2019).
Xiong, W., He, L., Lai, J., Dooner, H. K. & Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc Natl Acad Sci USA 111, 10263–10268 (2014).
Ou, S. & Jiang, N. LTR_FINDER_parallel: Parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob DNA 10 (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, (2008).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Su, W., Gu, X. & Peterson, T. TIR-Learner, a New Ensemble Method for TIR Transposable Element Annotation, Provides Evidence for Abundant New Transposable Elements in the Maize Genome. Mol Plant 12, 447–460 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Korf, I. Gene finding in novel genomes. http://www.biomedcentral.com/1471-2105/5/59 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9 (2008).
Drula, E. et al. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res 50, D571–D577 (2022).
Huerta-Cepas, J. et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46, D624–D632 (2018).
Bateman, A. et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489 (2021).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. in Methods in Molecular Biology 1962, 1–14 (Humana Press Inc., 2019).
Haas, B. J. et al. De novo transcript sequence recostruction from RNA-Seq: reference generation and analysis with Trinity. Nature protocols 8 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25517176 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25517175 (2023).
Landi, M. et al. The genome information of African cassava cultivar TMEB117 genome (Hap1). GenBank. https://identifiers.org/ncbi/insdc:JAWPHJ000000000 (2023).
Landi, M. et al. The genome information of African cassava cultivar TMEB117 genome (Hap2). GenBank. https://identifiers.org/ncbi/insdc:JAWPHK000000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25537339 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25537340 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25537338 (2023).
Landi, M. et al. Genome annotation of African cassava cultivar TMEB117 genome. Zenodo. https://zenodo.org/doi/10.5281/zenodo.10013084 (2023).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit - interactive quality assessment of genome assemblies. G3: Genes, Genomes, Genetics 10, 1361–1374 (2020).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21 (2020).
Aubry, S., Kelly, S., Kümpers, B. M. C., Smith-Unna, R. D. & Hibberd, J. M. Deep Evolutionary Comparison of Gene Expression Identifies Parallel Recruitment of Trans-Factors in Two Independent Origins of C4 Photosynthesis. PLoS Genet 10 (2014).
Acknowledgements
This work was funded by the Swedish Research Council (VR) funded projects, EpiCass (Identification of epigenetic variations influencing viral resistance and yield in cassava (Manihot esculenta) farmers’ fields) grant number 2020-04457 and Cassavanet4Dev (In a race to prevent hunger: C assava Bioinformatics for African cassava biotechnology) grant number 2021-05105. This study was supported by the SLU Bioinformatics Infrastructure (SLUBI), Scilifelab, NGI, and NBIS Uppsala, Sweden. We thank the Swedish University of Agricultural Sciences (SLU), Uppsala, Sweden, and the International Institute of Tropical Agriculture (IITA) for providing research facilities and other support for this study. ML acknowledges using the High-Performance Computing services of the International Institute of Tropical Agriculture (IITA) and the International Livestock Research Institute (ILRI).
Author information
Authors and Affiliations
Contributions
A.G. and E.B.R. conceived the idea and led the grant proposal writing. L.S. handled the preparation of the plant materials and sequencing. ML analyzed the data and wrote the original draft of the manuscript. T.S., L.F. and A.N. provided oversight throughout the process. The final manuscript underwent review and approval by all authors. Correspondence and requests for materials should be addressed to M.L. and A.G.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Landi, M., Shah, T., Falquet, L. et al. Haplotype-resolved genome of heterozygous African cassava cultivar TMEB117 (Manihot esculenta). Sci Data 10, 887 (2023). https://doi.org/10.1038/s41597-023-02800-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02800-0
