
Abstract
Zoonotic Salmonella causes millions of human salmonellosis infections worldwide each year. Information about the source of the bacteria guides risk managers on control and preventive strategies. Source attribution is the effort to quantify the number of sporadic human cases of a specific illness to specific sources and animal reservoirs. Source attribution methods for Salmonella have so far been based on traditional wet-lab typing methods. With the change to whole genome sequencing there is a need to develop new methods for source attribution based on sequencing data. Four European datasets collected in Denmark (DK), Germany (DE), the United Kingdom (UK) and France (FR) are presented in this descriptor. The datasets contain sequenced samples of Salmonella Typhimurium and its monophasic variants isolated from human, food, animal and the environment. The objective of the datasets was either to attribute the human salmonellosis cases to animal reservoirs or to investigate contamination of the environment by attributing the environmental isolates to different animal reservoirs.
Measurement(s) | sequence_assembly • DNA |
Technology Type(s) | sequence assembly process • DNA sequencing • Paired-End Sequencing |
Factor Type(s) | geographic location • Salmonella Typhimurium isolate source |
Sample Characteristic - Organism | Salmonella |
Sample Characteristic - Location | Kingdom of Denmark • Germany • United Kingdom • French Republic |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.11816748
Similar content being viewed by others

Background & Summary
The datasets described in this descriptor were collected in the context of Work Package 4/7 of the Collaborative Management Platform for detection and Analyses of (Re-) emerging and foodborne outbreaks in Europe (COMPARE, Horizon2020 research project grant number 643476). This research network aims to develop general analytical workflows for population-based disease surveillance, outbreak detection and epidemiological modeling of foodborne infections. A specific task therein was to develop methods for source attribution applying whole genome sequencing (WGS)-based surveillance and outbreak data for foodborne pathogens. In brief, source attribution models estimate the number and/or proportion of (human) cases of a specific foodborne illness that can be attributed to specific food categories and animal reservoirs1. Source attribution models require data from humans and potential sources that are: (1) representative of what the human population is exposed to, (2) related in time and space2, (3) harmonized regarding categorization of the sources1 and (4) analysed using a discriminatory subtyping method1. Data collected through integrated surveillance systems of humans, food and animals complies with these requirements2.
The potential of applying sequence data for source attribution purposes based on a machine learning approach has recently been reported3,4 and discussed5. These agree on the potential of the machine learning method to discriminate between different sources and applicability to trace foodborne outbreaks. In comparison to the datasets used by Zhang et al. (2019) and Lupolova et al. (2018), our datasets consist of food and animal data collected in a narrow time frame (3–5 years) and specific geographical area (country) and to the extent possible include a large representative sample of all human Salmonella Typhimurium infections reported in the country in the same time period. With such datasets, we believe we can make inferences on the relative contribution of the included sources (food and animals) to the number of human infections in the study period. In the study by Zhang et al. (2019), 51 human cases were attributed (or predicted) to animal reservoirs. However, the isolates from food, animals and humans were collected over a much wider time span and therefore more appropriate for studying the evolution of Salmonella Typhimurium in livestock sources and the relation to humans, than for quantifying the contribution of specific sources to human infections occurring in a shorter time period. If applied regularly on surveillance data, such quantification of the contribution of specific sources to human infections can be used to inform food-safety decision-making and to monitor the effect of control initiatives.
A benchmarking study was established to venture further into this subject and explore and assess different bioinformatics analyses and source attribution models based on sequencing data and to provide recommendations on how to evaluate and select the best approach for a given dataset. The results of the benchmark study will be reported elsewhere. In this paper, we describe in detail the four datasets that were applied including the sampling frameworks, metadata, sequencing techniques and quality control analyses (Fig. 1). Three of the datasets, representing data from Denmark, Germany and the United Kingdom, consist of strains of Salmonella enterica serovar Typhimurium incl. monophasic variants from humans and different animal reservoirs and food including pigs/pork, cattle/beef, broilers/chicken meat and laying hens. The fourth dataset from France includes a selection of environmental strains as well as strains from animal reservoirs. A total of 1,781 strains from humans (n = 943), animals (n = 804) and environment (n = 34) were collected as part of this study (Table 1). A number of the UK human data was outbreak cases and therefore excluded.

Flow chart of data generation, data management and quality control. dcc_vivaldi8: a private datahub (https://www.ebi.ac.uk/ena/pathogens/login) set up for data sharing and hosted at the European Nucleotide Archive (ENA). Computerome: a local server used by DTU Food (https://www.computerome.dk). Prokka: Rapid Prokaryotic genome annotation32. Roary: Rapid prokaryotic genome annotation33. cgMLST and wgMLST obtained using the Enterobase scheme11 in BioNumerics version 7.6 (Applied Maths, Sint Martens Latem, Belgium).
As the use of genomics data for source attribution is still nascent6 we consider the datasets valuable for other researchers who seek to explore new approaches or for comparing the performance of their own models and datasets with those presented here. Partners involved in this project were microbiologists, epidemiologists and bioinformaticians from the following institutes: The National Food Institute (DTU Food), Statens Serum Institute (SSI), Robert Koch Institute (RKI), German Federal Institute for Risk Assessment (BfR), French Agency for Food, Environmental and Occupational Health & Safety (ANSES), French Research Institute for Exploitation of the Sea (Ifremer), University of Bologna (UNIBO), Animal & Plant Health Agency (APHA), and Public Health England (PHE).
Methods
Sampling procedure
A representative set of sequenced isolates of Salmonella Typhimurium strains, including its monophasic variants, from humans and different environmental and animal sources, locations and years were collected from four different countries: Denmark, France, Germany, and the United Kingdom. The isolates were available through national surveillance/monitoring/control programs/research projects or larger surveys conducted between 2010 and 2016. Isolates from animals and food represent the major food animal reservoirs and thus reflect what humans are exposed to through consumption of food. Only one isolate per farm or food batch was included, and clinical isolates of animals were not considered. As a minimum, the following animal species and food types were covered: broilers/chicken meat (fresh), pigs/pork (fresh) and cattle/beef (fresh). The minimum sample size was initially set to 25 isolates per animal species per country per year7. However, this number of isolates was not always available, due to low Salmonella occurrence and low sample size of some sources. If available, isolates from other animal species and/or their related meat type, from fruit and vegetables and environment were included as well with a minimum sample size of 10 samples per category per country per year. The minimum sample size for humans was 100 isolates per country per year.
Data sharing
Metadata
Metadata was shared among partner institutions via a database in Microsoft Excel format set up for the purpose and shared via the COMPARE share site. Dropdown menus defining categories for variables that could be standardized, such as “Primary source” and “Patient travel”, were set up using the Microsoft Excel function “Data validation” facilitating standardized registration of metadata. Each partner added sequence information and epidemiological data to the database (Table 2). A unique “sample ID” was provided for each sample. Validation of metadata was performed by DTU by assuring data was added in the correct format. In case of errors, the data provider was asked for correction.
Sequences
Raw reads were shared in a private datahub called dcc_Vivaldi8 (https://www.ebi.ac.uk/ena/pathogens/login) set up for the purpose and hosted at the European Nucleotide Archive (ENA). An accession number was linked to the single sequences upon uploading to ENA. Personal access credentials to dcc_Vivaldi datahub were granted by ENA whenever consent was obtained from a representative of each institution involved in the COMPARE Work Package 4/7. A subset of sequences was already publicly available and obtained from the Sequence Read Archive (SRA), these are designated with SRR IDs. Metadata and sequences were linked via the unique sample ID and accession number.
Before analyzing, the sequence data was transferred from the dcc_Vivaldi datahub to a local server such as Computerome (https://www.computerome.dk) used by DTU Food. Transferring these data required a stable internet connection during the transfer. All isolates were sequenced using Illumina chemistry producing paired end reads.
Assemblies
Assemblies were generated by the in-house software called FoodQCPipeline (https://bitbucket.org/RolfKaas/foodqcpipeline) and shared on a password-protected FTP server set up by DTU Food. Reads were de novo assembled using SPAdes 3.11.09 in last step of the pipeline. FoodQCPipeline trimmed the raw reads using bbduk2 (part of BBMap version 36.49, https://jgi.doe.gov/data-and-tools/bbtools/) according to the following: (1) length of read must be higher or equal to 50 base pairs (bp), otherwise were excluded, (2) phred score per base higher or equal to 20 and (3) filter away adapters based on an internal database with Illumina adapters that was created and maintained by DTU Food. FastQC10 version 0.11.5 was applied to the reads before and after trimming generating a quality control report for every sample. The quality of the de novo assemblies were assessed using Quast version 4.5.
Core genome Multi-locus sequence typing (cgMLST) and whole genome Multi-locus sequence typing (wgMLST) analysis were used to generate input to different source attribution methods developed in the COMPARE project. cgMLST and wgMLST were obtained using the Enterobase scheme11 in BioNumerics version 7.6 (Applied Maths, Sint Martens Latem, Belgium). The cgMLST is based on 3,002 loci and the wgMLST on 21,065 loci with one single locus having several allele variations11. cgMLST allele calls were accepted for strains with a core genome coverage higher than 95% (2,852) of 3,002 core genomes alleles and a detection of mixed sequence alleles lower than 50 alleles.
Quality control
A 2-step quality control was applied to all sequences. First, outcomes from the FoodQC pipeline were assessed. Secondly, outcomes resulting from the quality control that is part of BioNumerics11 were assessed. The applied inclusion criteria are listed below.
From the FoodQC pipeline the following were assessed (1) fewer than 500 contigs (where contigs are >500 bp each), with an N50 value preferable larger than 30,000 bp, (2) total number of base pairs in contigs larger than 500 bp summed to approximately 5,000,000 according to the size of the Salmonella bacterial genome, (3) depth of coverage equal to or higher than 30x calculated as (base pairs sequenced)/(base pairs in assembly (from (2)) and (4) phred score equal to or higher than 20, which is calculated as part of the trimming process.
From BioNumerics the following were assessed based on the cgMLST calls (5) core genome coverage higher than 95% of 3,002 core genomes alleles and (6) mixed sequence alleles detected lower than 50 alleles.
Final datasets
All sequences with acceptable quality were included in the final datasets. Table 1 outlines the sequences collected, number of isolates that passed the quality control and the final datasets included in the study.
Applicability of the datasets for source attribution purposes
The phylogeny of all four individual datasets was analysed in order to examine the applicability of the four different datasets to develop new source attribution models. Datasets were assumed applicable when human salmonellosis cases or environmental strains were intermixed with the food and animal sources. Maximum likelihood phylogenetic trees were constructed from sequence variations in the genome shared between strains included in the given analysis using FastTree (gtr + cat model12). Sequence variations were defined as the single nucleotide polymorphisms (SNPs) within the genome shared between the strains included in the given analysis. SNPs were identified using the CSI phylogeny13,14, freely available from the Center for Genomic Epidemiology (www.genomicepidemiology.org) and described in more details in the following. Trimmed paired-end reads of each isolate included in the given analysis were aligned against the COMPARE reference genome, Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 (AE006468.2, 4,857,432 base pairs)15 using Burrows-Wheeler Aligner (BWA) version 0.7.216. The SNPs were identified using ‘mpileup’ module in SAMtools version 0.1.1817. SNPs fulfilling the following criteria were selected: (1) a minimum distance of 15 bps between each SNP (pruning), (2) a minimum of 10% of the average depth, (3) mapping quality above 30, (4) the SNP quality was more than 20, and (5) all indels were excluded. The selected SNPs from each genome were concatenated into a single pseudo alignment corresponding to the position of the reference genome. Phylogenetic trees were annotated and visualized using iTOL18 and distances between isolates equivalent to the amount of SNPs between them.
Communication
The datasets including the metadata, quality control criteria and strategies for sharing of data were continuously discussed at regular face-to-face meetings. In between the face-to-face meetings, continuous email dialogue and tele conferences were held to inform about progress and clarify any misunderstandings.
Data and sampling
This section lists the reasons for choosing Salmonella Typhimurium and its monophasic variants as study organism and describes the sampling plan for all four European datasets. The Danish, German and British datasets were collected with the objective of attributing the number of reported sporadic human salmonellosis cases to animal reservoirs and food. The French dataset was collected with the objective of attributing environmental Salmonella Typhimurium strains to animal reservoirs.
Data
Salmonella Typhimurium including its monophasic variants is the second most prevalent serotype in humans in EU and most EU member states19. During the last few years, monophasic variants of Salmonella Typhimurium have been dominating human cases and have also repeatedly been involved in food-borne outbreaks20. Salmonella Typhimurium is a major serotype in pigs, but is also commonly found in a number of other food-animal reservoirs (e.g. poultry and cattle) and environmental samples. This is in contrast to other Salmonella serotypes such as Salmonella Enteritidis and Salmonella Dublin which are mainly associated with poultry and bovines, respectively. It was, therefore, decided to focus on attribution of human infections caused by Salmonella Typhimurium and its monophasic variants.
Data sampling Denmark
Samples from human surveillance included in this dataset
Clinical cases of Salmonella in humans in Denmark are notifiable through the laboratory surveillance systems at Statens Serum Institut (SSI). SSI receives isolates from the Danish hospitals and is responsible for the pheno- and genotyping of clinical Salmonella isolates. Information regarding travelling abroad before disease onset was obtained from either travel interviews or general practitioner, and information about outbreak cases was available from SSI and registered in the Food- and Waterborne Outbreak Database21.
Samples from food and animal surveillance included in this dataset
All major food animals and food of animal origin are monitored for Salmonella through national surveillance programs. In addition, results from centrally coordinated studies supplement the surveillance programs, particularly regarding data on imported food of animal origin. The surveillance and monitoring programs are regularly revised and their contents are described in detail in the Annual Reports on Zoonoses in Denmark22,23. The following describes the content of the programs as they were during the years 2013–2014. Samples from animal and food were analysed at authorized private laboratories, the Danish Food and Veterinary Administration’s laboratory and the Technical University of Denmark (DTU). The National Food Institute at DTU performed serotyping, WGS and antimicrobial resistance testing.
Every commercial flock of layers was tested every 9 weeks before 1/10/2013 and every two weeks thereafter. All commercial flocks of broilers were tested two times at approximately three weeks and again one week before slaughter. All commercial flocks of turkeys and ducks were tested approximately three weeks before slaughter. There were no samples from flocks of ducks available in 2014. Every batch of broiler carcasses was tested after slaughter by the examination of a pool of neck skin samples. Pork and beef were sampled as pooled carcass swabs. The number of samples collected from each slaughterhouse was proportional to the number of animals slaughtered. Samples of imported pork and chicken meat were collected randomly at importers’ premises throughout the year. Finally, through centrally coordinated surveys, samples of both imported and Danish beef and duck meat were obtained at the retail level. All Salmonella surveillance and monitoring programmes, from which the reported data originates, are described in the Annual Report on Zoonoses in Denmark22,23.
In total, 325 samples of Salmonella Typhimurium and its monophasic variants isolated from domestic and imported food and animals were collected via the national Salmonella surveillance programs for animals and food during 2013 and 2014. Of these, 65% were available for sequencing and thus included in the dataset for this specific study. In total, 764 samples of Salmonella Typhimurium and its monophasic variants were isolated from humans in 2013 and 2014. Of these, 18% were available for sequencing and included in the human dataset for this specific study. All isolates were sequenced using an Illumina HiSeq, NextSeq or MiSeq sequencing machine. The isolates in the Danish dataset presented here originate from a quite intensive sampling as described above and the dataset per se is thus representative of Salmonella Typhimurium and its monophasic variants. Few isolates from Danish cattle and layer flocks reflects a very low prevalence.
This Danish dataset was used to attribute the number of reported human salmonellosis cases to animal reservoirs and food.
Data sampling Germany
Samples from human surveillance included in this dataset
In Germany, the detection of Salmonella indicating an acute human salmonellosis is notifiable. Laboratories have to report this diagnostic finding to the respective local health authority of the district where the patient lives. Local health authorities then forward all the information relevant for surveillance via the state health authorities to the Robert Koch Institute as the national public health institute. Laboratories can also send a Salmonella isolate to the National Reference Center (NRC) for Salmonella and other bacterial enteric pathogens which is located at the Robert Koch Institute in Wernigerode, Germany, for further typing. However, forwarding of Salmonella isolates to the NRC is not mandatory. The NRC estimates receiving about 20% of isolates of all notified cases. Of all the Salmonella Typhimurium or the monophasic variants confirmed isolates which have been submitted to the NRC between the years 2014 and 2016, a random selection was chosen: 100 human isolates per year. For known outbreaks, only one isolate per outbreak was included.
Samples from food and animal monitoring included in this dataset
Food and animal isolates which originate either from official sampling or companies’ self-monitoring can be send to the National Reference Laboratory for the Analysis and Testing of Zoonoses (NRL Salmonella) located at the German Federal Institute for Risk Assessment (BfR) for further typing. Sending is not mandatory which means that the isolates at the NRL are not necessarily representative for all food and animal isolates in Germany. In accordance to the human isolates, a random selection was chosen according to the minimum sample size per source and year (25 isolates). Isolates originating from the same farm were excluded.
After checking the comparability of sequence quality between NRC and NRL, food and animal isolates were sequenced at the BfR. For library preparation, the Nextera XT kit was used and sequencing was performed on an Illumina MiSeq benchtop sequencer. Sequence data were then send to the NRC for further analysis.
This German dataset was used to attribute the number of reported human salmonellosis cases to animal reservoirs and food.
Data sampling UK
Samples from human surveillance included in this dataset
Sequences from 177 Salmonella Typhimurium isolates from human infections collected and sequenced by Public Health England as part of the notifiable routine surveillance of human infections through the National Health Service were included in this study. Information regarding foreign travel before disease onset was obtained based on declaration from the sending physician.
Samples from food monitoring included in this dataset
The Animal and Plant Health Agency (APHA) receives reports of all Salmonella isolates from cattle, deer, goats, horses, pigs, rabbits, sheep, chickens, turkeys, ducks, geese, guinea fowl, partridges, pheasants, pigeons and quail, as required by the Zoonoses Order 1989 (http://www.legislation.gov.uk/uksi/1989/285/made). Under the 1989 Order the responsibility for reporting the isolation of Salmonella is placed on the laboratory carrying out the examination or in the case of examinations elsewhere, the person carrying out the examination. In practice, all reports of Salmonella isolations must be made to a Veterinary Officer at one of the Veterinary Investigation Centres (VICs) of the Animal and Plant Health Agency (APHA) or to a Regional Veterinary Lead in Scotland. A culture of the organism must be made available. Many isolations of Salmonella from livestock are not associated with clinical disease or occur on farm premises where Salmonella has been isolated from a group of animals rather than an individual. Further information on the reporting of Salmonella, Salmonella culture methods, phage typing and antimicrobial sensitivity testing methods is available in The Salmonella in Livestock production in GB reports available on the APHA website: https://www.gov.uk/government/publications/salmonella-in-livestock-production-in-great-britain-2017.
The objective of this dataset was to attribute the number of reported human salmonellosis cases to animal reservoirs and food.
Data sampling France
Samples from food and animal monitoring included in this dataset
The dataset of strains used to characterize reservoirs mainly comes from a targeted national epidemiological surveillance system, called “The Salmonella network” and coordinated by ANSES’s Laboratory of Food Safety (based in Maisons-Alfort). “The Salmonella network” was established in 1997 in order to monitor Salmonella strains of non-human origin isolated from all stages of the food chain24. Salmonella strains and serotyping data are submitted to ANSES on a voluntary basis from public and private laboratories spread across the whole country. In the ANSES’s laboratories, serological and/or molecular typing (e.g. PCR, MLVA, PFGE and sequencing for the characterization of Salmonella Typhimurium variants) are performed on Salmonella strains, which are isolated from a variety of matrices such as sick and healthy animals, human food and feed and environments (natural environment, farm and processing plants). The typing data are generally associated with epidemiological data (e.g. country of isolation, the sample type, the sampling site, the context of isolation, etc.).
For the aim of this study, a dataset of 69 strains of Salmonella Typhimurium and its monophasic variants, isolated from different reservoirs between 2010–2014, were collected and whole genome sequenced.
A total of 49 strains of Salmonella were isolated from pigs, 14 from poultry (layers, broilers, turkeys and ducks) and 6 from ruminants (cattle, sheep and goat).
Samples from environment included in this dataset
Twenty eight (28) strains of Salmonella were isolated from environment (from fresh or brackish water as well as soil isolates) from both ANSES and IFREMER with the collaboration of University of Caen (France). The 15 IFREMER’s environmental isolates originate from a research project25. Institute Pasteur (France) determined the serotype (isolates from IFREMER). ANSES environmental isolates have been collected by the Salmonella network’s passive surveillance. They were isolated from soils (three strains) and fresh water (ten strains).
One additional strain isolated in crustacean (specifically, shellfish) was associated to the environmental dataset.
The objective of this French dataset was to attribute environmental Salmonella Typhimurium strains and its monophasic variants to animal reservoirs.
Data Records
The final datasets are described in Tables 3, 4, 5 and 6. All sequences included are available for download through ENA (DK dataset26,27,28, DE dataset27, FR dataset27 and UK animal data29) and SRA (UK human30), where they are located. Sequences from SRA were publicly available before the beginning of this study. Assemblies, associated metadata and quality control of sequences with acceptable quality are available at figshare31.
Technical Validation
All sequences of the four individual datasets were quality controlled to confirm the quality of the sequences further described in the Methods section. Isolates for which the quality was unacceptable (n = 403) were excluded from further studies as were isolates excluded for other reasons (n = 119). Output of the quality control per isolate with acceptable quality (n = 1,259) are included in the files at figshare31. Mean, minimum and maximum value of parameters of interest stratified for the different datasets are included in Table 7.
Phylogeny
The applicability of the four datasets to develop new source attribution models was assessed by examining the population structure obtained from the phylogenetic analysis (Figs. 2, 3, 4 and 5).

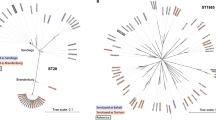
Phylogeny of the Danish dataset. //: Branch length to outgroup ST36 reduced by 30 from 0.71421 to 0.023807. Isolates are annotated by source. Light red: domestically produced pigs, Pigs(DK), light pink: imported pigs, Pigs(Import), yellow; imported ducks, Ducks(Import), blue: domestically produced broilers, Broilers(DK), turquoise: domestically produced eggs, Layers(DK), light green: domestically produced cattle, Cattle(DK), dark green: imported cattle, Cattle(Import), dark grey: Danish human salmonellosis cases, Humans(DK).

Phylogeny of the German dataset. //: Branch length to outgroup ST36 reduced by 30 from 0.73924 to 0.0246413333. Isolates are annotated by source. Light red: domestically produced pigs, Pigs(DE), red: domestically birds, Birds(DE), blue: domestically produced broilers, Broilers(DE), turquoise: domestically produced eggs, Layers(DE), dark green: domestically produced cattle, Cattle(DE), purple: domestically game, Game(DE), dark grey: German human salmonellosis cases, Human(DE).

Phylogeny of the British dataset. Isolates are annotated by source. Light red: domestically produced pigs, Pigs(UK), light green: domestically produced turkey, Turkey(UK), blue: domestically produced broilers, Broilers(UK), turquoise: domestically produced eggs, Layers(UK), dark green: domestically produced cattle, Cattle(UK), brown: domestically produced sheep, Sheep(UK), red: Game(UK), yellow: reptiles(UK), purple: other mammals, dark grey: British human salmonellosis cases.

Phylogeny of the French dataset. Isolates are annotated by source. Light red: domestically produced pigs, Pigs(FR), green: domestically produced turkey, Turkey(FR), light blue: domestically produced ducks, Ducks(FR), blue: domestic produced broilers, Broilers(FR), orange: domestically produced eggs, Layers(FR), dark green: domestically produced cattle, Cattle(FR), brown: domestically produced sheep goats, SheepGoat(FR), purple: domestically produced crustaceans, Crustaceans(FR), dark grey: French environment samples, Environment(FR).
From the population structure of the Danish, German and British datasets, human isolates were seen to be intermixed with the potential food and animal sources. From the population structure of the French dataset environmental isolates were seen to be intermixed with the potential sources of contamination. It was therefore concluded that all four datasets were applicable for development of new source attribution models.
Usage Notes
All four datasets and a selection of the metadata provided here are available for download through ENA26,27,28,29 or SRA30. Accession numbers are listed in figshare31. The accession numbers are associated with larger projects and consequently include isolates not discussed in this descriptor. The curated datasets presented in this descriptor are described in figshare31.
These datasets were collected and sequenced with the purpose of developing new source attribution models based on sequencing data. This descriptor argues that all four datasets are applicable for this purpose. The four datasets can be used to further develop and benchmark source attribution models using new and emerging bioinformatics analysis and mathematical models. Next step in developing the new source attribution models could therefore be to apply further bioinformatics analyses to the datasets, such as core genome MLST, whole genome MLST and distance matrices and test these as input to statistical models such as machine learning and Bayesian based models as well as to population genetic methods such as the asymmetric island model, STRUCTURE and Network Analysis. This extensive work has already been initiated by the WP4/7 of COMPARE as conceptualized in Fig. 1.
Code availability
No code per se was developed for this article, as available tools were applied. The quality control pipeline used is available at bitbucket: https://bitbucket.org/RolfKaas/foodqcpipeline/ The CSI Phylogeny pipeline is available as a webtool through Center for Genomic Epidemiology (www.genomicepidemiology.org). Following options were used: Select min. depth at SNP positions: 10x. Select min. relative depth at SNP positions: 10%. Select minimum distance between SNPs (prune): 10. Select min. SNP quality: 30. Select min. read mapping quality: 25. Select min. Z-score: 1.96.
References
Pires, S. M. et al. Attributing the human disease burden of foodborne infections to specific sources. Foodborne Pathog. Dis. 6, 417–24 (2009).
EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards), E. P. Scientific Opinion on the evaluation of molecular typing methods for major food-borne microbiological hazards and their use for attribution modelling, outbreak investigation and scanning surveillance: Part 1 (evaluation of methods and applications). EFSA J. 11, 3502 (2013).
Zhang, S. et al. Zoonotic source attribution of Salmonella enterica serotype typhimurium using genomic surveillance data, United States. Emerg. Infect. Dis. 25, 82–91 (2019).
Lupolova, N., Dallman, T. J., Holden, N. J. & Gally, D. L. Patchy promiscuity: machine learning applied to predict the host specificity of Salmonella enterica and Escherichia coli. Microb. Genomics 3, 1–10 (2017).
Wheeler, N. E. Tracing outbreaks with machine learning. Nature Reviews Microbiology 17, 269 (2019).
Mughini-Gras, L. et al. Source attribution of foodborne diseases: Potentialities, hurdles, and future expectations. Frontiers in Microbiology 9, 1983, 1–5 (2018).
Nielsen, E. M. et al. Closing gaps for performing a risk assessment on Listeria monocytogenes in ready‐to‐eat (RTE) foods: activity 3, the comparison of isolates from different compartments along the food chain, and from humans using whole genome sequencing (WGS) analysis. EFSA Support. Publ. 14, 170pp (2017).
Amid, C. et al. The COMPARE Data Hubs. Database 2019, 1–14 (2019).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477 (2012).
Andrews, S. Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. Available at, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed: 3rd April 2019) (2010).
Alikhan, N. F., Zhou, Z., Sergeant, M. J. & Achtman, M. A genomic overview of the population structure of Salmonella. Plos Genetics 14, e1007261 (2018).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 - Approximately maximum-likelihood trees for large alignments. Plos One 5(3), e9490, 1–10 (2010).
Leekitcharoenphon, P. et al. snpTree–a web-server to identify and construct SNP trees from whole genome sequence data. BMC Genomics 13, S6 (2012).
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M. & Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. Plos One 9, e104984 (2014).
McClelland, M. et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413, 852–856 (2001).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–5 (2016).
EFSA. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J. 16, 262 pp (2018).
Hazards, E. B. P. On B. Scientific Opinion on monitoring and assessment of the public health risk of “Salmonella Typhimurium-like” strains. EFSA J. 8, 48 (2010).
Anonymous. Annual Report on Zoonoses in Denmark 2005. (2006).
Anonymous. Annual Report on Zoonoses in Denmark, 2013. (2014).
Anonymous. Annual Report on Zoonoses in Denmark, 2014. (2015).
Leclerc, V. et al. Le réseau Salmonella, un dispositif de surveillance des salmonelles sur la chaîne alimentaire: bilan 2015. (2015).
Rincé, A. et al. Occurrence of Bacterial Pathogens and Human Noroviruses in Shellfish-Harvesting Areas and Their Catchments in France. Front. Microbiol. 9, 2443 (2018).
ENA European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB15201 (2019).
ENA European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB16326 (2019).
ENA European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB14853 (2019).
ENA European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB18442 (2019).
NCBI, Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP042645 (2019).
Munck, N. et al. Four European Salmonella Typhimurium datasets collected to develop WGS-based source attribution methods. Figshare, https://doi.org/10.6084/m9.figshare.c.4748825 (2020).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Acknowledgements
As part of the COMPARE project, this work received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 643476. COMPARE is a Horizon 2020 EU project with the intention to speed up the detection of and response to disease outbreaks among humans and animals worldwide through the use of new genome technology. The aim is to reduce the impact and cost of disease outbreaks. We would also like to acknowledge Philipp Kirstahler from DTU Food for developing a script to retrieve sequences from dcc_Vivaldi to institution-based servers.
Author information
Authors and Affiliations
Contributions
N. Munck executed all stages of the study, manuscript write up, revision, submission and assisted the coordination of the benchmark study. L. Guillier and F. Palma contributed to the quality check of the French dataset and to revising the manuscript. E. Litrup performed cgMLST and wgMLST analyses, organized the collection of metadata, organized the sharing of sequences, performed and assessed the quality control of all datasets and edited the article. A. Meinen organized and performed the random selection of the German human, food and animal samples and added the associated metadata to the shared Microsoft Excel database. B. Malorny and M. Borowiak provided Salmonella Typhimurium from food and animals in Germany and were responsible for sequencing of these isolates. Moreover, they were involved in critical reading of the manuscript. M. Gourmelon provided Salmonella Typhimurium and variants from environment in France and was responsible for sequencing these isolates. S. Simon selected the NRC isolates for the German human dataset, supervised and organized the sequencing. S. Banerji registered the metadata of the German datasets into the shared Microsoft Excel database, uploaded the sequence reads to ENA. P. Leekitcharoenphon organized Danish data, submitted Danish dataset to ENA, contributed to the quality checking of all dataset, and contributed to the planning of the study. R. Kaas critical reviewed the sections regarding bioinformatics. T. Dallman provided human strains and metadata for the UK dataset and described the associated sampling plan. L. Petrovska-Holmes and Y. Tang provided animal strains and metadata for the British dataset and described the associated sampling plan. They contributed to the quality check of the British dataset and to revising the manuscript. T. Hald coordinated the overall planning and design of the benchmark study, drafted the protocol and metadata sheets, contributed to the selection of the Danish data and the description of the Danish data, supervised and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Munck, N., Leekitcharoenphon, P., Litrup, E. et al. Four European Salmonella Typhimurium datasets collected to develop WGS-based source attribution methods. Sci Data 7, 75 (2020). https://doi.org/10.1038/s41597-020-0417-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0417-7
