
Abstract
The Network for Pancreatic Organ donors with Diabetes (nPOD) is the largest biorepository of human pancreata and associated immune organs from donors with type 1 diabetes (T1D), maturity-onset diabetes of the young (MODY), cystic fibrosis-related diabetes (CFRD), type 2 diabetes (T2D), gestational diabetes, islet autoantibody positivity (AAb+), and without diabetes. nPOD recovers, processes, analyzes, and distributes high-quality biospecimens, collected using optimized standard operating procedures, and associated de-identified data/metadata to researchers around the world. Herein describes the release of high-parameter genotyping data from this collection. 372 donors were genotyped using a custom precision medicine single nucleotide polymorphism (SNP) microarray. Data were technically validated using published algorithms to evaluate donor relatedness, ancestry, imputed HLA, and T1D genetic risk score. Additionally, 207 donors were assessed for rare known and novel coding region variants via whole exome sequencing (WES). These data are publicly-available to enable genotype-specific sample requests and the study of novel genotype:phenotype associations, aiding in the mission of nPOD to enhance understanding of diabetes pathogenesis to promote the development of novel therapies.
Similar content being viewed by others


Background & Summary
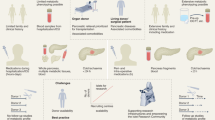
Genetic predisposition to risk for or protection from type 1 diabetes (T1D) is highly polygenic, with the total possible set of disease-associated variants yet to be fully defined1. Genome-wide association studies (GWAS) have identified population-level risk loci (minor allele frequency (MAF) >1%), dominated by Human Leukocyte Antigen (HLA) class II and insulin, and accompanied by 77 additional regions, which in total cover over 3,600 predicted causal moderate effect size variants (odds ratio (OR) <2) associated with genes thought to impact leukocyte and pancreatic β-cell function2,3. While a combination of many such population-level variants may contribute to the development of “classical” T1D2,3 and latent autoimmune diabetes in adults (LADA)4, we are beginning to appreciate that rare (MAF ≤1%) larger effect size (OR ≥2) variants may explain the “missing heritability” of autoimmune diabetes5 (Fig. 1a). In support of this notion, rare variants with large effect size are associated with monogenic autoimmune forms of diabetes including immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), signal transducer and activator of transcription 3 (STAT3)-, and cytotoxic T-lymphocyte protein 4 (CTLA4)-associated diabetes6, in addition to non-autoimmune forms of diabetes such as maturity-onset diabetes of the young (MODY) and cystic fibrosis-related diabetes (CFRD)7.

Complementary methods for detecting genetic variants associated with T1D. (a) The effect size of any given variant on T1D risk is inversely related to the frequency of the variant5. (b) To detect more commonly observed variants, DNA samples from nPOD donors were probed for the presence of SNPs previously reported in T1D GWAS2 efforts using the UFDIchip, yielding CEL microarray image files (modified from Affymetrix Axiom website: https://www.affymetrix.com/products_services/arrays/specific/axiom_mydesign.affx). (c) To detect rare or novel variants, whole exome sequencing (WES) was performed on DNA from nPOD donors (modified from Roche NimbleGen SeqCap EZ Exome Library workflow68). Diagrams created in BioRender.
The Network for Pancreatic Organ donors with Diabetes (nPOD)8, founded in 2007, has become the largest biorepository of human pancreata, pancreatic lymph nodes, and spleen from organ donors with T1D, MODY, CFRD, T2D, gestational diabetes, non-diabetic islet autoantibody positive (AAb+) donors, and non-diabetic autoantibody-negative (AAb-) control donors9. nPOD provides worldwide distribution of biospecimens to researchers working to elucidate T1D pathogenesis in order to promote the development of new strategies for prevention and treatment. To date (September 2022), nPOD has supplied biosamples to > 280 independent research projects studying β-cell physiology, β-cell differentiation, immunology, T1D biomarkers, technology development, T1D pathology, and diabetes etiology (https://www.jdrfnpod.org/publications/current-npod-projects/, accessed October 21, 2021). A major goal of nPOD, in addition to biosample distribution, is the sharing of de-identified donor data from multiple core laboratories to facilitate discovery efforts. The nPOD Data Portal provides approved investigators with access to donor clinical and demographic information, serum HbA1c and C-peptide levels, islet AAb status (insulin, glutamic acid decarboxylase [GAD], insulinoma-associated antigen-2 [IA-2], zinc transporter 8 [ZnT8])10, pancreas weights, and histopathology reviews8,11 (https://portal.jdrfnpod.org/, accessed October 21, 2022). Whole slide scans from hematoxylin and eosin-stained (H&E) sections are available for online viewing via the Online Pathology portal (https://aperioeslide.ahc.ufl.edu/, accessed October 21, 2022) for access to cross-sectional pancreas morphology as well as multiplex immunohistochemistry (IHC)-stained sections for insulin, glucagon, somatostatin, and pancreatic polypeptide (PP) to visualize endocrine β-, α-, δ-, and PP cells, respectively. Multiplex IHC staining panels are also available for markers including, but not limited to, Ki67, CD3, insulin, and/or glucagon for quantification of cell proliferation and immune cell infiltration in pancreatic endocrine and exocrine compartments. Histopathology reports summarizing blinded assessment of H&E- and IHC-stained sections from pancreas and other available organs are provided to detail islet parameters and major abnormalities. In terms of genetic data, the standard operating procedures (SOPs) for nPOD donors were previously limited to the collection of high-resolution four-digit HLA genotypes8,12. The availability of additional high-parameter genotyping data has therefore been a high priority that is now realized with the data release described herein.
Our approach for characterizing nPOD donor genetics was twofold: donors were genotyped with 1) the University of Florida Diabetes Institute (UFDI) custom single nucleotide polymorphism (SNP) microarray (UFDIchip)13 and 2) the University of California San Francisco (UCSF) standardized whole exome sequencing (WES) pipeline14. Specifically, nPOD cases (N = 372)— comprised of AAb- no diabetes controls (N = 147), AAb + without T1D (N = 26), T1D (N = 111), T1D medalists15,16 (N = 2), T1D recipients of pancreas transplant (N = 5), type 2 diabetes (T2D, N = 38), gastric bypass (N = 2), gestational diabetes (N = 4), monogenic diabetes (N = 4), cystic fibrosis (CF, N = 5), other diabetes (N = 12), other no diabetes (N = 12), and pregnant without diabetes (N = 4)— were genotyped using the UFDIchip13 custom AxiomTM array (Fig. 1b). All nPOD donors with available DNA or tissue were evaluated for population-level variants via UFDIchip. We prioritized the selection of T1D, AAb + without T1D, gestational diabetes, monogenic diabetes, and other diabetes donors in addition to including a few no diabetes donors as controls for WES-based characterization of rare diabetes-associated variants that may not have been powered for detection by previous GWAS studies. Specifically, nPOD donors (N = 207)— including AAb- no diabetes controls (N = 13), AAb + without T1D (N = 34), T1D (N = 135), T1D recipients of pancreas transplant (N = 6), gestational diabetes (N = 4), monogenic diabetes (N = 4), and other diabetes (N = 11)— were queried for rare known and novel coding region variants in autoimmune and MODY-associated genes via WES14 (Fig. 1c). Data emanating from these assays were used to provide individual genotypes, infer relatedness17 and genetic ancestry18, impute HLA19, and calculate a combined T1D genetic risk score (GRS)20,21,22 per donor.
These genotyping data have been generated and made accessible to enable genotype-selected sample requests and the study of novel genotype:phenotype associations by the international community of nPOD investigators. We anticipate that the diversity of nPOD donor genetics may be partly responsible for inter-donor heterogeneity observed in islet health, insulitis composition, age at T1D onset, islet AAb status, and other endotype-related characteristics23,24,25. Importantly, beyond explaining diabetes heterogeneity, the findings facilitated by these data are expected to inform precision medicine strategies for prevention or suspension of the pathogenesis of T1D as well as other forms of diabetes.
Methods
Donor tissues
Transplant-quality organs, including pancreas and up to 13 other tissues, were recovered from cadaveric organ donors by United States (U.S.) organ procurement organizations (OPOs, http://www.jdrfnpod.org//for-partners/npod-partners/, accessed October 15, 2021) in accordance with federal guidelines, then processed by the nPOD Organ Processing and Pathology Core (OPPC) according to University of Florida (UF) Institutional Review Board (IRB) approved protocol IRB201600029, as previously described8,11. Studies conducted using organ donor tissue samples from the nPOD biobank are classified as minimum risk research, as study participants are no longer living. However, informed consent for research participation is obtained from family members via both written and verbal communication prior to organ donation, with the consent processes undertaken by qualified personnel affiliated with the U.S. OPO network. All subject information is de-identified in accordance with HIPAA regulations. For each donor, clinical and demographic information, were obtained via medical chart review and OPO-conducted interview with the donor’s family. High-resolution four-digit HLA typing was performed by Next Generation Sequencing (NGS) as previously described8,12 at the Barbara Davis Center for Childhood Diabetes HLA Core (University of Colorado Anschutz Medical Campus). nPOD donors were categorized by diabetes type, verified by UF endocrinologist review of the de-identified terminal medical records (including diagnosis and duration of diabetes, history or clinical data for diabetic ketoacidosis, medications, and BMI), donor metadata (e.g., age, sex, reported race and ethnicity), and additional data (serum C-peptide levels, islet AAb status10, hemoglobin A1c [HbA1c], and high-resolution HLA8,12). Unique research resource identifiers (RRIDs) were assigned to each organ donor, in order to facilitate the provenance and reproducibility of results26.
DNA isolation
DNA was extracted from frozen spleen or, for a limited number of cases in which spleen was unavailable, frozen pancreas, pancreatic lymph node, or small intestine were used. DNA isolation was performed using the Qiagen DNeasy Blood and Tissue DNA isolation kit according to the manufacturer’s instructions. Purity and concentration of extracted DNA were assessed with the Epoch Microplate Spectrophotometer (BioTek).
UFDIchip design
372 nPOD donors (Table 1, Phenotype_data.txt27) were genotyped at 985,971 unique loci on a custom SNP array termed the UFDIchip13 (Fig. 1b). The base array is the AxiomTM Precision Medicine Research Array (Thermo Fisher Scientific), to which all content from the ImmunoChip v228 was added, as well as all previously reported credible T1D risk variants3 (Fig. 2, UFDIchip_library_file.xlsx27). The array also includes dense coverage of the highly polymorphic HLA region, which allows for accurate imputation of HLA haplotypes to 4-digit resolution.

University of Florida Diabetes Institute (UFDI) chip design. The UFDIchip contains probes from the following modules of the AxiomTM Precision Medicine Research Array (PMRA, Thermo Fisher Scientific): imputation genome-wide association study (GWAS) grid, human leukocyte antigens (HLA), killer cell immunoglobulin-like receptors (KIR), expression quantitative trait loci (eQTL), and fingerprinting/sample tracking. Custom additions to the UFDchip included single nucleotide polymorphisms (SNPs) from the following categories: ImmunoChip v2, credible type 1 diabetes (T1D) causal variants3, and investigator-initiated markers of interest.
Genotype processing and analysis
UFDIchip plates were processed on an Affymetrix GeneTitan instrument with external sample handling on a BioMek FX dual arm robotic workstation. Axiom™ Analysis Suite software (v3.0, Thermo Fisher Scientific) was used to process raw CEL file data to plink text files. The software includes quality control (QC) procedures at the sample, plate, and SNP levels. These QC threshold parameters were set to Axiom™ Analysis Suite default stringency (“Best Practices Workflow” using “Human.legacy.v5” settings). Under these settings, samples were included in analysis if dish QC (DQC) ≥ 0.82 and if QC call rate ≥ 97%. Plates were considered acceptable for analysis if average QC call rate ≥ 98.5% for passing samples. Best probe set was identified per SNP, with the SNP call rate threshold set to 95%. A screen for discordance from reported sex via X chromosome heterozygosity was then performed using plink v1.929. All data passed these QC screens and raw CEL files and a binary plink file containing processed data (GRCh37/hg19) from all cases are stored in the database of Genotypes and Phenotypes (dbGaP)27. Subsequent analyses included relatedness estimation using KING30, genetic ancestry imputation using ADMIXTURE18, HLA imputation using AxiomTM HLA Analysis Software19, imputation to 300 M SNPs and indels using the Trans-Omics for Precision Medicine (TOPMed) reference cohort with the Michigan Imputation Server31, and calculation of a T1D GRS21,22.
Validation of technical replicates
DNA from 24 nPOD donors were run in duplicate on the UFDIchip. SNP call rates were compared between technical replicates using Bland-Altman analysis. Reproducibility of genotype calls between technical replicates were evaluated by kinship coefficient using KING30 (v2.1.2) software.
Relatedness
Genotyping data from the nPOD cohort with unknown and from the 1000 Genomes phase 3 cohort32 with known family relationships were merged and analyzed for genetic relatedness using KING30 (v2.1.2) software. The integrated relationship inference command was used to infer up to third-degree relatives. Relationships between nPOD case pairs and between 1000 Genomes pairs were represented by plotting estimated kinship coefficients. Kinship coefficients of unrelated 1000 Genomes pairs were randomly downsampled to the number of nPOD subject pairs to allow for data visualization.
Genetic ancestry
Data from unrelated subjects from the 1000 Genomes phase 3 cohort32 were filtered for SNPs that overlap with the UFDIchip array using plink v1.929. The data were pruned for linkage disequilibrium (LD) by removing SNPs with R2 > 0.1, screening within a 50 SNP block and proceeding by steps of 10 SNPs. This yielded 1000 Genomes genotypes for 320,005 SNPs, which were used to run an unsupervised analysis using ADMIXTURE software18 (v1.3.0) with k set to five populations. Each of the five groups represented a unique continental population from 1000 Genomes and as such, were assigned: 1) African (AFR), 2) Admixed American (AMR), 3) East Asian (EAS), 4) European (EUR), and 5) South Asian (SAS)32. The 372 nPOD cases were then projected onto the reference population to estimate ancestry proportions. Dimensionality reduction of the resulting Q-values (ancestry proportions) was performed using principal component analysis (PCA) to enable visualization.
HLA Imputation
AxiomTM HLA Analysis Software (v1.2.0.38)19 was used to impute 2-digit and 4-digit HLA genotypes, along with probability scores for the imputed calls. Concordance with nPOD HLA typing results8 was assessed at HLA-A, HLA-DRB1, HLA-DQA1, and HLA-DQB1. The typed result was considered ground truth when the imputed result was discordant.
Imputation accuracy for each of these loci [Acc(L)] was calculated as previously reported33, substituting the dosage for the probability score that is provided by AxiomTM HLA Analysis Software19:
where Pi is the probability for imputed alleles A1i,L and A2i,L for donor i at locus L. Imputed alleles were considered concordant when they were included in the donor’s set of typed alleles at locus L, and discordant when they were not in the set of typed alleles at locus L. For discordant alleles, Pi was set to 0. The summation of probabilities for the total number of donors assessed, n, was then divided by the total number of alleles tested, 2n. The accuracy score ranges from 0, for no concordant calls, to 1, for complete concordance with probabilities of 1 for all alleles.
Concordance was calculated at the 2-digit and 4-digit level for genotypes related to T1D risk or protection, as determined in primarily White cohorts34,35,36. These included HLA-A*02:01, HLA-A*24:02, HLA-DRB1*03:01 (DR3), HLA-DQA1*05:01–HLA-DQB1*02:01 (DQ2), HLA-DRB1*04:xx (DR4), HLA-DQA1*03:01–HLA-DQB1*03:02 (DQ8), HLA-DRB1*08:01 (DR8), HLA-DQA1*04:01–HLA-DQB1*04:02 (DQ4), HLA-DRB1*15:xx (DR15), HLA-DQA1*01:02–HLA-DQB1*06:02 (DQ6), and HLA-DQA1*03:01–HLA-DQB1*03:01 (DQ7), where xx is any sub-allele. The following formula was used:
where the number of imputed alleles A1i,L and A2i,L matching the genotype of interest for donor i at locus L was summed across all donors, n, and divided by the number of typed alleles A1t,L and A2t,L matching the genotype of interest for donor i at locus L summed across all donors. The accuracy score ranges from 0, for no concordant calls, to 1, for complete concordance.
Donor-level imputation accuracy [Acc(S)] was calculated as:
where Pj is the probability for imputed alleles A1j,S and A2j,S at each HLA locus j of donor S. Concordance was determined as described above, and Pj was set to 0 for discordant alleles. The total number of loci tested, n, was 4 per donor (HLA-A, HLA-DRB1, HLA-DQA1, and HLA-DQB1). The accuracy score ranges from 0, for no concordant calls, to 1, for complete concordance with probabilities of 1 for both alleles at each locus for donor S.
T1D GRS calculation
We computed polygenic T1D genetic risk scores, referred to as GRS121,22, GRS237, and African-Ancestry (AA)-GRS38. GRS1 is calculated using dosages of risk genotypes for 30 T1D-associated SNPs21. Genotypes were obtained by imputing to the TOPMed (v r2)31 panel (R2 > 0.97). rs2187668 was not found in TOPMed, thus, a suitable proxy SNP from GRS237, rs9273369, was used instead. The HLA component of GRS1 was calculated using the Polygenic Risk Score (PRS) Toolkit for HLA (v0.22a) developed by Sharp et al.37. The non-HLA component of GRS1 was then calculated via weighted sum, using odds ratios from Oram et al.21. The HLA and non-HLA scores were summed and normalized as described in Oram et al.21. GRS2 is calculated using dosages of risk genotypes for 67 T1D-associated SNPs37. Genotypes were obtained by imputing to the TOPMed (v r2)31 panel (R2 > 0.97). rs2476601, rs1281934, rs9273342, rs9271346, rs1233320, rs16822632, rs116522341, rs559242105, and rs371250843 were not found in TOPMed, thus, suitable proxy SNPs rs6679677, rs1281943, rs9273032, rs9271347, rs1233320, rs17840116, rs9268500, rs3129197, and rs9266268 were respectively used instead. The HLA component of GRS2 was calculated using the PRS Toolkit for HLA (v0.22a) developed by Sharp et al.37. The non-HLA component of GRS2 was then calculated via weighted sum, using odds ratios from Sharp et al.37 and added to the HLA component. AA-GRS is calculated using dosages of risk genotypes for 7 T1D-associated SNPs38. Genotypes were obtained by imputing to the TOPMed (v r2)31 panel (R2 > 0.96). rs2187668 and rs34303755 were not found in TOPMed; thus, suitable proxy SNPs rs9273369 and rs9268838 were respectively used instead. The AA-GRS was then calculated via weighted sum, using odds ratios from Onengut-Gumuscu et al.38.
WES
For 207 nPOD donors (Table 2, Phenotype_data.txt27), WES libraries were generated as previously described39 (Fig. 1c) using the Agilent SureSelect Human All Exon kit (Agilent Technologies, CA, USA). Procedures and quality control (QC) measures were performed following manufacturer’s recommendations. Briefly, 180–280 bp fragments were generated from genomic DNA by sonication (Covaris) with exonuclease and polymerase subsequently utilized to convert remaining overhangs into blunt ends. The DNA fragments were adenylated on the 3′ ends followed by ligation of adapter oligonucleotides. Successfully ligated DNA fragments were enriched by PCR. Following hybridization with biotin-labelled probes, exons were captured with streptavidin-coated magnetic beads. After a wash, probes were digested. Libraries were enriched and index tags added by PCR. Amplified exon libraries were purified using AMPure XP (Beckman Coulter), quantified by Agilent high sensitivity DNA kit using an Agilent Bioanalyzer 2100, then sequenced via Illumina Novaseq. 6000 (Illumina, CA, USA). Burrows-Wheeler Aligner (BWA, v0.7.17) was utilized to map the paired-end clean reads to the GRCh37/hg19 human reference genome40. Genome Analysis Toolkit (GATK, v4.1.2.0) was employed for SNP/InDel detection41. Annotate Variation (ANNOVAR, v20191024) was used for variant annotation42. Other variant annotations were performed using American College of Medical Genetics (ACMG) Classification43, Sorting Intolerant from Tolerant (SIFT) Function Prediction (SIFT4G)44, PolyPhen-2 Function Prediction (v 2.2.2)45, Combined Annotation Dependent Depletion (CADD, v1.6) Score46, Genome Aggregation Database (gnomAD, v2.1.1) frequency47, Human Gene Mutation Database (HGMD professional 2020.2)48, ClinVar (accessed August 31, 2020)49 and Centogene Mutation Database (CentoMD, v5.8)50. All data passed these QC screens and are stored in dbGaP27.
UFDIchip and WES comparison
For 167 nPOD donors, both UFDIchip- (Table 1, Phenotyp_data.txt27) and WES-based (Table 2, Phenotype_data.txt27) genotyping were performed. Biallelic autosomal variants detected in both assays and with at least one minor allele count (MAC) in the WES data were filtered using plink v1.929, resulting in 27,852 variants for comparison. Per-SNP intra-assay concordance levels were calculated across all subjects.
Data Records
UFDIchip array data are stored in dbGaP27 as raw CEL files and compiled processed data from all donors deposited as binary plink files (hg19). All genotyped donors, as well as their age, sex, reported race, diabetes status and duration, are provided in Phenotype_data.txt27 with additional donor information available on the nPOD Data Portal (https://portal.jdrfnpod.org/, accessed October 21, 2022).
WES data are stored in dbGaP27, including raw exome sequencing data files (fastq format) or hg19 aligned exome sequencing data (bam format), in addition to processed variant files (vcf format). A spreadsheet listing variants and associated annotations per donor, as described in the methods, was also submitted (csv format). All donors subjected to WES, as well as their age, sex, reported race, diabetes status and duration, are listed in Phenotype_data.txt27 with additional donor information available on the nPOD Data Portal (https://portal.jdrfnpod.org/, accessed October 21, 2022).
Technical Validation
Quality control assessment of the UFDIchip genotype array
As of this report, 372 nPOD donors have been genotyped on the UFDIchip and the resulting data are accessible on dbGaP (see Data Records). All array results were subjected to basic QC analyses that assessed donor-level DQC; donor-, plate-, and SNP-level call rate; and sex concordance. Donor DQC or call rate failures were re-processed with freshly extracted DNA when necessary. nPOD samples were batch-processed with data from living donors20 to facilitate calling of low-frequency variants51, resulting in 942,466 high quality genotypes passing the SNP call rate threshold. The nPOD cohort demonstrated SNP call rates of 99.58 [99.19–99.84] (median [interquartile range (IQR)]). All nPOD samples were assessed for concordance between reported and imputed sex according to level of X chromosome heterozygosity using plink v1.929. For all nPOD cases, imputed sex matched reported sex. Thus, all 372 nPOD samples passed basic QC measures. Additionally, 24 nPOD samples were run in technical replicate to assess assay reproducibility. Call rates between the technical replicates differed minimally, with 0.087 ± 0.640% bias (mean ± standard deviation, Fig. 3a). Importantly, the kinship coefficients between the 24 technical replicates were 0.499 [0.496–0.499] (median [IQR]), suggesting near identical genotype calls (Fig. 3b).

UFDIchip data are highly replicable and nPOD donors are unrelated. (a) Bland-Altman plot showing average vs. difference in QC call rates for n = 24 technical replicates. Horizontal dashed lines indicate 95% limits of agreement. (b) Relatedness analysis was performed using KING software30 for genotyping data from 372 nPOD donors, 24 of which were run in technical replicate, along with 2,504 1000 Genomes phase 3 cohort32 subjects, known to include some closely related individuals. Relatedness assessed via estimated kinship coefficients. Kinship coefficients from nPOD donor pairs were compared to those from 1000 Genomes subject pairs including inferred parent-offspring (PO), first-degree siblings (FS), second-degree relatives (2nd), third-degree relatives (3rd), and unrelated (UN). Bars represent median and interquartile range (IQR). Horizontal dashed lines indicate lower cutoffs for duplicate samples (kinship coefficient = 0.354) and second-degree relatives (kinship coefficient = 0.0884). Kruskal-Wallis test with Dunn’s multiple comparisons test for nPOD & nPOD (different subjects) versus all inferred 1000 Genomes subject relationship types or nPOD technical replicates.
Relatedness estimation
Next, relatedness between donors was assessed. Due to the nature of donor organ procurement, it is highly improbable, although not impossible, that nPOD donors may be related. A relatedness analysis of the 372 nPOD donors (69,006 possible pair combinations) using KING software30 found that all of these donor pairs were inferred to be unrelated (>third-degree relatives). For comparison, we also assessed the relatedness of 2,504 1000 Genomes phase 3 cohort32 subjects. While this set was designed to consist of unrelated individuals, it unintentionally included a few first-, second-, and third-degree relatives32. When relatedness between nPOD donor pairs was compared to relatedness between 1000 Genomes32 subject pairs, nPOD donor pairs showed significantly smaller kinship coefficients than inferred parent-offspring (PO), full sibling (FS), 2nd degree relative, and 3rd degree relative pairs from 1000 Genomes (Fig. 3b), suggesting that nPOD donors are not closely related. Note that nPOD donor pairs had significantly larger kinship coefficients than inferred unrelated (UN) pairs from 1000 Genomes32 (Fig. 3b), potentially due to increased similarity in genetic ancestry52 between subjects in the nPOD cohort than in the 1000 Genomes cohort, which was specifically designed to sample individuals with diverse genetic ancestry. Beyond confirming expected relatedness in the nPOD cohort, this validates that users of this resource may employ population-level quantitative trait locus (QTL) analysis methods with these genetic data.
Alignment with genetic ancestry
To further validate the UFDIchip data, we used the 1000 Genomes phase 3 cohort32 to build a reference model for genetic ancestry using ADMIXTURE software18 (Fig. 4a,b), projected all 372 nPOD donors onto this model to impute ancestry (Fig. 4c), and compared those results with reported race. Using methods modified from Kaddis, et al.53, we plotted PCA results of ancestry proportions and observed that each of the five major continental populations in the 1000 Genomes cohort (AFR, AMR, EAS, EUR, and SAS)32 clustered to occupy distinct PC space (Fig. 4b). This suggested that the five ancestry populations computed by ADMIXTURE were representative of the five continental populations from 1000 Genomes32. The ancestry proportions of 1000 Genomes32 continental populations were almost entirely represented by a single ancestry group, with the exception of admixed populations including Admixed Americans (AMR), as well as the subcontinental populations, Americans of African ancestry in SW USA (ASW) and African Caribbeans in Barbados (ACB, Fig. 4a,b), as previously observed32. Next, the nPOD cohort was projected onto the 1000 Genomes reference, revealing overlap with AFR, AMR, EAS, and EUR groups in PC space (Fig. 4c). Donors were then assessed for agreement between reported race and genetic ancestry, showing that the highest AFR, AMR, EAS, and EUR ancestry proportions were observed in donors reported as Black/African American, Hispanic/Latinx, Asian, and White/Caucasian respectively (UFDIchip_admixture.xlsx27), which is consistent with other U.S.-based admixture studies54,55. Notably, racial identity is complex and the method of estimating proportions of continental genetic ancestries may not adequately reflect genetic diversity56. With this limitation in mind, these analyses accomplish the aims of: 1) ADMIXTURE model validation using UFDIchip array data and 2) qualification of the genetic ancestry results as an alternative or additional covariate to reported race for users of this resource (UFDIchip_admixture.xlsx27)57.

Diverse genetic ancestries of nPOD donors. (a) An unsupervised ADMIXTURE model for five population classes was built with the 1000 Genomes phase 3 cohort32 using linkage disequilibrium-pruned SNPs that overlap with the UFDIchip. Stacked bar plots of the proportion of the ancestry group assigned to each 1000 Genomes donor are shown grouped by major continental populations [African (AFR): yellow; Admixed American (AMR): red; East Asian (EAS): green; European (EUR): blue; South Asian (SAS): violet]. Four genetic ancestry classes were assigned to 1000 Genomes continental populations based on their high proportion in those groups (AFR, EAS, EUR, SAS). The fifth class was almost entirely found in the Admixed American group and was thus assigned to AMR. (b) While all five continental populations occupy distinct principal component (PC) space, a portion of AMR subjects are in proximity to EUR, suggesting shared genetic ancestry. (c) All 372 nPOD donors (black) were projected onto the 1000 Genomes reference. Americans of AFR Ancestry in SW USA (ASW, n = 112); African Caribbeans in Barbados (ACB, n = 123); Gambian in Western Divisions in the Gambia (GWD, n = 180); Mende in Sierra Leone (MSL, n = 128); Yoruba in Ibadan (YRI, n = 186); Esan in Nigeria (ESN, n = 173), Luhya in Webuye, Kenya (LWK, n = 116); Puerto Ricans from Puerto Rico (PUR, n = 115); Mexican Ancestry from Los Angeles, USA (MXL, n = 107); Colombians from Medellin, Colombia (CLM, n = 115); Peruvians from Lima, Peru (PEL, n = 130); Japanese in Tokyo, Japan (JPT, n = 105); Han Chinese in Beijing, China (CHB, n = 108); Southern Han Chinese (CHS, n = 171); Chinese Dai in Xishuangbanna, China (CDX, n = 109); Kinh in Ho Chi Minh City, Vietnam (KHV, n = 124); Finnish in Finland (FIN, n = 105); Utah Residents (CEPH) with Northern and Western European Ancestry (CEU, n = 183); British in England and Scotland (GBR, n = 107); Iberian Population in Spain (IBS, n = 162); Toscani in Italia (TSI, n = 112), Punjabi from Lahore, Pakistan (PJL, n = 158); Gujarati Indian from Houston, Texas (GIH, n = 113); Bengali from Bangladesh (BEB, n = 114); Indian Telugu from the UK (ITU, n = 118); Sri Lankan Tamil from the UK (STU, n = 128). Figure adapted from Kaddis, et al.53.
HLA imputation accuracy and concordance
The nPOD cohort was HLA typed using next generation sequencing (NGS) at HLA-A, HLA-DRB1, HLA-DQA1, and HLA-DQB18 to identify donors with genotypes that are associated with T1D risk or protection34,35,36. This enables an extra level of QC and validation of the UFDIchip array data by comparing typed to genetically imputed HLA genotypes. Imputation accuracy at each locus, Acc(L), was calculated assuming typed results were correct if discordant with imputed results. Overall, Acc(L) was >0.93 for low-resolution HLA (2-digit) and >0.90 for high-resolution HLA (4-digit) for the four loci tested (UFDIchip_HLA_imputation_accuracy.xlsx27).
Next, we assessed concordance between typed and imputed HLA for T1D risk (A2, A24, DR3, DQ2, DR4, DQ8, DR8, and DQ4) or protective (DR15, DQ6, and DQ7) genotypes34,35,36 (Table 3). At 2-digit resolution, all tested loci were greater than 93% concordant (median [IQR]: 98.5% [97.4%–99.8%], Table 3). At 4-digit resolution, HLA concordance was predictably lower (median [IQR]: 97.1% [92.5%–99.3%]), with notable discordance in the less common HLA-DRB1*04:xx genotypes (Table 3). Importantly, 4-digit genotypes that convert 2-digit risk to protective genotypes, such as HLA-DRB1*04:03 and HLA-DQB1*03:01, were accurately imputed with greater than 97.9% concordance (Table 3).
Data validation at the sample level was assessed using a sample imputation accuracy score, Acc(S), for 2-digit HLA at the four typed loci. Acc(S) was 0.984 [0.946–0.998] (median [IQR]), indicating high performance of the imputation methodology per sample (Fig. 5a). HLA imputation may be inaccurate when rare HLA genotypes and ancestrally diverse populations are underrepresented in the reference cohort33,58,59. In agreement with this notion, a breakdown of nPOD donors by reported race or by top genetic ancestry proportion suggests that imputation accuracy could potentially be improved with greater reference cohort diversity (Fig. 5b,c). Donors with reported race of White had significantly higher HLA imputation accuracy than those reported as Black or Hispanic/Latinx (Fig. 5b). Similarly, donors whose highest genetic ancestry proportion were EUR had higher imputation accuracy than donors whose were AFR or AMR (Fig. 5c). Notably, 4-digit HLA imputation showed 100% concordance for the 24 nPOD subjects run in technical replicate on the UFDIchip.

HLA imputation accuracy is decreased in non-European donors. Donor-level HLA imputation accuracy [Acc(S)] was calculated for HLA-A, HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci by comparing to next generation sequencing (NGS)-based HLA typing. Acc(S) is shown for (a) all 372 nPOD donors, (b) for donors grouped by reported race, and (d) for donors grouped by highest proportion of genetic ancestry. African (AFR), Admixed American (AMR), East Asian (EAS), European (EUR), South Asian (SAS). Bars represent median and interquartile range (IQR). Kruskal-Wallis test with Dunn’s multiple comparisons test.
T1D polygenic GRS performance using UFDIchip data
Polygenic risk scores summarize genetic risk for T1D as a continuous value by aggregating estimated risk at HLA and non-HLA loci21,22,37,38. The original reports of GRS1 described its utility for discerning T1D from other forms of diabetes including T2D21,60 and MODY22. We previously observed that a similar GRS robustly discriminated living controls from T1D subjects reported as White but was less effective for non-White subjects, highlighting a need for diversity in risk modeling20,53. Shortly thereafter, GRS2 was developed to incorporate the impact of interactions between HLA haplotypes on T1D risk, showing improved discrimination of European ancestry T1D from control subjects37. Additionally, an AA-GRS was created to account for ancestry-specific T1D risk loci, with enhanced performance at distinguishing T1D from control subjects in AFR populations38. We therefore attempted to validate these previous findings regarding the ability of GRS1, GRS2, and AA-GRS to differentiate controls from T1D subjects by using the 372 nPOD cases subjected to genotyping. Indeed, White T1D donors (0.287 [0.264–0.303], median [IQR]) had significantly higher GRS1 values than White No Diabetes donors (0.231 [0.195–0.256], Fig. 6a). While Hispanic/Latinx T1D donors (0.283 [0.274–0.303]) also showed significantly higher GRS1 values than Hispanic/Latinx No Diabetes donors (0.233 [0.223–0.257]), Black T1D and No Diabetes donors were unable to be distinguished by GRS1 due to low scores in T1D donors (0.250 [0.234–0.261], Fig. 6a). In contrast, GRS2 values were significantly higher in Black T1D donors (11.62 [10.05–12.78]) than Black No Diabetes donors (8.83 [6.75–10.36]), although Black T1D donor GRS2 values remained significantly lower than those of White T1D donors (14.38 [12.94–15.16], Fig. 6b). While the AA-GRS likewise succeeded at differentiating Black T1D donors (5.634 [4.061–8.001]) from Black No Diabetes donors (1.751 [0.804–3.964]), no significant differences remained between Black T1D and White T1D donors (Fig. 6c). Taken together, these results indicate that the nPOD cohort UFDIchip array data represent a validated resource for genetic studies of T1D. Additionally, we provide GRS1, GRS2, and AA-GRS genotypes and calculated scores to the community for future studies (GRS1_GRS2_AAGRS_TOPMed_Imputed.xlsx27). Note that these scores differ from those provided in Kaddis, et al.53, due to updating the reference cohort for imputation from the Haplotype Reference Consortium (HRC)61 cohort, with predominantly European ancestry, to the TOPMed31 reference, with diverse genetic ancestry.

GRS2 and AA-GRS show improved performance over GRS1 at distinguishing T1D from no diabetes in non-White donors. (a) GRS1, (b) GRS2, and (c) AA-GRS of all 372 nPOD donors grouped by self-reported White/Caucasian (blue), Hispanic/Latinx (red), and Black/African American (yellow) no diabetes versus T1D donors. Bars represent median and interquartile range (IQR). Two-way ANOVA with Sidak’s multiple comparisons test.
WES
207 nPOD donors were also queried for rare variants using WES and associated data are accessible on dbGaP (see Data Records). Standard QC measures were performed to minimize adapter contamination, low-quality reads, error rate, and sequencing bias. To further validate data quality, we measured concordance between genotype calls from the UFDIchip and WES (N = 167 donors). Indeed, 27,852 autosomal biallelic variants with at least one minor allele count (MAC) in the WES data showed a concordance of 98.8% [92.2%–99.7%] (median [IQR]) with UFDIchip calls.
Six of the nPOD donors were previously reported to have genetic variants with possible clinical impact in KCNJ11, LMNA, HNF1A, GLIS3, INSR, and GATA6 using a custom-designed NGS panel that included 140 genes implicated in monogenic diabetes62. These genetic variants were validated with WES (Table 4) and visual exploration of the data using the Integrative Genomics Viewer (IGV)63 confirmed reads for each variant (Fig. 7). WES captures genomic DNA sequence in exons and the intronic sequence adjacent to exons. This enables the discovery of variants that directly alter the protein coding portion of mRNA (missense, nonsense, insertion/deletions) and also some regulatory intronic sequences, such as splice sites. Variants in genes associated with autoimmune diabetes (AIRE, FOXP3, IL2RA, ITCH, LRBA, SKAP2, STAT1, and STAT3) or MODY and neonatal diabetes (GCK, HNF1A, HNF4A, HNF1B, ABCC8, KCNJ11, and INS)39,64,65,66 were observed in 141 nPOD cases with T1D (Fig. 8a). Monogenic forms of diabetes are rare, and the vast majority of the detected variants are not expected to have functional or clinical consequences.

WES data identify previously published monogenic diabetes-associated gene variants in nPOD donors. Six nPOD donors were previously identified to have variants in monogenic diabetes-associated genes67: (a) KCNJ11, (b) LMNA, (c) HNF1A, (d) GLIS3, (e) INSR, and (f) GATA6. Alignment data viewed using the Integrative Genomics Viewer (IGV) software63. See Table 4 for specific gene variant information. Note that the WES reads are sometimes of the complement strand depending on gene orientation.

Variants in genes associated with monogenic diabetes observed in nPOD donors with T1D. Variants in genes associated with autoimmune diabetes (orange: AIRE, FOXP3, IL2RA, ITCH, LRBA, SKAP2, STAT1, and STAT3), or MODY and neonatal diabetes (blue: GCK, HNF1A, HNF4A, HNF1B, ABCC8, KCNJ11, and INS)39,64,65,66 were observed from WES data of 141 nPOD cases with T1D. (a) Distribution of variant types. (b) Frequency of variants in the Genome Aggregation Database (gnomAD)47. (c) Variants with a Combined Annotation Dependent Depletion (CADD) score > 2046, predicting the variant is deleterious to protein function. (d) Combining these tools can help identify variants that are predicted to be both rare (<0.01%) and deleterious. Total number of variants shown above bars.
There are several databases and tools available to help with the identification and interpretation of genetic variants. For example, the frequency of a variant in the general population can be estimated using the gnomAD, which contains data from 140,000 + exomes and genomes from unique, unrelated individuals spanning six global ancestries47. Additionally, the CADD score can be used to predict severity of impact of the variants based on a variety of criteria such as sequence context, evolutionary constraint, and functional predictions46. As expected, the variants observed in T1D cases were distributed across a spectrum of functional classes, with only a few predicted to be both rare (frequency < 0.01%) and deleterious (CADD score ≥ 20, Fig. 8b–d). As an example of how these tools can be used, variants in the monogenic diabetes genes HNF1A and STAT1 were analysed in the nPOD donors classified as T1D. One variant for each gene was predicted to be rare and deleterious based on the thresholds set for the gnomAD frequency and CADD score (Fig. 9, Table 5). The thresholds set for these and other bioinformatic tools are determined by each investigator, and are often informed by the clinical phenotype of the patient and previous knowledge about the gene’s disease association. Other variant annotations from tools including ACMG Classification43, SIFT Function Prediction44, PolyPhen-2 Function Prediction45, HGMD48, ClinVar49, and CentoMD50 are available for all 207 nPOD donors on dbGaP (see Data Records). A suggested workflow for evaluating genetic variants for potential clinical significance is shown in Fig. 10. Importantly, while computational tools facilitate interpretation, confidence in the functional or clinical relevance of the genetic variants reported herein requires rigorous experimentation.

HNF1A and STAT1 gene variants observed in nPOD donors with T1D. STAT1 (orange, Autoimmune diabetes gene) and HNF1A (blue, MODY/Neonatal diabetes gene)39,64,65,66 variants observed from WES data of 141 nPOD cases with T1D. (a) Variant types, (b) frequency distribution of variants, (c) variants with CADD score ≥ 20, and (d) variants with CADD score ≥ 20 and gnomAD frequency < 0.01%. Note that there are two potential monogenic diabetes variants in (d), one in HNF1A and one in STAT1. Total number of variants shown above bars.

Suggested workflow for evaluating variants in monogenic diabetes genes observed in WES data. Note that functional studies are needed for potential monogenic diabetes variants that have not already been previously validated.
Usage Notes
The associated data are openly available with unrestricted access.
Code availability
No custom code or scripts were used for the curation and validation of the dataset.
References
Grant, S. F. A., Wells, A. D. & Rich, S. S. Next steps in the identification of gene targets for type 1 diabetes. Diabetologia 63, 2260–2269, https://doi.org/10.1007/s00125-020-05248-8 (2020).
Robertson, C. C. et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet 53, 962–971, https://doi.org/10.1038/s41588-021-00880-5 (2021).
Onengut-Gumuscu, S. et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 47, 381–386, https://doi.org/10.1038/ng.3245 (2015).
Chen, Y. & Chen, G. New genetic characteristics of latent autoimmune diabetes in adults (LADA). Ann Transl Med 7, 81, https://doi.org/10.21037/atm.2019.01.01 (2019).
Pang, H. et al. Emerging roles of rare and low-frequency genetic variants in type 1 diabetes mellitus. J Med Genet 58, 289–296, https://doi.org/10.1136/jmedgenet-2020-107350 (2021).
Strakova, V. et al. Screening of monogenic autoimmune diabetes among children with type 1 diabetes and multiple autoimmune diseases: is it worth doing? J Pediatr Endocrinol Metab 32, 1147–1153, https://doi.org/10.1515/jpem-2019-0261 (2019).
Porter, J. R. & Barrett, T. G. Acquired non-type 1 diabetes in childhood: subtypes, diagnosis, and management. Arch Dis Child 89, 1138–1144, https://doi.org/10.1136/adc.2003.036608 (2004).
Campbell-Thompson, M. et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 28, 608–617, https://doi.org/10.1002/dmrr.2316 (2012).
Insel, R. A. et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 38, 1964–1974, https://doi.org/10.2337/dc15-1419 (2015).
Wasserfall, C. et al. Validation of a rapid type 1 diabetes autoantibody screening assay for community-based screening of organ donors to identify subjects at increased risk for the disease. Clin Exp Immunol 185, 33–41, https://doi.org/10.1111/cei.12797 (2016).
Pugliese, A. et al. The Juvenile Diabetes Research Foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) Program: goals, operational model and emerging findings. Pediatric Diabetes 15, 1–9, https://doi.org/10.1111/pedi.12097 (2014).
Noble, J. A. et al. The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 59, 1134–1148 (1996).
Williams, M. D. et al. Genetic Composition and Autoantibody Titers Model the Probability of Detecting C-Peptide Following Type 1 Diabetes Diagnosis. Diabetes 70, 932–943, https://doi.org/10.2337/db20-0937 (2021).
Moore, P. C. et al. Elastase 3B mutation links to familial pancreatitis with diabetes and pancreatic adenocarcinoma. J Clin Invest 129, 4676–4681, https://doi.org/10.1172/JCI129961 (2019).
Yu, M. G. et al. Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. J Clin Invest 129, 3252–3263, https://doi.org/10.1172/JCI127397 (2019).
Keenan, H. A. et al. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 59, 2846–2853, https://doi.org/10.2337/db10-0676 (2010).
Manichaikul, A. et al. Robust Relationship Inference in Genome-Wide Association Studies. Bioinformatics (Oxford, England) 26, 2867–2873, https://doi.org/10.1093/bioinformatics/btq559 (2010).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19, 1655–1664, https://doi.org/10.1101/gr.094052.109 (2009).
Dilthey, A. et al. Multi-population classical HLA type imputation. PLoS Comput Biol 9, e1002877, https://doi.org/10.1371/journal.pcbi.1002877 (2013).
Perry, D. J. et al. Application of a Genetic Risk Score to Racially Diverse Type 1 Diabetes Populations Demonstrates the Need for Diversity in Risk-Modeling. Sci Rep 8, 4529, https://doi.org/10.1038/s41598-018-22574-5 (2018).
Oram, R. A. et al. A Type 1 Diabetes Genetic Risk Score Can Aid Discrimination Between Type 1 and Type 2 Diabetes in Young Adults. Diabetes Care 39, 337–344, https://doi.org/10.2337/dc15-1111 (2016).
Patel, K. A. et al. Type 1 Diabetes Genetic Risk Score: A Novel Tool to Discriminate Monogenic and Type 1 Diabetes. Diabetes 65, 2094–2099, https://doi.org/10.2337/db15-1690 (2016).
Campbell-Thompson, M. et al. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 65, 719–731, https://doi.org/10.2337/db15-0779 (2016).
Battaglia, M. et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes care 43 https://doi.org/10.2337/dc19-0880 (2020).
Arif, S. et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 63, 3835–3845 DB_140365 [pii] db14-0365 [pii] https://doi.org/10.2337/db14-0365 (2014).
Bandrowski, A. et al. The Resource Identification Initiative: A Cultural Shift in Publishing. Neuroinformatics 14, 169–182, https://doi.org/10.1007/s12021-015-9284-3 (2016).
Perry, D. J. et al. dbGaP https://identifiers.org/dbgap:phs002861.v1.p1 (2022).
Cortes, A. & Brown, M. A. Promise and pitfalls of the Immunochip. Arthritis Res Ther 13, 101, https://doi.org/10.1186/ar3204 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics 81, 559–575, https://doi.org/10.1086/519795 (2007).
A, M. et al. Robust Relationship Inference in Genome-Wide Association Studies. Bioinformatics (Oxford, England) 26 https://doi.org/10.1093/bioinformatics/btq559 (2010).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299, https://doi.org/10.1038/s41586-021-03205-y (2021).
1000 Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PloS One 8, e64683, https://doi.org/10.1371/journal.pone.0064683 (2013).
Noble, J. A. et al. HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 59, 2972–2979. db10-0699 [pii] https://doi.org/10.2337/db10-0699 (2010).
Erlich, H. et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57, 1084–1092. db07-1331 [pii] https://doi.org/10.2337/db07-1331 (2008).
Noble, J. A., Johnson, J., Lane, J. A. & Valdes, A. M. HLA class II genotyping of African American type 1 diabetic patients reveals associations unique to African haplotypes. Diabetes 62, 3292–3299, https://doi.org/10.2337/db13-0094 (2013).
Sharp, S. A. et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care 42, 200–207, https://doi.org/10.2337/dc18-1785 (2019).
Onengut-Gumuscu, S. et al. Type 1 Diabetes Risk in African-Ancestry Participants and Utility of an Ancestry-Specific Genetic Risk Score. Diabetes Care 42, 406–415, https://doi.org/10.2337/dc18-1727 (2019).
Rutsch, N. et al. Diabetes With Multiple Autoimmune and Inflammatory Conditions Linked to an Activating SKAP2 Mutation. Diabetes Care 44, 1816–1825, https://doi.org/10.2337/dc20-2317 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11 10 11–11 10 33, https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164, https://doi.org/10.1093/nar/gkq603 (2010).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814, https://doi.org/10.1093/nar/gkg509 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7, Unit7 20, https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315, https://doi.org/10.1038/ng.2892 (2014).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443, https://doi.org/10.1038/s41586-020-2308-7 (2020).
Stenson, P. D. et al. The Human Gene Mutation Database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum Genet 139, 1197–1207, https://doi.org/10.1007/s00439-020-02199-3 (2020).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067, https://doi.org/10.1093/nar/gkx1153 (2018).
Trujillano, D. et al. A comprehensive global genotype-phenotype database for rare diseases. Mol Genet Genomic Med 5, 66–75, https://doi.org/10.1002/mgg3.262 (2017).
Chierici, M., Miclaus, K., Vega, S. & Furlanello, C. An interactive effect of batch size and composition contributes to discordant results in GWAS with the CHIAMO genotyping algorithm. Pharmacogenomics J 10, 355–363, https://doi.org/10.1038/tpj.2010.47 (2010).
Thornton, T. et al. Estimating kinship in admixed populations. Am J Hum Genet 91, 122–138, https://doi.org/10.1016/j.ajhg.2012.05.024 (2012).
Kaddis, J. et al. Improving the Prediction of Type 1 Diabetes Across Ancestries. Diabetes Care, dc211254 https://doi.org/10.2337/dc21-1254 (2022).
Bryc, K., Durand, E., Macpherson, J., Reich, D. & Mountain, J. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. American Journal of Human Genetics 96, 37–53, https://doi.org/10.1016/j.ajhg.2014.11.010 (2015).
Dai, C. L. et al. Population Histories of the United States Revealed through Fine-Scale Migration and Haplotype Analysis. Am J Hum Genet 106, 371–388, https://doi.org/10.1016/j.ajhg.2020.02.002 (2020).
Lewis, A. et al. Getting genetic ancestry right for science and society. Science 376, 250–252, https://doi.org/10.1126/science.abm7530 (2022).
Borrell, L. et al. Race and Genetic Ancestry in Medicine - A Time for Reckoning with Racism. The New England Journal of Medicine 384, 474–480, https://doi.org/10.1056/NEJMms2029562 (2021).
Abi-Rached, L. et al. Immune diversity sheds light on missing variation in worldwide genetic diversity panels. PloS One 13, e0206512, https://doi.org/10.1371/journal.pone.0206512 (2018).
Luo, Y. et al. A high-resolution HLA reference panel capturing global population diversity enables multi-ancestry fine-mapping in HIV host response. Nature Genetics 53, 1504–1516, https://doi.org/10.1038/s41588-021-00935-7 (2021).
Carr, A. L. J. et al. Histological validation of a type 1 diabetes clinical diagnostic model for classification of diabetes. Diabet Med 37, 2160–2168, https://doi.org/10.1111/dme.14361 (2020).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–1283, https://doi.org/10.1038/ng.3643 (2016).
Sanyoura, M. et al. Pancreatic Histopathology of Human Monogenic Diabetes Due to Causal Variants in KCNJ11, HNF1A, GATA6, and LMNA. J Clin Endocrinol Metab 103, 35–45, https://doi.org/10.1210/jc.2017-01159 (2018).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26, https://doi.org/10.1038/nbt.1754 (2011).
Riddle, M. C. et al. Monogenic Diabetes: From Genetic Insights to Population-Based Precision in Care. Reflections From a Diabetes Care Editors’ Expert Forum. Diabetes Care 43, 3117–3128, https://doi.org/10.2337/dci20-0065 (2020).
Johnson, M. B., Hattersley, A. T. & Flanagan, S. E. Monogenic autoimmune diseases of the endocrine system. Lancet Diabetes Endocrinol 4, 862–872, https://doi.org/10.1016/S2213-8587(16)30095-X (2016).
Husebye, E. S., Anderson, M. S. & Kampe, O. Autoimmune Polyendocrine Syndromes. N Engl J Med 378, 1132–1141, https://doi.org/10.1056/NEJMra1713301 (2018).
Sanyoura, M. et al. Pancreatic Histopathology of Human Monogenic Diabetes Due to Causal Variants in KCNJ11, HNF1A, GATA6, and LMNA. J Clin Endocrinol Metab https://doi.org/10.1210/jc.2017-01159 (2017).
Chen, R., Im, H. & Snyder, M. Whole-Exome Enrichment with the Roche NimbleGen SeqCap EZ Exome Library SR Platform. Cold Spring Harb Protoc 2015, 634–641, https://doi.org/10.1101/pdb.prot084855 (2015).
Acknowledgements
This research was performed with the support of the Network for Pancreatic Organ donors with Diabetes (nPOD; RRID:SCR_014641), a collaborative type 1 diabetes research project supported by JDRF (nPOD: 5-SRA-2018-557-Q-R) and The Leona M. & Harry B. Helmsley Charitable Trust (Grant#2018PG-T1D053, G-2108-04793). The content and views expressed are the responsibility of the authors and do not necessarily reflect the official view of nPOD. Organ Procurement Organizations (OPO) partnering with nPOD to provide research resources are listed at http://www.jdrfnpod.org/for-partners/npod-partners/. Additional funding was provided by the National Institutes of Health (NIH, P01 AI042288, UC4 DK108132, U01 DK112217) and The Leona M. & Harry B. Helmsley Charitable Trust (G-2018PG-T1D018, G-2003-04376). MRS was supported by a JDRF Postdoctoral Fellowship (3-PDF-2022-1137-A-N). LDP was supported by NIH predoctoral training grants (T32 DK108736 and F31 DK129004-01A1).
Author information
Authors and Affiliations
Contributions
Daniel J. Perry: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing—original draft preparation, Writing—reviewing and editing, Visualization. Melanie R. Shapiro: Software, Validation, Formal analysis, Investigation, Data curation, Writing—original draft preparation, Writing—reviewing and editing, Visualization. Sonya W. Chamberlain: Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing—original draft preparation, Writing—reviewing and editing, Visualization. Srikar Chamala: Writing—reviewing and editing, Irina Kusmartseva: Investigation, Writing— reviewing and editing, Supervision, Project administration. Leandro Balzano-Nogueira: Investigation, Data curation, Writing— reviewing and editing. Mingder Yang: Data curation, Writing— reviewing and editing, Supervision, Project administration. Jason Brant: Investigation, Writing— reviewing and editing. Maigan Brusko: Investigation, Writing— reviewing and editing. MacKenzie D. Williams: Investigation, Data curation, Writing— reviewing and editing. Kieran M. McGrail: Investigation, Writing— reviewing and editing. James McNichols: Investigation, Writing— reviewing and editing. Leeana D. Peters: Investigation, Writing— reviewing and editing. Amanda L. Posgai: Writing—original draft preparation, Writing— reviewing and editing. John S. Kaddis: Software, Writing— reviewing and editing. Clayton E. Mathews: Writing— reviewing and editing. Clive H. Wasserfall: Writing— reviewing and editing, Supervision, Project administration. Bobbie-Jo Webb-Robertson: Writing— reviewing and editing. Martha Campbell-Thompson: Investigation, Writing— reviewing and editing, Supervision, Project administration. Desmond Schatz: Investigation, Writing— reviewing and editing, Supervision, Project administration. Carmella Evans-Molina: Writing— reviewing and editing, Supervision, Project administration. Alberto Pugliese: Writing— reviewing and editing, Supervision, Project administration. Patrick Concannon: Conceptualization, Writing— reviewing and editing, Supervision, Project administration, Funding acquisition. Mark S. Anderson: Conceptualization, Writing— reviewing and editing, Supervision, Project administration. Michael S. German: Conceptualization, Writing— reviewing and editing, Supervision, Project administration. Chester E. Chamberlain: Conceptualization, Writing— reviewing and editing, Supervision, Project administration. Mark A. Atkinson: Conceptualization, Resources, Writing— reviewing and editing, Supervision, Project administration, Funding acquisition. Todd M. Brusko: Conceptualization, Writing— reviewing and editing, Supervision, Project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Perry, D.J., Shapiro, M.R., Chamberlain, S.W. et al. A genomic data archive from the Network for Pancreatic Organ donors with Diabetes. Sci Data 10, 323 (2023). https://doi.org/10.1038/s41597-023-02244-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02244-6
