
Abstract
Bumblebees (Hymenoptera: Apidae) are important pollinating insects that play pivotal roles in crop production and natural ecosystem services. Although protein-coding genes in bumblebees have been extensively annotated, regulatory sequences of the genome, such as promoters and enhancers, have been poorly annotated. To achieve a comprehensive profile of accessible chromatin regions and provide clues for all possible regulatory elements in the bumblebee genome, we performed ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) on Bombus terrestris samples derived from four developmental stages: egg, larva, pupa, and adult, respectively. The ATAC-seq reads were mapped to the B. terrestris reference genome, and its accessible chromatin regions were identified and characterized using bioinformatic methods. We identified 36,390 chromatin accessible regions in total, including both shared and stage-specific chromatin accessible signals. Our study will provide an important resource, not only for uncovering regulatory elements in the bumblebee genome, but also for expanding our understanding of bumblebee biology throughout development.
Measurement(s) | open_chromatin_region |
Technology Type(s) | ATAC-seq |
Factor Type(s) | developmental stage |
Sample Characteristic - Organism | Bombus terrestris |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13047905
Similar content being viewed by others


Background & Summary
Bumblebees (Hymenoptera: Apidae) are important pollinating insects that play pivotal roles in crop production and natural ecosystem services1,2. They were well known as excellent pollinators of greenhouse tomato crops, decreasing the cost of labour and improving the yield and quality of fruit1,3. Now, their usage has been expanded to other crops, including those grown in poly-tunnels and open fields4. Also, bumblebees are ecologically important pollinators, with a large number of wild plants being pollinated predominantly or exclusively by bumblebees1,5. Because they are holometabolous insects that undergo four developmental stages (egg, larva, pupa, adult), they are also useful models to study mechanisms underlying developmental signalling and plasticity6. Regulatory elements play a major role in controlling the temporal and spatial expression of genes, through which they control the development and physiology of an organism7. To date, the protein-coding sequences of bumblebees have been extensively annotated8,9. However, regulatory elements, such as promoters, enhancers, and silencers, have been poorly annotated in the bumblebee genome.
ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) is a fast and highly-sensitive method that can determine accessible chromatin regions across the genome10,11, from which regulatory sequences can be inferred genome-wide. This technique not only requires less starting material, but also produces more precise results than previous approaches12,13. Furthermore, ATAC-seq can detect chromatin accessibility using whole animal preparations (containing mixtures of tissues or organs) with high sensitivity14.
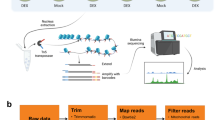
In this study, we used ATAC-seq to perform a genome-wide survey of accessible chromatin regions in Bombus terrestris, the most widely used commercial bumblebee species globally3. To achieve a comprehensive profile of open chromatin regions and provide clues for all possible regulatory elements in the bumblebee genome, we generated eight chromatin accessibility datasets for B. terrestris samples derived from its four developmental stages: egg, larva, pupa, and adult, respectively, with two biological replicates for each stage (Fig. 1a). In this experiment, we used whole animals, containing multiple cell types, for ATAC-seq. Therefore, our approach generates an atlas of open chromatin in each developmental stage, which does not necessarily mean that the focal chromatin region is open in each cell type. Our integrative ATAC-seq bioinformatic analysis workflow is shown in Fig. 1b. The accessible chromatin regions identified by this study will provide important resources for uncovering promoters, enhancers and other regulatory elements in the bumblebee genome. A total of 5,694, 4,850, 13,126 and 12,720 chromatin accessible regions were identified for developmental stage of egg, larva, pupa, and adult, respectively (Table 1).

An overview of study design and data analysis workflow. (a) The four developmental stages of bumblebee used for ATAC-seq. (b) The analysis workflow of ATAC-seq data.
To understand the position of accessible chromatin regions relative to their nearest gene, we compared the coordinates of ATAC-seq peaks with that of B. terrestris protein-coding genes. Using this approach, if we consider 2 kb upstream of TSS putative promoter regions, at least 35% of accessible chromatin regions are located in promoter regions. Open chromatin regions could also be found in exons, introns, and distal intergenic regions (Fig. 3b–e). Enhancers could be found far from their target genes or even within exons and introns15,16, thus the identified open chromatin in such regions represents the best candidates for enhancers in the bumblebee genome. In addition, the information obtained will expand our understanding of bumblebee biology, generally, and facilitate the cloning of bumblebee genes that control important traits.
The accessibility of regulatory elements is crucial and strongly associated with gene transcriptional activity, which reveals real-time gene expression during developmental and physiological processes17. Differences in accessible chromatin signals among different developmental stages could lay the foundation for further metamorphosis research. We deposited the identified differential ATAC-seq peaks between developmental stages in Figshare18.
Methods
Sample collection
Commercial B. terrestris colonies were bought from Koppert China (http://www.koppert.cn). Worker bee samples were collected from each of the four developmental stages: egg, larva, pupa, and adult, respectively, with two biological replicates for each stage (as listed in Table 1). Each sample was comprised of either an individual adult worker bee or pooled tissue of another developmental stage (egg, whole larva or whole pupa) equivalent to the volume of an adult worker bee. The eggs we collected were straight and smooth; all larvae had a C-shape curve; pupae had visible compound eye pigmentation and clear head-thorax-abdomen segmentation, but their wings were not developed yet; adult bees were bright with dense hair, and could flap their wings (Fig. 1a). All samples were frozen in liquid nitrogen after collection.
ATAC-seq protocol
ATAC-seq was performed by BGI-Shenzhen (https://en.genomics.cn), following previously published protocols10,11. Briefly, about 50,000 intact and homogenous cells were harvested and counted for each developmental stage, which were then centrifuged for 5 min at 500 × g, 4 °C. After discarding supernatant, the pellet was gently re-suspended with 50 µL of cold 1x PBS buffer, followed by 5 min of centrifuging at 500 × g, 4 °C. After removing supernatant, the pellet was gently pipetted and resuspended in 50 µL of cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) to release nuclei. After lysis, the suspension was spun at 500 × g for 10 minutes, 4 °C. After centrifugation, the pellet was immediately resuspended in the transposase reaction mix (25 μL 2x TD buffer, 2.5 μL Transposase (Illumina) and 22.5 μL of nuclease free water. The purified products were amplified in a 50 µL of reaction mixture containing the purified transposed DNA, 1x NEBnext High-Fidelity PCR master mix and 1.25 μM of custom Nextera PCR primers, with the following PCR program: (1) 72 °C, 5 minutes; (2) 98 °C, 30 seconds; (3) 98 °C, 10 seconds; (4) 63 °C, 30 seconds; (5) 72 °C, 1 minute; (6) Repeat steps 3–5 for 4 times; (7) Hold at 4 °C. After amplification, the PCR products obtained were purified by Qiagen MinElute PCR Purification Kit, with the purified PCR products being eluted in 20 µL Elution Buffer (10 mM Tris Buffer, pH 8). Next, the purified PCR products were used to produce single-strand DNA circles, from which DNA nanoballs were generated by rolling circle replication as previously described19. Finally, the DNA nanoballs were sequenced on the BGISEQ-500 sequencing platform, generating paired-end reads with a read length of 50 bp.
ATAC-seq data analysis
Raw reads were filtered first to remove low-quality reads and adaptor sequences by SOAPnuke20. Clean reads were mapped to the reference genome of B. terrestris (GenBank: GCF_000214255.1) using Bowtie221. The fragment length distribution of ATAC-seq was determined by the “fragSizeDist” function of R package ATACseqQC (Version: 1.12.3)22. The read coverages for genomic regions were computed and genome-wide similarities were assessed between the two replicates of the same developmental stage using deepTools23, with “multiBamSummary” and “plotCorrelation” function. We used MACS2 to call peaks (open chromatin regions) with parameters as reported previously14,24,25. The Irreproducible Discovery Rate (IDR) analysis was used to evaluate the reproducibility of high-throughput experiments by measuring consistency between the two biological replicates of the same developmental stage26. Peaks passing the suggested threshold (IDR < = 0.05) were reproducible peaks between the two replicates, which were retained for further analyses. The frequencies of peaks around transcription start sites (TSSs) were plotted by the “plotAvgProf” function of R package Chipseeker (Version: 1.24.0)27 based on B. terretris genome annotation file (Bter_1.0.46.gff3.gz on http://metazoa.ensembl.org/Bombus_terrestris/Info/Index). DEseq2 was employed to identify differential peaks between developmental stages using R package DiffBind (Version: 2.16.0)28. Peaks with FDR < 0.05 were treated as differential peaks between each two developmental stages. To retrieve the nearest genes around reproducible peaks of each developmental stage and differential peaks between different developmental stages, the coordinates of peaks were compared with the annotation of B. terrestris genome with the following priority order: promoter (−2kb, TSS), 5′UTR, 3′UTR, exon, intron, downstream (TES, 3 kb) and distal intergenic region. The distribution of accessible regions was plotted in pie charts by ChIPseeker27.
Data Records
Accession numbers are listed in Table 1. All ATAC-seq reads and peak files have been submitted to the NCBI Gene Expression Omnibus (NCBI GEO)29, which are accessible through GEO Series accession number GSE15185830. The sequencing data for every developmental stage (in fastq format) have been linked to the Sequence Read Archive of NCBI under the accession number SRP26609431. Reproducible peaks of each developmental stage, as well as differential peaks between developmental stages, have been deposited in Figshare18.
Technical Validation
Raw reads of ATAC-seq data were first filtered to remove adaptor sequences, contamination and low-quality reads. Clean reads were mapped to the reference genome of B. terrestris (Table 1). Based on the mapping results, we inferred the fragment size distribution. As expected, while a majority of fragments were shorter than one nucleosome length (approximately 150 bp), there were also significant number of fragments longer than this length and displayed periodicity (Fig. 2a,b; figures in Figshare18). Pearson correlation analysis was used to calculate and visualize pairwise correlation values between the two replicates of the same developmental stage, and results showed that correlation coefficients of each group were all greater than 0.95 (Fig. 2c; figure in Figshare18). Peaks (potential accessible chromatin regions) were called by MACS2 for each replicate of the four developmental stages. Most peaks are with peak score (−log10 (P value)) > 20 (Fig. 2e,f; figure in Figshare18), indicating the high reliability of peak calling. IDR method was applied to find reproducible peaks between replicates of the same developmental stage (Table 1; Fig. 2d; figure in Figshare18), and on average, 45% of peaks could pass the threshold (IDR < = 0.05) for each developmental stage. The intensity of ATAC-seq signal corresponds to the level of chromatin accessibility and can be used to identify poised and active regulatory regions genome-wide. We plotted chromatin accessible signals around genes for each developmental stage (using reproducible peaks for each stage), and as expected, the regions around transcription start sites were enriched for these signals (Fig. 3a). Open chromatin regions could also be found in exons, introns, and distal intergenic regions (Fig. 3b–e), therefore, except for protomers, our ATAC-seq dataset could also be used to identify other types of regulatory elements in bumblebee genome32. Peaks obtained from each developmental stage were used to identify differential open chromatin sites among the four developmental stages. Only peaks with FDR < 0.05 (based on DEseq2 method) were treated as differential sites, which can be used for further analysis of metamorphosis in bumblebees.

The quality metrics of ATAC-seq data. (a,b) Fragment size distribution of ATAC-seq reads for developmental stage larva (sample ID: L1 and L2, respectively). (c) Pearson correlation of the two biological replicates of stage larva. (d) IDR analysis of ATAC-seq peaks obtained from stage larva. The scatter plot shows one point for every peak, with its location representing in rank in each replicate. Peaks that pass the specified IDR threshold are coloured in black. (e,f) Peak score (-log10 (P value)) distribution for sample L1 (e) and L2 (f), respectively.

Features of ATAC-seq data in bumblebee. (a) The profile of chromatin accessible signals around bumblebee genes for each developmental stage. TSS represents transcription start site. (b–e) Reproducible peaks from egg, larva, pupa and adult stage, respectively, were annotated and classified based on their relative locations to nearest genes.
Bumblebees in different developmental stages exhibit differential morphological features and behave differently due to the temporal and spatial regulation of gene expression. We used ATAC-seq to perform a genome-wide survey of accessible chromatin regions in bumblebee (B. terrestris) by integrating data obtained from its four distinct developmental stages. The identified open chromatin regions will provide resources to uncover candidate regulatory sequences, such as promoters, enhancers and other cis-regulatory elements in the bumblebee genome. Also, the identified differential ATAC-seq peaks between developmental stages will be useful to identify genes or pathways involved in insect metamorphosis.
Code availability
SOAPnuke: Version: 2.1.2. Parameters: filter -l 5 -q 0.5 -n 0.1 -Q 2 –5 1 -c 50.
Bowtie2: Version: 2.2.5. Parameters: -q --phred64 --sensitive --dpad 0 --gbar 99999999 --mp 1,1 --np 1 --score-min L,0, -0.1 -I 1 -X 1000 -p 16 -k 200.deepTools: Version: 3.4.3. Parameters: --corMethod pearson --whatToPlot scatterplot --skipZeros --removeOutliers.MACS2: Version:2.2.5. Parameters: --nomodel --extsize 200 --shift -100 -- format BAM --gsize 2.17e8 -- call-summits.IDR: Version: 2.0.3. Parameters: --input-file-type narrowPeak --rank p.value --plot --log-output-file.
References
Fontaine, C., Dajoz, I., Meriguet, J. & Loreau, M. Functional diversity of plant–pollinator interaction webs enhances the persistence of plant communities. Plos Biol. 4, e1 (2006).
Garibaldi, L. A. et al. Wild pollinators enhance fruit set of crops regardless of honey bee abundance. Science. 339, 1608–1611 (2013).
Velthuis, H. H. W. & van Doorn, A. A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie. 37, 421–451 (2006).
Martin, C. D., Fountain, M. T. & Brown, M. J. F. Varietal and seasonal differences in the effects of commercial bumblebees on fruit quality in strawberry crops. Agric Ecosyst Environ. 281, 124–133 (2019).
Goulson, D., Lye, G. C. & Darvill, B. Decline and conservation of bumble bees. Annu Rev Entomol. 53, 191–208 (2008).
Tian, L. & Hines, H. M. Morphological characterization and staging of bumble bee pupae. PeerJ. 6, e6089 (2018).
Narlikar, L. & Ovcharenko, I. Identifying regulatory elements in eukaryotic genomes. Brief Funct Genomic Proteomic. 8, 215–230 (2009).
Sadd, B. M. et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16, 76 (2015).
Sun, C. et al. Genus-wide characterization of bumblebee genomes provides insights into their evolution and variation in ecological and behavioral traits. Mol Biol Evol, https://doi.org/10.1093/molbev/msaa240 (2020).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10, 1213–1218 (2013).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 109, 21.29.1–21.29.9 (2015).
Tsompana, M. & Buck, M. J. Chromatin accessibility: a window into the genome. Epigenetics Chromatin. 7, 33 (2014).
Lai, W. K. M. & Pugh, B. F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol. 18, 548–562 (2017).
Daugherty, A. C. et al. Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 27, 2096–2107 (2017).
Birnbaum, R. Y. et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 22, 1059–68 (2012).
Pennacchio, L. A., Bickmore, W., Dean, A., Nobrega, M. A. & Bejerano, G. Enhancers: five essential questions. Nat Rev Genet. 14, 288–95 (2013).
Li, B., Carey, M. & Workman, J. L. The role of chromatin during transcription. Cell. 128, 707–719 (2007).
Zhao, X., Su, L., Xu, W. & Sun, C. Genome-wide identification of accessible chromatin regions in bumblebee. figshare https://doi.org/10.6084/m9.figshare.c.5006981.v1 (2020).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience. 6, 1–9 (2017).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 7, 1–6 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9, 357–359 (2012).
Ou, J. et al. ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics. 19, 169 (2018).
Ramirez, F. et al. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Cusanovich, D. A. et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature. 555, 538–542 (2018).
Li, Q., Brown, J. B., Huang, H. & Bickel, P. J. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 5, 1752–1779 (2011).
Yu, G., Wang, L. & He, Q. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 31, 2382–2383 (2015).
Stark, R. & Brown, G. DiffBind: differential binding analysis of ChIP-Seq peak data. Bioconductor http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Zhao, X., Su, L., Xu, W. & Sun, C. Genome-wide identification of accessible chromatin regions in bumblebee by ATAC-seq. Gene Expression Omnibus https://identifiers.org/geo:GSE151858 (2020).
Zhao, X., Su, L., Xu, W. & Sun, C. Genome-wide identification of accessible chromatin regions in bumblebee by ATAC-seq. NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP266094 (2020).
Bozek, M. et al. ATAC-seq reveals regional differences in enhancer accessibility during the establishment of spatial coordinates in the Drosophila blastoderm. Genome Res. 29, 771–783 (2019).
Acknowledgements
This work was supported by the Elite Youth Program of Chinese Academy of Agricultural Sciences [to CS], and National Natural Science Foundation of China [31971397].
Author information
Authors and Affiliations
Contributions
C.S. and X.Z. conceived the study. X.Z. and W.X. collected samples and are involved in ATAC-seq. X.Z. and L.S. performed bioinformatics analysis. C.S., S.S. and X.Z. wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Zhao, X., Su, L., Xu, W. et al. Genome-wide identification of accessible chromatin regions in bumblebee by ATAC-seq. Sci Data 7, 367 (2020). https://doi.org/10.1038/s41597-020-00713-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00713-w
