
Abstract
Real-world observational datasets that record and quantify pressure-stressor-response linkages between effluent discharges and natural aquatic systems are rare. With global wastewater volumes increasing at unprecedented rates, it is urgent that the present dataset is available to provide the necessary information about microbial community structure and functioning. Field studies were performed at two time-points in the Austral summer. Single-species and microbial community whole effluent toxicity (WET) testing was performed at a complete range of effluent concentrations and two salinities, with accompanying environmental data to provide new insights into nutrient and organic matter cycling, and to identify ecotoxicological tipping points. The two salinity regimes were chosen to investigate future scenarios based on a predicted salinity increase at the study site, typical of coastal regions with rising sea levels globally. Flow cytometry, amplicon sequencing of 16S and 18S rRNA genes and micro-fluidic quantitative polymerase-chain reactions (MFQPCR) were used to determine chlorophyll-a and total bacterial cell numbers and size, as well as taxonomic and functional diversity of pelagic microbial communities. This strong pilot dataset could be replicated in other regions globally and would be of high value to scientists and engineers to support the next advances in microbial ecotoxicology, environmental biomonitoring and estuarine water quality modelling.
Measurement(s) | water composition • total dissolved solids • conductivity of water • pH • concentration of oxygen in water • Total Organic Carbon • dinitrogen • phosphorus atom • ammonia • nitrate • nitrite • biological oxygen demand • chlorophyll a • Cell Density • Algae • Bacteria • hydrogen sulfide • Toxicity • rRNA_16S • rRNA_18S • abundance of nutrient cycling genes • abundance of antibiotic resistance genes |
Technology Type(s) | water quality unit • water testing suite • dilution method • autofluorescence • flow cytometry method • whole effluent toxicity testing • DNA sequencing • microfluidic quantitative polymerase chain reaction |
Factor Type(s) | effluent concentration • salinity levels |
Sample Characteristic - Organism | Bacteria • algae |
Sample Characteristic - Environment | waste water • estuary • sea coast • saline water • fresh water body |
Sample Characteristic - Location | Hunter River |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12221738
Similar content being viewed by others

Background & Summary
The world is facing a global water quality crisis1,2. The vast majority (more than 80%) of global wastewater is released directly into natural waterways, resulting in widespread pollution3. The most frequent contaminants are domestic waste (~2 million tonnes per day), industrial wastes and chemicals, agricultural pesticides and fertilizers2,4. The implications of wastewater discharges include, but are not limited to: degraded aquatic ecosystems5,6; decreased biodiversity7; increased greenhouse gas emissions8,9; and a wide range of detrimental impacts to human health10. Global wastewater volumes are increasing at unprecedented rates as a result of population growth, rapid urbanisation and economic development, and these drivers are concentrated in coastal regions11,12,13,14. This worldwide trend poses immediate management challenges if we are to prevent further damage to sensitive aquatic ecosystems, human health and water security15.
Globally, comprehensive datasets that characterise the impacts of wastewater discharges to water quality in natural aquatic environments are generally lacking2. Based on a comprehensive review of data published in 181 countries11, the authors found that only 55 countries had any data available on wastewater generation, treatment and use, and much of this information was dated (i.e., pre-2008). Significant data gaps exist on the linkages between the physical, chemical and biological characteristics of many urban surface water environments that receive wastewater discharges16,17,18. In highly developed countries where the largest percentage of treated domestic wastewater is currently discharged directly to natural waterways – for example: Australia (85%; 1,234 treatment plants), North America (75%; 14,748 treatment plants) and Europe (71%; >18,000 treatment plants) – an understanding of the impacts of effluent discharges on ecosystem health is still in its relative infancy11. These pressure-stressor-response relationships are particularly difficult to disentangle in estuaries, due to highly variable physio-chemical conditions19. To effectively address increasing concerns regarding wastewater discharge to natural aquatic systems worldwide, comprehensive data is urgently needed from real-world observations. These data can be used to investigate and understand the pressure-stressor-response linkages between treated effluent discharges and natural aquatic environments.
Pelagic microbial communities are extremely sensitive to rapid changes in their environment making them ideal indicators of water quality processes and functioning19,20,21. They are also ubiquitous in natural aquatic environments and play an important role in nutrient and organic matter cycling22. Traditional microbial ecotoxicological studies have relied on the combination of single algal species toxicity testing and chemical surveys to ascertain the aggregate toxic effect of whole effluent wastewater discharge on microalgae, rather than attempting to quantify both diversity and function of entire microbial communities at the same time21. The combination of community-level testing and environmental ‘omics moves beyond the scenario of single species toxicity testing and provides the opportunity to determine real-world community interactions and shifts in response to wastewater.
In this study we have incorporated recent advances in water quality science and assessment techniques to characterise the ecotoxicological response of tertiary-level treated effluent following discharge in temperate estuarine environments. The integrated field and laboratory assessments were completed on the Hunter River estuary located on the New South Wales (NSW) coastline in southeast Australia. The dataset obtained is specifically targeted at the dynamics and health of pelagic microbial communities at a practical scale, with these novel observations having the potential to provide new insights and understanding of nutrient and organic matter cycling. Whole Effluent Toxicity (WET) testing was performed to highlight the effects of mixing treated effluent within freshwater and saltwater environments and to identify potential tipping points that both inhibit and stimulate the growth of microalgae and microbial communities. Two salinity regimes were chosen to include future scenarios based on a predicted increasing salinity of the Hunter River with rising sea levels. All water samples were subjected to sequencing for 16S and 18S rRNA genes to measure changes in microbial community structure. Flow cytometry was used to enumerate chlorophyll-a and total bacterial cells and to estimate their size. The abundance of genes associated with nutrient cycling, antibiotic resistance and the identification of pathogens that would be harmful to human health were determined using microfluidic quantitative polymerase-chain reactions (MFQPCR) at the microbial community-level.
Methods
Study area
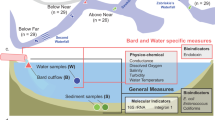
The Hunter River estuary (151.8°E, 32.9°S) is situated on the temperate southeast coastline of NSW, Australia (Fig. 1). The Hunter River estuary is a typical wave-dominated, mature barrier estuary23 with a large tidal pool that extends 60 km inland to its tidal limits. The Hunter River estuary has semi-diurnal tides and a mean tidal range of approximately 1.2 m. Catchment inflows to the estuary via the Hunter River and its two main tributaries – the Paterson and Williams Rivers – are regulated by dams and weirs.

Hunter River estuary study area and WWTP river discharge sites for Kurri Kurri, Farley, Morpeth, Raymond Terrace and Shortland shown as circled numbers. River water collection sites used for WET test dilutions are shown as boxed letters.
The Hunter River catchment covers an area over 22,000 km2 and is typical of many other developed coastal regions globally in that it has been extensively modified by human activity and multiple land uses24. The upper catchment is predominately agricultural land, whereas the lower catchment around the Port of Newcastle includes extensive urban and industrial areas, entrance dredging and training, the world’s largest coal export terminal and a growing multi-purpose cargo hub. Despite these pressures, the Hunter River estuary still supports significant areas of estuarine habitat such as mangroves, saltmarsh, inter-tidal and sub-tidal soft sediment shoals, as well as a Ramsar listed wetland of international significance25.
The Hunter River estuary receives diffuse water pollution and nutrients from the catchment, as well as high nutrient point loads from several major wastewater treatment plants (WWTP) capable of servicing a population of approximately 200,000 people across the floodplain. Generally, these WWTP provide tertiary-level wastewater treatment designed to remove excess nutrients of nitrogen and phosphorus. Unallocated treated effluent is discharged either directly, or indirectly via tributary channels, to the tidal zone of the Hunter River. The tributary channels in this study are freshwater creek systems as they are excluded from tidal flows via one-way floodgates and form part of the expansive Lower Hunter Flood Mitigation Scheme.
The study sampling points were located in the upper, mid and lower portions of the Hunter River estuary (Fig. 1). In the upper Hunter River estuary, the Swamp-Fishery-Wallis Creek system near Maitland, receives unallocated treated effluent from the townships of Kurri Kurri (3.4 ML/day) and Farley (5.6 ML/day). Kurri Kurri WWTP discharges into Swamp Creek which flows into Wentworth Swamp, a large, low-lying permanent waterbody, which in turn discharges into Fishery Creek. Farley WWTP discharges into Fishery Creek downstream of its confluence with Wentworth Swamp, and this flows into Wallis Creek which discharges into the Hunter River estuary (Fig. 1, river discharge site 1). Unallocated treated effluent from Morpeth WWTP (10 ML/day) is discharged directly into the Hunter River, approximately 3 km downstream of Wallis Creek (Fig. 1, river discharge site 2). Unallocated treated effluent from the Raymond Terrace WWTP (7.3 ML/day) is discharged to the Hunter River via Grahamstown Drain and Windeyers Creek (Fig. 1, river discharge site 3). The Shortland WWTP (9.6 ML/day) discharges unallocated treated effluent directly to the Hunter River South Arm (Fig. 1, river discharge site 4). A description of site characteristics for each WWTP river discharge site (Fig. 1) is provided in Table 1.
Microbial field surveys
Water samples were taken on two (2) separate occasions in the Austral summer (November 2016 and February 2017) from five (5) WWTP outfalls along the Hunter River estuary, including Kurri Kurri, Farley, Morpeth, Raymond Terrace and Shortland. Water samples were collected upstream and downstream of each outfall at different distances totalling 20 sampling points (see Table 2). Note that water at Shortland was only sampled from the river immediately upstream of the Shortland WWTP outfall – downstream sampling was not logistically possible and not relevant as the WWTP was not discharging at the time of the investigation. All 20 sites were sampled within the same two-day time window on both sampling occasions with three (3) replicates collected per site for sequencing of microbial (prokaryotic and eukaryotic) communities and a single replicate for water quality measurements.
Specifically, surface water was collected in 2 L sterile Whirl-Paks® stored on ice in the dark until filtering within 24 hours. To capture all microbial cells and fragments in the water samples for DNA extraction and sequencing, samples were homogenised by repeated inversion and 500 mL of water was filtered through a 0.22 µm Express PLUS Polyethersulfone membrane (Millipore) using a hydraulic pump. Filter units were sterilised before use and rinsed with ethanol between water samples. In some cases, when the water samples contained excess organic material, the filters were clogged before 500 mL could be filtered, and the volume that had been filtered was noted for later adjustment of the data. Filters were rolled, inserted into bead tubes from the DNeasy PowerWater Kit (Qiagen) and frozen at −80 °C until DNA extraction and sequencing.
For water quality measurements, water samples were simultaneously obtained using standard bottles provided by a NATA accredited facility for analysis of total suspended solids (TSS), total organic carbon (TOC), total nitrogen (TN), total phosphorous (TP), ammonia (NH3), nitrate and nitrite (NOx), biological oxygen demand (BOD), chlorophyll-a, and hydrogen sulphide (H2S). Water samples were stored on ice in the field and then sent for testing on the same day they were sampled. Additional environmental data, including pH, dissolved oxygen (DO), and electrical conductivity (EC) were measured using a calibrated water quality unit (Horiba) at each site. The water quality unit was calibrated before and after each sampling trip.
Whole Effluent Toxicity (WET) testing
Whole Effluent Toxicity (WET) testing was conducted within the laboratory facilities of the Sydney Institute of Marine Science (SIMS) in Chowder Bay, Sydney, Australia. WET tests were completed for algal single-species and whole microbial communities in a fully-crossed experimental design created using UV-disinfected effluent from the five (5) WWTP sampled. Following standard protocols for WET testing as published by the US EPA and the ANZECC/ARMCANZ water quality guidelines (2000), five effluent concentrations were selected along with a control. The concentrations comprising 0% effluent (control), 0.1% effluent, 1% effluent, 10% effluent, 50% effluent, 90% effluent and 100% effluent.
Sample collection, preparation and testing was completed over three (3) weeks in May 2017. Disinfected effluent from each WWTP and river water samples were collected on the first day of each test week and stored in a dark constant temperature (25 °C) room O/N to allow for water temperature adjustment to the testing conditions. On each sampling occasion, a total of 30 L of disinfected effluent from each WWTP and 60 L from each river site was collected to create the dilutions for the WET tests.
Single species
The single-species WET tests were completed based on standard procedures – US EPA Test Method 1003.026 and the Environment Canada test method27 – using a 4 to 7-day old culture of the freshwater unicellular green algae Raphidocelis subcapitata (formerly also known as Selenastrum capricornutum and Pseudokirchneriella subcapitata) obtained from the NSW Department of Planning, Industry and Environment (formerly NSW Office of Environment and Heritage). Prior to WET testing, cells of R. subcapitata were washed three (3) times with artificial soft-water to remove culture media and extracellular substances. For this step, the algal culture was centrifuged at 700 g for seven (7) minutes, the supernatant discarded, and the pellet resuspended in artificial soft-water. Algal cells were counted using a haemocytometer and each test sample was inoculated with approximately 3 × 104 microalgal cells/mL.
For the single-species WET tests, 200 mL dilutions were prepared using UV-disinfected effluent (filtered at 0.45 µm) from all five (5) WWTP and artificial soft-water (filtered at 0.22 µm). Artificial soft-water used for the tests was created by the addition of sodium bicarbonate (NaHCO3), calcium sulfate dihydrate (CaSO4·2H2O), magnesium sulfate heptahydrate (MgSO4·7H20), and potassium chloride (KCl) to milli-Q water. The artificial soft-water was adjusted to the hardness of each effluent water type on the starting day of the WET tests. Total hardness as CaCO3 for effluents from each WWTP included: Kurri Kurri (106 mg/L); Farley (257 mg/L); Morpeth (78 mg/L); Raymond Terrace (65 mg/L); and Shortland (168 mg/L). Three (3) replicates of 50 mL each were added to 100 mL conical flasks and one (1) replicate of 50 mL was added to a 100 mL glass beaker. The flasks and beakers were covered with a 0.4 mm mesh to avoid any airborne particles contaminating the samples. Further, 200 µL of 2.1 g/L sodium nitrate (1.5 mg \(N{O}_{3}^{-}\)/L) and 200 µL of 0.22 g/L potassium dihydrogen phosphate (0.15 mg \(P{O}_{4}^{3-}\)/L) was added to each sample to provide sufficient nutrients for exponential algal growth.
The test samples were incubated for 72 hours at continuous daylight conditions. During the tests, full-spectrum daylight fluorescent lighting (36 W) provided a light intensity of 71.6 W/m2. The shelves used for the tests were lined with aluminium foil and samples were randomised once a day to maximise light exposure. At the time of inoculation (time point 0 h), the concentrations of NH3, NOx, TKN, TN, and TP, as well as water hardness were measured in each effluent type using standard inorganics and nutrients water testing suites. At the end of every nominal 24-hour period, test samples were mixed by swirling (conical flasks) or with pipette tips (beakers). 100 µL from each conical flask was taken to determine cell counts using flow cytometry, and water quality microsensors (Unisense) were used to measure pH and DO levels in the beakers.
Whole microbial community
For the community-level microbial WET tests, dilutions were prepared using unfiltered UV-disinfected effluent from all five (5) WWTP and ulfiltered river water from two (2) locations in the Hunter River estuary, including a freshwater site (salinity < 1 PSU, river site ‘F’ in Fig. 1) and a saltwater site (salinity > 30 PSU, river site ‘S’ in Fig. 1). These two (2) salinity regimes were chosen to include future scenarios based on a predicted increasing salinity of the Hunter River with rising sea levels. The community-level WET tests were run in triplicate 2 L plastic beakers for 72 hours at a 12/12 hour day/night light cycle. During the tests, LED-600 aquarium lights (BeamsWork) provided conditions suitable for the growth of photoautotrophic microbes, having a light frequency of 10,000 K (white LED) and 460 nm (blue LED), equivalent to 1,340 lumens and a light intensity of 65.1 W/m2 (assuming a luminous efficacy of 40.98 lm/W). As for the single-species tests, the shelves used for the tests were lined with aluminium foil and the beaker positions on the shelves were randomised once a day to maximise the light exposure in each beaker.
At the time of inoculation (time point 0 h), concentrations of NH3, NOx, TKN, TN, and TP were measured in each effluent type and river site sample. At the end of every nominal 24-hour period, beakers were mixed with a glass stirring rod and samples from each beaker were taken for further analysis, including 1 mL of water for microbial cell counts using flow cytometry, and approximately 500 mL of water, collected in sterile Whirl-Paks and stored on ice in the dark until filtering, for DNA extraction and sequencing. Note that filtering for DNA extraction during the WET tests followed the same protocol previously described for the microbial field surveys. Further, water quality microsensors (Unisense) were used to measure pH, DO and H2S at the sampling times (i.e. every 24 hours) for each effluent type and dilution, except Shortland WWTP due to technical difficulties. Note that DO at time point 0 h was only measured for Raymond Terrace and Morpeth effluent types due to a temporary fault in the DO sensor.
Biological oxygen demand
At the end of the community-level WET tests (time point 72 h), water was sampled from two (2) replicates of each effluent type and dilution for nutrient (NH3, NOx, TKN, TN, TP) analysis, and 200 mL of the remaining water was used to estimate the BOD of the microbial communities. The water sampled for the BOD tests were capped in standard BOD bottles and incubated for 2.5 hours in the dark. DO measurements were taken at the start of the BOD tests (T0) and after 2.5 hours incubation in the dark (T1).
Flow cytometry
Staining optimization
The nucleic acid dye SYTO9 (ThermoFisher Scientific), which has been shown to preferentially stain live and dead total bacteria cells, was tested at varying dilutions (1:80, 1:500, 1:1000 v/v of the dye’s stock solution and dimethyl sulfoxide (DMSO))28,29. All dilutions were tested using triplicate positive (stained) and negative controls (no stain). After stain optimisation using an analogue river water sample, all aliquots were subsequently stained using 0.2 µL of SYTO9 (1:1000 v/v dilution of commercial stock solution with DMSO) and incubated for 15 minutes in the dark at room temperature prior to analysis28. Note the differentiation and quantification of both live and dead cells, using Propidium Iodide, was purposely omitted, since sequencing typically incorporates both live and dead cells, and the results were aimed at determining the total number of bacteria cells (live and dead) present in the sample.
Flow cytometry analysis
All cell count samples were analysed using a LSRFortessa SORP (BD Biosciences) flow cytometer with the FACSDiva software (v8.0.1) in the Mark Wainwright Analytical Centre (Flow Cytometry Facility) at UNSW Sydney. Cell count samples from the single-species and microbial community WET tests were stored on ice until flow cytometry analysis. The samples analysed in May 2017 (‘Fresh’) were left unstained and the autofluorescence from chlorophyll-a present in the live algal cells was used to determine cell densities. The chlorophyll-a fluorescence assays were designed to compare the growth response of single-celled algae between the two (2) WET setups. Additional water samples from the WET tests were stored at −20 °C and later used to quantify the total bacterial cell densities in February and March 2019 (‘Frozen’). This was done to distinguish between the bacterial and algal (chlorophyll-a) cell populations in the water samples.
All cell count samples were separated into 200 μL aliquots across eight (8) microplates (96-wells) by effluent type (Kurri Kurri and Farley, Morpeth and Raymond Terrace) and sampling time (0 h, 24 h, 48 h, 72 h). The Frozen samples were thawed quickly (at 37 °C) to avoid cell loss during the thawing process30, and after thawing, were mixed via manual shaking for 10 s. The defrosted 200 μL aliquots in the microplate wells were stained and analysed automatically in standard-throughput mode.
A threshold of 200 cell counts was set on the side-scattered (SSC) light detector to exclude noise from non-algal and unstained bacterial cell particles. All readings were collected as logarithmic signals at a flow rate of 2.0 µL/s. Maximum events were set to 10,000,000 to ensure the counting of all cells within a 60 µL sample (100 µL for the Fresh samples). Before cell counting, all samples were mixed twice-through by mechanical pipetting up and down of 100 µL of sample at a mixing rate of 180 µL/s. Note there was no cell count data available for the community-level WET tests containing the Shortland effluent type due to complications with the flow cytometer at the time of the analysis.
Enumeration and size
The 488 nm laser was used for excitation of both SYTO9 stain and chlorophyll-a fluorescence. The detection was measured using emission filters for green (530 nm ± 15 nm) and red (780 nm ± 60 nm) fluorescence. Determination of the region within the SSC vs chlorophyll bivariate density plot, which included live algal cells containing chlorophyll-a, was done prior to the WET tests, using algal cell cultures and negative controls. Further, determination of the region defined for total bacteria within the SSC vs green fluorescence plot, was done during staining optimisation, using positive (stained) and negative (unstained) controls. Example cytograms of bacterial and chlorophyll-a populations resolved by fluorescence for 50% dilution of Kurri Kurri effluent type in freshwater (replicate 1) at 48 hours are provided in Fig. 2. Figure 2a shows the total population which is separated into quadrants within the green vs red fluorescence plot (Fig. 2b). The region defined for total bacteria is labelled “Bacteria”, while the region for cells containing chlorophyll and SYTO9 stain is labelled “Double positive”. The double positives were counted as bacteria due to the separation shown in SSC vs green florescence (SYTO9 stain) (Fig. 2d). Final cell counts were determined as the number of events within the previously selected regions. Presentation of the data as bivariate density plots (Fig. 2) enabled the best distinction between stained bacteria and chlorophyll-a. Following enumeration of the microbial cells, size was estimated using CountBright absolute counting beads (200 nm, 500 nm, 800 nm, 1 um, 3 um, 6 um) as volumetric standards. There may be some variation in the size estimates caused by the way cells are placed during the analysis.

Example cytograms of bacterial and chlorophyll-a populations resolved by fluorescence for 50% dilution of Kurri Kurri effluent type in freshwater (replicate 1) at 48 hours, February and March 2019 dataset. A bivariate density plot of SSC vs red fluorescence (chlorophyll) shows the total population (a). Regions on the green vs red fluorescence plot were defined for bacteria (Bacteria), cells containing chlorophyll and SYTO9 stain (Double positive), cells containing only chlorophyll (Chlorophyll-a) and all other cells (Other) (b). Double positives were counted as total bacteria due to the separation of cells shown in SSC vs green fluorescence (d).
DNA extraction and sequencing
DNA was extracted using the DNeasy PowerWater Kit (Qiagen) following manufacturer protocols and stored at −80 °C until sequencing. Bacterial communities were identified using the 27f (5′-3′: AGAGTTTGATCMTGGCTCAG31) and 519r (5′-3′: GWATTACCGCGGCKGCTG32) primers for the V1-V3 region of the 16 S rRNA gene and Illumina™ MiSeq. 2 × 300 base pair (bp), paired-end v2 sequencing runs across two (2) lanes. Eukaryotic composition was determined using the 1391 f (5′-3′: GTACACACCGCCCGTC) and EukBr (5′-3′: TGATCCTTCTGCAGGTTCACCTAC) primer set33 for the V9 region of the 18S rRNA gene and Illumina™ MiSeq. 2 × 150 bp paired-end v3 sequencing runs across two (2) lanes. All sequencing was done at the Ramaciotti Centre for Genomics (UNSW Sydney). All raw sequence data are publicly available through the National Center for Biotechnology Information (NCBI)34 under SRA study accession SRP224901. The SRA data record includes 1,596 experiments derived from 1,476 samples.
Microfluidic quantitative polymerase chain reaction (MFQPCR)
The absolute abundance of nutrient (carbon, nitrogen, phosphorus and sulfur) cycling genes and genes associated with pathogens and antibiotic resistance were determined using microfluidic quantitative polymerase chain reaction (MFQPCR) on the Fluidigm platform. Like traditional qPCR, this method enables the determination of gene abundances through the measurement of fluorescence after each PCR cycle, but eliminates much of the manual pipetting from the sensitive qPCR reaction. Its use in ecological research in various environments has significantly increased in recent years, with a focus on fast and reliable detection of pathogens35,36,37.
Gene abundances were measured in two (2) out of three (3) replicates collected for microbial analyses from both the field surveys and WET tests. Suitable primers, as defined by38, were selected from the literature. Five (5) different gBlock gene fragments (Integrated DNA Technologies39) were designed as standards for the assays using targeted sequences for each primer pair sourced from NCBI (refer to Table 3) using Primer-BLAST40. Standard curves were generated using a dilution series of 101 to 107 copies/µL. Exact DNA concentrations of the samples were determined spectrophotometrically using the PicoGreen double-strand DNA kit (Life Technologies) on the ClarioSTAR® microplate reader (BMG Labtech) and samples were diluted to a final DNA concentration of approximately 7–8 ng/µL. To alleviate sample-specific inhibition41, DNA extracts were mixed with T4 gene 32 protein (New England Biolabs®), to a final concentration of 80 ng/uL. Specific target amplification (14 cycles) and MFQPCR were conducted at the UNSW Sydney Ramaciotti Centre for Genomics, as per 38 using Evagreen® chemistry and the 96.96 Fluidigm Dynamic Array™ Integrated Fluidic Circuit. Thermocycling consisted of 95 °C for 1 min, followed by 35 cycles of 96 °C for 5 s and 60 °C for 20 s or 25 s, followed by melt curve analysis for 60–95 °C at a ramp rate of 1 °C/3 s. Reactions were conducted in triplicate in 6.7 nL volumes with a final primer concentration of 700 nM.
Data Records
The data format and metadata for the full archived datasets34,42 is provided in Tables 4–6. The design table in each dataset provides the lookup reference for each sample analysed for raw 16S and 18S rRNA gene sequences uploaded to the NCBI SRA database.
Technical Validation
Single-species WET tests
The singles-species WET tests fulfilled the standard validity criteria43, including less than 20% coefficient of variation; greater than one (1) doublings per day of algal cells in control treatments; and less than 1-unit change in pH over the test duration. Note a reference test using a range of concentrations of copper sulfide (Cu2S: 2 µg/L, 4 µg/L, 8 µg/L, 16 µg/L, 32 µg/L and 64 µg/L) was also completed to ensure normal reactivity of the microalgae, and to provide a base for potential comparisons with tests conducted at different facilities. These concentrations were achieved by adding aliquots of a CuSO4 stock solution (2 mg/L) to soft-water of moderate hardness (100–120 mg CaCO3/L). The reference test using Cu2S showed a standard dose response of the algae, thus confirming the validity of the test conditions.
Flow cytometry
Between samples, fluidics lines were cleared with a wash volume of 500 µL undiluted phosphate-buffered saline (PBS) solution. Additional wash steps using wells containing bleach and then milli-Q water were included between effluent types on each microplate and at the end of each microplate to reduce sample carryover. Positive (stained) and negative (unstained) control samples containing aliquots of river water were also analysed at the end of each microplate for protocol validation.
DNA extraction
Extracted DNA was quantified using a NanoDrop Spectrophotometer (ThermoFisher Scientific) with concentrations greater than 5–10 ng/ul. To test if the 16S rRNA gene in the different samples could be amplified, PCRs were conducted using the same primer set that was later used for amplicon sequencing (27 f/519r). The PCR reaction mixture (50 μL) contained 10 μL of 5-fold MyTaq Reaction Buffer (Bioline, Alexandria, NSW), 0.4 μM of each primer, 1 U of MyTaq DNA Polymerase (Bioline), and approximately 10 ng of DNA as a template. The following thermal cycling scheme was used: initial denaturation at 95 °C for 2 min, 28 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s, followed by extension at 72 °C for 1 min. The final extension was carried out at 72 °C for 2 min. Negative controls were performed by using the reaction mixture without template. Genomic DNA from Escherichia coli DH5α was used in the positive control. PCR products were checked with agarose gel electrophoresis. All samples amplified and amplicon size matched the length of the amplified 16 S rRNA gene (approximately 500 bp).
MFQPCR
The PCR efficiencies of both standards and samples were calculated for each gene using the program LinRegPCR (v2017.1). Only genes with a PCR efficiency of greater than 80% for both standards and samples were further analysed. Further, only those genes with similar PCR efficiencies for the standard and samples (±10%) were used. Afterwards, MFQPCR results were analysed using the Fluidigm Real-Time PCR Analysis program (v4.3.1) using recommended settings (peak sensitivity 7, peak ratio threshold 0.7, quality threshold 0.65). The first analysis step included a manual linear derivative baseline correction. This correction was developed by Fluidigm to correct for baseline drift in real-time PCR data. Ranges of desired melt curve peaks for each primer set were determined manually based on the melt curves of the gBlock standards. Genes with multiple distinguishable melt curve peaks, indicating multiple PCR products due to non-specificity of the primer pair, were excluded from further analyses. Moreover, only samples within the desired melt curve peaks for each primer set were used for further analysis.
Samples were run across two separate 96.96 IFCs; Survey and Ecotox, and quality thresholds were applied separately to each IFC run. A total of 15 primer pairs for genes involved in carbon fixation (rubisco_synecho), nitrogen cycling (napA_3, narG_1, narG, nifD_33, nifH_32 and nirK), sulphur cycling (dsrA, phsA and sir), phosphorous uptake (Acc_ppk1) and pathogen identification (ciaB, ftsZ and stx2) passed the above standard validity tests for the Survey IFC. In addition, the primer pair for the 18S rRNA gene (18S) passed the validity criteria and was included in the analyses as a proxy for the abundance of eukaryotic cells in the samples. However, for genes ciaB and stx2, only single data points (sample-primer combinations) passed the validity tests, thus excluding these two (2) pathogen-related genes from further analyses. For the Ecotox IFC, 12 primer pairs passed the standard validity tests, encompassing the nitrogen cycle (AOA_8, AOB_1, napA_14, napA_3, narG_1, narG, nirS_ef, nrfA_2), sulphur cycling (dsrA, phsA), phosphorous cycle (phoD) and eukaryotes (18s).
Usage Notes
In this study, bacterial cell densities were quantified in the microbial WET tests from frozen samples several months after the initial flow cytometry was done using fresh samples. Previous studies have reported decreases in bacterial44 cell densities between natural and frozen samples. Therefore, these data are recommended for assessment of relative differences between samples since they may reflect a slight underestimation of the bacterial relative to algal cell chlorophyll-a contributions.
While several studies have found that MFQPCR produces copy number estimates that are directly comparable to those produced with traditional qPCR45,46, variations in reaction efficiency are common between samples from different sites47, and in MFQPCR these differences in efficiency may be substantial38. Therefore, whilst copy number tables presented here are suitable for intra-study comparisons and modelling, it is recommended that raw data files are utilised in studies intending to combine inter-study datasets, and uniform efficiency cut-offs be applied prior to further analysis.
References
Corcoran, E. C. et al. Sick Water? The Central Role of Wastewater Management In Sustainable Development. A Rapid Response Assessment. (United Nations Environment Programme, UN-Habitat, GRID-Arendal., 2010).
UN-Water. Wastewater Management - Analytical Brief. (United Nations World Water Assessment Programme, 2015).
UN-WWAP. The United Nations World Water Development Report - Wastewater: The Untapped Resource., (United Nations World Water Assessment Programme, Paris, 2017).
UN-WWAP. Water For People, Water For Life - The United Nations World Water Development Report. (United Nations Educational, Scientific and Cultural Organisation (UNESCO) and Berghahn Books, 2003).
Oelsner, G. P. & Stets, E. G. Recent trends in nutrient and sediment loading to coastal areas of the conterminous U.S.: Insights and global context. Sci. Total Environ. 654, 1225–1240, https://doi.org/10.1016/j.scitotenv.2018.10.437 (2019).
Wurtsbaugh, W. A., Paerl, H. W. & Dodds, W. K. Nutrients, eutrophication and harmful algal blooms along the freshwater to marine continuum. Wiley Interdiscip. Rev.: Water 6, e1373, https://doi.org/10.1002/wat2.1373 (2019).
Drury, B., Rosi-Marshall, E. & Kelly, J. Wastewater treatment effluent reduces the abundance and diversity of benthic bacterial communities in urban and suburban rivers. Appl. Environ. Microbiol. 79, 1897–1905, https://doi.org/10.1128/aem.03527-12 (2013).
Koutsou, O. P., Gatidou, G. & Stasinakis, A. S. Domestic wastewater management in Greece: Greenhouse gas emissions estimation at country scale. J. Cleaner Prod. 188, 851–859, https://doi.org/10.1016/j.jclepro.2018.04.039 (2018).
Nguyen, T. et al. Insight into greenhouse gases emissions from the two popular treatment technologies in municipal wastewater treatment processes. Sci. Total Environ. 671, 1302–1313, https://doi.org/10.1016/j.scitotenv.2019.03.386 (2019).
Zimmer, A., Winkler, I. T. & De Albuquerque, C. Governing wastewater, curbing pollution, and improving water quality for the realization of human rights. Waterlines 33, 337–356, https://doi.org/10.3362/2046-1887.2014.034 (2014).
Sato, T. et al. Global, regional, and country level need for data on wastewater generation, treatment, and use. Agric. Water Manage. 130, 1–13, https://doi.org/10.1016/j.agwat.2013.08.007 (2013).
Small, C. & Nicholls, R. J. A Global Analysis of Human Settlement in Coastal Zones. J. Coast. Res. 19, https://doi.org/10.2307/4299200 (2003).
UN-DESAP. World Population Prospects 2019 - Highlights. (United Nations, Department of Economic and Social Affairs, Population Division, 2019).
Clark, G. & Johnston, E. Australia State of the Environment 2016: Coasts, Independent Report to the Australian Government Minister for Environment and Energy. (Australian Government Department of the Environment and Energy, 2017).
Schindler, D. W. Recent advances in the understanding and management of eutrophication. Limnol. Oceanogr. 51, 356–363, https://doi.org/10.4319/lo.2006.51.1_part_2.0356 (2006).
Mara, D. Domestic Wastewater Treatment In Developing Countries (2013).
Palaniappan, M. et al. Clearing The Waters - A Focus On Water Quality Solutions. (United Nations Environment Programme (UNEP), 2010).
Hassett, B. A. et al. Pulling apart the urbanization axis: patterns of physiochemical degradation and biological response across stream ecosystems. Freshw. Sci. 37, 653–672, https://doi.org/10.1086/699387 (2018).
Kieft, B. et al. Microbial community structure-function relationships in Yaquina Bay estuary reveal spatially distinct carbon and nitrogen cycling capacities. Front. Microbiol. 9, 1282–1282, https://doi.org/10.3389/fmicb.2018.01282 (2018).
Jeffries, T. C. et al. Bacterioplankton dynamics within a large anthropogenically impacted urban estuary. Front. Microbiol. 6, https://doi.org/10.3389/fmicb.2015.01438 (2016).
Birrer, S. C., Dafforn, K. A. & Johnston, E. L. Microbial Community Responses to Contaminants and the Use of Molecular Techniques in Microbial Ecotoxicology (eds. Cravo-Laureau, C., Cagnon, C., Lauga, B. & Duran, R.), 165–183 (Springer International Publishing, 2017).
Carney, R. L. et al. River bacterioplankton community responses to a high inflow event. Aquat. Microb. Ecol. 75, 187–205, https://doi.org/10.3354/ame01758 (2015).
Roy, P. S. et al. Structure and function of south-east Australian estuaries. Estuarine Coastal Shelf Sci. 53, 351–384, https://doi.org/10.1006/ecss.2001.0796 (2001).
Ferguson, A., Potts, J., Dawson, G. & Mitrovic, S. M. Biogeochemical Function Of The Hunter River Tidal System - An Empirically Based Conceptual Model. (NSW Government, Office of Environment and Heritage (OEH), 2018).
Russell, K., Erskine, J. & Glamore, W. C. Tomago Wetland Rehabilitation Project: Integrated, Innovative Approaches. NSW Coastal Conference (Kiama, NSW, 2012).
US EPA. Short-Term Methods for Estimating Chronic Toxicity of Effluents and Receiving Waters to Freshwater Organisms. 4th edn (United States Environment Protection Agency, 2002).
Environment Canada. Biological Test Method: Growth Inhibition Test Using A Freshwater Alga. 2nd edn (2007).
Brown, M. R. et al. A flow cytometry method for bacterial quantification and biomass estimates in activated sludge. J. Microbiol. Methods 160, 73–83, https://doi.org/10.1016/j.mimet.2019.03.022 (2019).
Duhamel, S. & Jacquet, S. Flow cytometric analysis of bacteria- and virus-like particles in lake sediments. J. Microbiol. Methods 64, 316–332, https://doi.org/10.1016/j.mimet.2005.05.008 (2006).
Marie, D., Partensky, F., Vaulot, D. & Brussaard, C. Enumeration of phytoplankton, bacteria, and viruses in marine samples. Curr. Protoc. Cytom. 10, https://doi.org/10.1002/0471142956.cy1111s10 (1999).
Lane, D. J. 16S/23S rRNA sequencing in Nucleic acid techniques in bacterial systematics (eds. Stackebrandt, E. & Goodfellow, M.), 115-175 (John Wiley and Sons, New York, NY, 1991).
Lane, D. et al. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 82, 6955–6959, https://doi.org/10.1073/pnas.82.20.6955 (1985).
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W. & Huse, S. M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PloS one 4, https://doi.org/10.1371/journal.pone.0006372 (2009).
Ruprecht, J. E. et al. Next-generation sequencing of 16S and 18S rRNA genes in Hunter River estuary. NCBI Sequence Read Archive, https://identifiers.org/insdc.sra:SRP224901 (2019).
Ishii, S. et al. Water quality monitoring and risk assessment by simultaneous multipathogen quantification. Environ. Sci. Technol. 48, 4744–4749, https://doi.org/10.1021/es500578s (2014).
Ishii, S., Segawa, T. & Okabe, S. Simultaneous quantification of multiple food-and waterborne pathogens by use of microfluidic quantitative PCR. Appl. Environ. Microbiol. 79, 2891–2898, https://doi.org/10.1128/AEM.00205-13 (2013).
Zhang, Q. et al. Correlations between pathogen concentration and fecal indicator marker genes in beach environments. Sci. Total Environ. 573, 826–830, https://doi.org/10.1016/j.scitotenv.2016.08.122 (2016).
Crane, S. L. et al. Microfluidic qPCR enables high throughput quantification of microbial functional genes but requires strict curation of primers. Front. Environ. Sci. 6, https://doi.org/10.3389/fenvs.2018.00145 (2018).
Sadik, N. et al. Quantification of multiple waterborne pathogens in drinking water, drainage channels, and surface water in Kampala, Uganda, during seasonal variation. GeoHealth 1, 258–269, https://doi.org/10.1002/2017GH000081 (2017).
Ye, J. et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 13, 134, https://doi.org/10.1186/1471-2105-13-134 (2012).
Dandie, C. E. et al. Nitric oxide reductase-targeted real-time PCR quantification of denitrifier populations in soil. Appl. Environ. Microbiol. 73, 4250–4258, https://doi.org/10.1128/AEM.00081-07 (2007).
Ruprecht, J. E. et al. A novel real-world ecotoxicological dataset of pelagic microbial community responses to wastewater. Dryad, https://doi.org/10.5061/dryad.2fqz612km (2020).
OECD. Test No. 201: Freshwater Alga and Cyanobacteria, Growth Inhibition Test, https://doi.org/10.1787/9789264069923-en (2011).
Kamiya, E. et al. Effects of fixation and storage on flow cytometric analysis of marine bacteria. J. Oceanogr. 63, 101–112, https://doi.org/10.1007/s10872-007-0008-7 (2007).
Ishii, S. et al. Microfluidic quantitative PCR for simultaneous quantification of multiple viruses in environmental water samples. Appl. Environ. Microbiol. 80, 7505–7511, https://doi.org/10.1128/AEM.02578-14 (2014).
Byappanahalli, M. N., Nevers, M. B., Whitman, R. L. & Ishii, S. Application of a microfluidic quantitative polymerase chain reaction technique to monitor bacterial pathogens in beach water and complex environmental matrices. Environ. Sci. Tech. Lett. 2, 347–351, https://doi.org/10.1021/acs.estlett.5b00251 (2015).
Brankatschk, R., Bodenhausen, N., Zeyer, J. & Bürgmann, H. Efficiency of real-time qPCR depends on the template: a simple absolute quantification method correcting for qPCR efficiency variations in microbial community samples. Appl. Environ. Microbiol., 07878–07811, https://doi.org/10.1128/AEM.07878-11 (2012).
Maeda, H. et al. Quantitative real‐time PCR using TaqMan and SYBR Green for Actinobacillus Actinomycetemcomitans, Porphyromonas Gingivalis, Prevotella Intermedia, tetQ gene and total bacteria. FEMS Immunol. Med. Microbiol. 39, 81–86, https://doi.org/10.1016/S0928-8244(03)00224-4 (2003).
Zhu, F. et al. Mapping of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol. Ecol. 52, 79–92, https://doi.org/10.1016/j.femsec.2004.10.006 (2005).
He, S., Gall, D. L. & McMahon, K. D. “Candidatus Accumulibacter” population structure in enhanced biological phosphorus removal sludges as revealed by polyphosphate kinase genes. Appl. Environ. Microbiol. 73, 5865–5874, https://doi.org/10.1128/AEM.01207-07 (2007).
Zeng, W. et al. Community structures and population dynamics of “Candidatus Accumulibacter” in activated sludges of wastewater treatment plants using ppk1 as phylogenetic marker. J. Environ. Sci. 67, 237–248, https://doi.org/10.1016/j.jes.2017.09.001 (2018).
Fernando, D. M. et al. Detection of antibiotic resistance genes in source and drinking water samples from a first nations community in Canada. Appl. Environ. Microbiol. 82, 4767–4775, https://doi.org/10.1128/AEM.00798-16 (2016).
Ishii, S., Segawa, T. & Okabe, S. Simultaneous quantification of multiple food and waterborne pathogens by use of microfluidic quantitative PCR. Appl. Environ. Microbiol. 9, 2891–2898, https://doi.org/10.1128/AEM.00205-13 (2013).
Ben-Dov, E., Brenner, A. & Kushmaro, A. Quantification of sulfate-reducing bacteria in industrial wastewater by real-time polymerase chain reaction (PCR) using dsrA and apsA Genes. Microb. Ecol. 54, 439–451, https://doi.org/10.1007/s00248-007-9233-2 (2007).
Smith, C. J., Nedwell, D. B., Dong, L. F. & Osborn, A. M. Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Appl. Environ. Microbiol. 73, 3612–3622, https://doi.org/10.1128/AEM.02894-06 (2007).
López-Gutiérrez, J. C. et al. Quantification of a novel group of nitrate-reducing bacteria in the environment by real-time PCR. J. Microbiol. Meth. 57, 399–407, https://doi.org/10.1016/j.mimet.2004.02.009 (2004).
Henry, S. et al. Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR. J. Microbiol. Meth. 59, 327–335, https://doi.org/10.1016/j.mimet.2004.07.002 (2004).
Braker, G., Fesefeldt, A. & Witzel, K. P. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64, 3769–3775 (1998).
Henry, S. et al. Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl. Environ. Microbiol. 72, 5181–5189, https://doi.org/10.1128/AEM.00231-06 (2006).
Feng, T. et al. Examination of metabolic responses to phosphorus limitation via proteomic analyses in the marine diatom Phaeodactylum tricornutum. Sci. Rep. 5, 10373, https://doi.org/10.1038/srep10373 (2015).
Fraser, T. D. et al. Quantification of bacterial non-specific acid (phoC) and alkaline (phoD) phosphatase genes in bulk and rhizosphere soil from organically managed soybean fields. Appl. Soil Ecol. 111, 48–56, https://doi.org/10.1016/j.apsoil.2016.11.013 (2017).
Christensen, G. A. The Role Of Rex In Regulation Of Sulfate Reduction In Desulfovibrio Vulgaris Hildenborough, University of Missouri–Columbia (2014).
Selesi, D. et al. Quantification of bacterial RubisCO genes in soils by cbbL targeted real-time PCR. J. Microbiol. Meth. 69, 497–503, https://doi.org/10.1016/j.mimet.2007.03.002 (2007).
Selesi, D., Schmid, M. & Hartmann, A. Diversity of green-like and red-like ribulose-1,5-bisphosphate carboxylase/oxygenase large-subunit genes (cbbL) in differently managed agricultural soils. Appl. Environ. Microbiol. 71, 175–184, https://doi.org/10.1128/AEM.71.1.175-184.2005 (2005).
John, D. E. et al. Phytoplankton carbon fixation gene (RuBisCO) transcripts and air-sea CO2 flux in the Mississippi River plume. The ISME Journal 1, 517–531, https://doi.org/10.1038/ismej.2007.70 (2007).
Kettles, N., Kopriva, S. & Malin, G. Insights into the regulation of DMSP synthesis in the diatom Thalassiosira Pseudonana through APR activity, proteomics and gene expression analyses on cells acclimating to changes in salinity, light and nitrogen. PloS one 9, e94795, https://doi.org/10.1371/journal.pone.0094795 (2014).
Acknowledgements
This dataset was collected as part of a larger project on the Hunter River estuary funded by Hunter Water Corporation. JER was supported by an Australian Government Research Training Program Scholarship. The authors would like to thank all staff and scientists who helped with the ecotoxicology experiments at the Sydney Institute of Marine Science. Special thanks also go to Chris Brownlee and Dr Emma Johansson Beves at the Mark Wainwright Analytical Centre (Flow Cytometry Facility) at UNSW Sydney for their assistance with collection of the cell count data.
Author information
Authors and Affiliations
Contributions
J.E.R., W.C.G., S.C.B., K.A.D. and E.L.J. were involved with the conceptualisation and design of the data collection program and experiments. J.E.R., S.C.B., S.L.C., J.V.D. and F.W. were involved with data acquisition and analysis. J.E.R. lead the writing of the manuscript. All authors were involved with the interpretation of the data and review of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Ruprecht, J.E., Glamore, W.C., Dafforn, K.A. et al. A novel real-world ecotoxicological dataset of pelagic microbial community responses to wastewater. Sci Data 7, 158 (2020). https://doi.org/10.1038/s41597-020-0496-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0496-5
