
Abstract
Vertebrate gut microbiota provide many essential services to their host. To better understand the diversity of such services provided by gut microbiota in wild rodents, we assembled metagenome shotgun sequence data from a small mammal, the bank vole Myodes glareolus (Rodentia, Cricetidae). We were able to identify 254 metagenome assembled genomes (MAGs) that were at least 50% (n = 133 MAGs), 80% (n = 77 MAGs) or 95% (n = 44 MAGs) complete. As typical for a rodent gut microbiota, these MAGs are dominated by taxa assigned to the phyla Bacteroidetes (n = 132 MAGs) and Firmicutes (n = 80), with some Spirochaetes (n = 15) and Proteobacteria (n = 11). Based on coverage over contigs, Bacteroidetes were estimated to be most abundant group, followed by Firmicutes, Spirochaetes and Proteobacteria. These draft bacterial genomes can be used freely to determine the likely functions of gut microbiota community composition in wild rodents.
Measurement(s) | genome • DNA • gut microbiome measurement • metagenomic data |
Technology Type(s) | DNA sequencing |
Sample Characteristic - Organism | Bacteroidetes • Firmicutes • Spirochaetes • Proteobacteria • Myodes glareolus |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12936797
Similar content being viewed by others

Background & Summary
Vertebrate gut microbiota are often complex communities1,2 that are important determinants of their host’s health3,4, by providing essential nutrients and metabolites5,6, modulating the host’s immune system7 and by limiting the niche space available for colonisation by pathogens8,9. Laboratory rodents have provided compelling evidence that the gut microbiota has an important role in maintaining host health10,11. The diversity of species within a gut microbiota provides the host with potential access to thousands of novel, accessory genes1. Understanding the type and diversity of services that can be delivered by the gut microbiota to the host requires knowledge about microbial genomes.
Gut microbiota composition of wild rodents have been characterised using marker gene sequencing surveys. Within the superfamily Muroidea (containing mice, rats, voles, hamsters, gerbils and related taxa)12, for example, gut microbiota of wild caught mice Mus musculus domesticus are dominated by the bacterial phyla Firmicutes and Bacteroidetes13,14,15,16,17. Wild wood mice Apodemus sylvaticus show marked seasonal variation in gut microbiota, presumably reflecting a change in diet14; other studies have found an association between helminth infection and gut microbiota community in wild mice Apodemus flavicolus13. Moreover, the gut microbiota of bank voles Myodes glareolus (arvicoline voles within the Cricetidae) may be altered by anthropogenic environment impacts. For example, the gut microbiota of M. glareolus inhabiting areas contaminated by radionuclides (adjacent to the former nuclear power plant at Chernobyl, Ukraine) were characterised by an increase in Firmicutes and a reduction in Bacteroidetes16,17. Exposure to radionuclides is also associated with a reduction in (1) inter-individual variation and (2) the degree of temporal changes in bank vole gut microbiota community composition18.
While a change in microbiota composition can elicit a change in its function6, the impact of altered gut microbiota community composition in wild rodents is unknown because available genome/gene catalogue information is biased towards microbiota derived from laboratory animal models19,20 rather than wild animals. To better understand whether a change in gut microbiota could have some functional relevance, we used shotgun sequencing to construct metagenome assembled genomes (MAGs) for the gut microbiota of an arvicoline rodent, the bank vole M. glareolus. Bank voles are common in forest habitats of central and northern Europe and parts of Asia21. Also, bank voles are used as a model species in evolutionary ecology research22,23, for example with selection lines created to examine effects of physiology and diet on gut microbiota24.
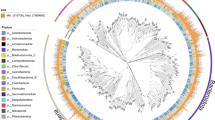
We generated shotgun sequence data for DNA extracted from the faeces of bank voles. Here we present a description of 254 draft bacterial genomes (MAGs) that we estimate to be 50% or more complete and with ≤10% contamination. Phylogenetic analysis of these MAGs indicates that they comprise a typical gut microbiota for rodents within the superfamily Muroidea13,14,15,16,17, being dominated by members of the Bacteroidetes (n = 132) and Firmicutes (n = 80), but with some Spirochaetes (n = 15) and Proteobateria (n = 11) (Fig. 1). Bacteroidetes were estimated to be most abundant (55% abundance based on coverage, with all taxa assigned to the Bacteroidales), followed by Firmicutes (24%, dominated by MAGs assigned to Clostridiales), Spirochaetes (12%, all MAGs assigned to Spirochaetales) and Proteobacteria (3%, predominantly Desulfovibrionales) (Table 1). Taxonomic placements for each MAG, as well as contig statistics and estimates of genome completeness and MAG abundance, are provided in Supplementary Table 3.

Phylogenetic diversity of metagenome assembled genomes (MAGs) from bank vole Myodes glareolus gut microbiota and reference genomes of Bacteria and Archaea available in RefSeq (listed in Supplementary Table 4). Presence of a MAGs from the bank vole gut microbiota is highlighted by the coloured outer ring, with black indicating the MAGs whose taxonomy was not be resolved (to phyla or better resolution) using either ANVI’O or CheckM. Midpoint rooted maximum likelihood tree was constructed from the concatenated alignments of 16 ribosomal proteins. Size of grey circles on branch midpoints indicate the level of bootstrap support (range 0.8–1.0).
These draft bacterial genomes to provide a useful resource to quantify the functions of the dominant members of wild rodent gut microbiota. Moreover, given the emerging interest in identifying the functions of relatively uncharacterised families of bacteria, such as the S24-7 that often dominate rodent gut microbiota25,26, these data should facilitate comparative genomic analyses of gut microbiota function and evolution across mammalian hosts. Also, the bank vole is a hyper-reservoir of zoonotic pathogens27,28,29,30. Given its role as a reservoir host of zoonotic pathogens, the potential link between gut microbiota and host health3,4, and evidence that infections associate with altered gut microbiota in rodents13,14, these data can provide detailed insights into the potential changes in services provided by gut microbiota that accompany infections in the bank vole. Draft genomes are deposited with NCBI Genbank under the BioProject accession PRJNA61338131, with metadata about the (1) bank vole samples and the (2) MAGs provided under the BioSample accessions SAMN14404158-SAMN14404199 and SAMN14407068-SAMN14407322 respectively.
Methods
Sampling and read data collection
Two faecal samples from 20 bank voles were collected from within the Chernobyl Exclusion Zone, Ukraine (51.30 N, 30.07 E) during May-June (faecal sample 1) and June-July 2016 (faecal sample 2). Full details of the live trapping procedures are provided in Lavrinienko et al.16,17. All procedures were performed in accordance with legal requirements and regulations from the Ukrainian authorities (957-i/16/05/2016) and the Animal Experiment Board in Finland (ESAVI/7256/04.10.07/2014). Metadata (e.g. location, body size, weight) associated with these samples are provided in Supplementary Table 1. Samples were transported (on dry ice) to Finland for DNA extraction based on the import permission from the Evira (3679/0460/2016). Total DNA was isolated from 0.1 g of faecal material using a PowerFecal DNA Isolation kit (MoBio Laboratories, Carlsbad, CA, USA) following the manufacturer’s instructions. Briefly, DNA extractions were performed in a dedicated laboratory space within a laminar flow hood using aseptic techniques (surface sterilisation, sterile plastic, aerosol barrier filter tips). We did not extract blank DNA samples as this procedure returns insufficient DNA for library construction and faecal samples contain high microbial biomass; rather we extracted DNA from two samples twice to assess to confirm that a sample’s microbiota profile was consistent (see Technical Validation). All the procedures were completed within 10 days by AL, using the same DNA isolation kit batch for all the samples (Supplementary Table 1). DNA concentrations were quantified with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA) (data provided in Supplementary Table 2).
NGS data were generated using Illumina HiSeq 4000 sequencing technology to generate 100 bp paired end read data at the Beijing Genomics Institute (BGI, www.bgi.com/global/). Read data are available at NCBI Genbank (SRA) under the accession numbers SRR11425428-SRR11425469 (Supplementary Table 1), and within the BioProject accession number PRJNA61338131.
Assembly
Read data were processed using ATROPOS32 v.1.1.5 (parameters: -q 15 --minimum-length 90), after which the reads were mapped (BOWTIE233 v.2.3.4, parameters: --very-sensitive) against a draft bank vole genome (GCA_001305785.1) to filter out reads (from *.bam files) derived from the host (SAMTOOLS v.1.4 view, parameters: view -f 12 -F 256) (https://www.htslib.org/doc/samtools.html). Assembly of metagenome read data to obtain draft bacterial genomes followed the approach used to recover draft genomes from the TARA oceans metagenomics data34. Individual samples were assembled using MEGAHIT35 v.1.1.1-2 (parameters: default) to reduce memory requirements and in attempt to avoid bubbles (unresolvable branches) due to genetic diversity among strains. Using MEGAHIT, we assembled a total of 1,057 million paired end reads into 4,721,549 primary contigs (5,916,721,003 bp). These primary contigs were filtered to retain only those contigs ≥ 2 kbp in length, which were then passed through CD-HIT-EST36 v.4.7.0 (parameters: -c 0.99 -n 11 -M 0) to merge the completely overlapping contigs (at 99% identity); this reduced set of primary contigs was then co-assembled using MINIMUS2 in AMOS37 v.3.1.0 (parameters: -D REFCOUNT = 0 OVERLAP = 100 MINID = 95) to combine overlapping contigs. After this procedure, we were left with 171,806 secondary contigs (39,092 contigs and 132,714 singletons) for binning.
Construction of metagenome assembled genomes (MAGs)
Host-filtered metagenomic reads were mapped against the secondary contigs using BOWTIE233 v.2.3.4 (parameters: --sensitive). Binning of contigs into MAGs was competed using BINSANITY38 v.0.2.7. First, we generated a coverage file using BINSANITY profile (parameters: scale; multiply by 100 and log transform). We then ran the secondary contigs through BINSANITY-LC workflow (parameter: -x 5000 -C 50 -p -5); use of a 5 kbp length cutoff for contigs (which meant that 66,480 secondary contigs were input into BINSANITY) and the BinSanity-lc script was required as there were too many contigs to complete the binning procedure using BinSanity-wf and/or all secondary contigs. BinSanity-lc attempts to overcome memory limitation problems associated with binning many contigs by creating subsets of contigs using K-means clustering prior to implementing affinity propagation for clustering. The quality and likely taxonomic identities of the putative MAGs was assessed using two softwares39,40,41,42. First, we used CHECKM39 that searches for the occurrence of a collection of lineage-specific markers genes in the putative bins. Second, we mapped read data to the contigs (constructed by MEGAHIT) using BOWTIE233 (parameters: --sensitive --no-unal), and the resulting *.sam files were processed using the metagenomic workflow implemented by ANVI’O v.5.240,41: ANVI’O also assesses completeness and contamination of MAGs by searching for presence of a collection of 139 bacterial single-copy core genes42. Bins were defined as ‘high completion’ when they met the following criteria with either ANVI’O or CHECKM: ≥95% complete with ≤10% redundancy (category 1), ≥80% complete with ≤5% redundancy (category 2), or ≥50% complete with ≤2% redundancy (category 3). ANVI’O (--anvi-summarize, parameters: default) was used to estimate the relative proportions of bacterial phyla and families based on the average coverages of MAGs (that had been divided by the overall average coverages in samples) (Table 1).
Using BINSANITY, contigs were placed into 900 bins, of which 775 (86%) contained putative bacterial DNA (based on the presence of the core panel of markers used by CHECKM). Two hundred and fifty-four (33%) of these bins were classified as high-quality bacterial metagenomes (MAGs), representing 16,395 contigs (24.7% of the 66,480 contigs used for binning). Of these MAGs, 45, 76 and 133 were defined in categories 1, 2 and 3 respectively. MAGs derived from multiple samples (in this case, from 42 faecal samples taken from 20 bank voles over two time points, and with two samples extracted twice – see Supplementary Information 1) often represent genomes of several taxa or strains, and further analysis and assembly will be required to generate complete bacterial genomes43. The remaining contigs, presumably representing a mixture of genetic material from the gut microbiota (such as viruses, plasmids, protists, some bacteria, etc.) as well as some read data derived from the bank vole host genome (potentially the fraction of highly repetitive DNA that could not be assembled in the draft genome) and also bank vole dietary material, are available for further analysis.
Phylogenetic assessment of MAGs
To assess the phylogenetic diversity of the MAGs we predicted coding sequences in the secondary contigs (that had been assigned to draft genomes) using PRODIGAL44 v.2.6.3 (parameters: -m -p meta), and then searched for the sequences of a core panel of phylogenetic markers: the phylogenetic markers used are 16 ribosomal proteins (L2, L3, L4, L5, L6, L14, L16, L18, L22, L24, S3, S8, S10, S17 and S19) that are often syntenic, and which have been used for a comprehensive phylogenetic assessment of Bacteria, Archaea and Eukarya45, as well as phylogenetic placement of MAGs34. To provide wider context to the phylogenetic diversity of our MAGs, we also downloaded more than 1,600 Bacterial and Archaeal complete/representative genomes from NCBI’s Refseq database46 (https://www.ncbi.nlm.nih.gov/refseq/: date accessed 11/09/2019) (see Supplementary Table 4 for list of reference genomes). We searched for the 16 phylogenetic markers from our MAGs and the reference genomes using HMMSEARCH47 in HMMER v.3.1b2 (parameters: -E 1e-5) and the Hidden Markov Models for the 16 ribosomal proteins that were downloaded from Pfam database48 (https://pfam.xfam.org). Genomes that lacked half or more of the phylogenetic markers were not used in the phylogenomics analysis. Sequence data for each marker were aligned using MUSCLE49 v.3.8.31 (parameters: --maxiters 16). Alignments were trimmed using TRIMAL50 v.1.2rev59 (parameters: -automated1) and concatenated using the CONCAT script implemented by BINSANITY38. A detailed protocol for this phylogenomic workflow can be found at ProtocolsIO (https://doi.org/10.17504/protocols.io.mp5c5q6). Following this protocol, we were able to construct a phylogenetic tree for 167 MAGs and 1,664 reference genomes using FASTTREE51 v2.1.9 (parameters: -gamma -lg), which was viewed and annotated using iTOL52 (https://itol.embl.de).
Data Records
Project data (host metadata, metagenome shotgun sequence data, MAGs) have been deposited in NCBI Sequence Read Archive, under the SRA study accession SRP25405631. The concatenated alignment of the 16 phylogenomic loci for MAGs and reference genomes and the associated phylogenetic tree (Newick format) have been deposited with Figshare53.
Technical Validation
Quality of the host-filtered and trimmed Illumina reads was quantified using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and observed to be very good: example quality plots for six samples are given in Supplementary Figure 1, and the basic information on the total number of reads and other statistics associated with reads is presented in Supplementary Table 7. Potential cross-contamination of samples was limited by following guidelines for analyses of microbiota communities54,55; for example, DNA extractions took place within a dedicated laboratory space under a laminar flow hood using aseptic techniques (such as, surface sterilisation, use of sterile plasticware, and use of aerosol barrier pipette tips). Sample processing was completed within 10 days by AL, using the same batch of DNA isolation kits for all samples. DNA concentrations were quantified with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA) and NanoDrop Spectrophotometer (ThermoFisher Scientific, MA, USA). Extracting total DNA from 0.1 g of faecal material using the PowerFecal DNA Isolation kit (MoBio Laboratories, Carlsbad, CA, USA) typically provided 20–100 ngµl−1 DNA (Supplementary Table 2) for library construction. To assess for potential contamination (e.g. from laboratory reagents, and/or contamination of samples), we extracted DNA from two samples twice, independently, and these samples were sequenced as separate libraries also (i.e. there were 42 libraries prepared) (Supplementary Table 1). We found low (r < 0.2) correlations (Supplementary Table 6) in the abundances of the MAGs among most pairs of samples (see Supplementary Table 5 for mean coverages per sample and MAG, which were calculated using ANVI’O v.5.239,40 as the ‘mean coverage of a contig divided by overall sample mean coverage’). Hence, these MAGs exhibit substantial variation in their abundance among individuals and timepoints (Supplementary Table 5), which is consistent with expected levels of inter-individual and temporal variation in rodent gut microbiota community composition14,16,17,18. The notable exceptions to these low pairwise correlations in MAG abundance were the comparisons of MAG abundances between the pairs of independent, replicate DNA extractions (r = 0.999 for samples 25 and T25, and r = 1.000 for samples 69 and T96) (Supplementary Table 6). The high similarity in MAG abundance among the independent, replicate extractions, but otherwise individual community profiles (that are typical rodent gut microbiota13,14,15,16,17) in each sample, implies that read data reflect the sample’s microbiota and that contamination of samples during DNA extraction was minimised55.
Metagenome data have been assembled and refined into MAGs using the automated quality control steps, assembly procedures and the thresholds described in the manuscript. In addition, contigs were passed through NCBI’s Contamination Screen to remove any residual adaptor contamination (21 out of 16,486 contigs contained within the MAGs had one NGS adaptor sequence that was removed; Supplementary Table 3). Technical validation of the taxonomic assignments, completeness and potential contamination of the putative bacterial MAGs was achieved using several softwares, but we recommend that further, independent technical validation be applied by users of these draft MAGs.
Change history
30 July 2021
The URL in reference 31, https://identifiers.org/insdc.sra:SRP254056 has been updated to, https://identifiers.org/ncbi/insdc.sra:SRP254056 in both the HTML and PDF versions of this Data Descriptor.
References
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Hird, S. M. Evolutionary biology needs wild microbiomes. Front. Microbiol. 8, 1–10 (2017).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 148, 1258–1270 (2012).
Marchesi, J. R. et al. The gut microbiota and host health: A new clinical frontier. Gut 65, 330–339 (2016).
Lee, W. J. & Hase, K. Gut microbiota-generated metabolites in animal health and disease. Nat. Chem. Biol. 10, 416–424 (2014).
Visconti, A. et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 10, 4505 (2019).
Belkaid, Y. & Hand, T. W. Role of microbiota in immunity and inflammation. Cell 157, 121–141 (2018).
Pickard, J. M., Zeng, M. Y., Caruso, R. & Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89 (2017).
Pickard, J. M. & Núñez, G. Pathogen Colonization Resistance in the Gut and Its Manipulation for Improved Health. Am. J. Pathol. 189, 1300–1310 (2019).
Nguyen, T. L. A., Vieira-Silva, S., Liston, A. & Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 8, 1–16 (2015).
Rosshart, S. P. et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171, 1015–1028 (2017).
Blanga-Kanfi, S. et al. Rodent phylogeny revised: analysis of six nuclear genes from all major rodent clades. BMC Evol. Biol. 9, 71 (2009).
Kreisinger, J., Bastien, G., Hauffe, H. C., Marchesi, J. & Perkins, S. E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos. T. Roy. Soc. B 370, 20140295 (2015).
Maurice, C. F. et al. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 9, 2423–2434 (2015).
Weldon, L. et al. The gut microbiota of wild mice. PLoS ONE 10, 1–15 (2015).
Lavrinienko, A., et al. Environmental radiation alters the gut microbiome of the bank vole Myodes glareolus. ISME J 12 (2018).
Lavrinienko, A., Tukalenko, E., Mappes, T. & Watts, P. C. Skin and gut microbiomes of a wild mammal respond to different environmental cues. Microbiome 6, 209 (2018).
Lavrinienko, A. et al. Applying the Anna Karenina principle for wild animal gut microbiota: temporal stability of the bank vole gut microbiota in a disturbed environment. J. Anim. Ecol. In press, https://doi.org/10.1111/1365-2656.13342 (2020).
Xiao, L. et al. A catalog of the mouse gut metagenome. Nat. Biotech. 33, 1103–1108 (2015).
Pan, H. et al. A gene catalogue of the Sprague-Dawley rat gut metagenome. GigaScience 7, 1–8 (2018).
Hutterer, R., et al. Myodes glareolus. The IUCN Red List of Threatened Species e.T4973A115070929 (2016); erratum (2017).
Lonn, E. et al. Balancing selection maintains polymorphisms at neurogenetic loci in field experiments. Proc. Natl. Acad. Sci. USA 114, 3690–3695 (2017).
Van Cann, J., Koskela, E., Mappes, T., Sims, A. & Watts, P. C. Intergenerational fitness effects of the early life environment in a wild rodent. J. Anim. Ecol. 88, 1355–1365 (2019).
Kohl, K. D., Sadowska, E. T., Rudolf, A. M., Dearing, M. D. & Koteja, P. Experimental evolution on a wild mammal species results in modifications of gut microbial communities. Front. Microbiol. 7, 1–10 (2016).
Ormerod, K. L. et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 1–17 (2016).
Lagkouvardos, I. et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7, 1–15 (2019).
Tonteri, E. J. et al. Tick-borne encephalitis virus in wild rodents in winter, Finland, 2008–2009. Emerg. Infect. Dis. 17, 72–75 (2011).
Vaheri, A. et al. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 23, 35–49 (2013).
Han, B. A., Schmidt, J. P., Bowden, S. E. & Drake, J. M. Rodent reservoirs of future zoonotic diseases. Proc Natl. Acad. Sci. USA 112, 7039–7044 (2015).
Van Duijvendijk, G., Sprong, H. & Takken, W. Multi-trophic interactions driving the transmission cycle of Borrelia afzelii between Ixodes ricinus and rodents: A review. Parasite. Vector. 8, 13–15 (2015).
Lavrinienko, A. et al. Two hundred and fifty-four metagenome-assembled bacterial genomes from the bank vole gut microbiota. NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP254056 (2020).
Didion, J. P., Martin, M. & Collins, F. S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Tully, B. J., Graham, E. D. & Heidelberg, J. F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 5, 1–8 (2018).
Li, D. et al. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Li, W. et al. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 13, 656–668 (2012).
Sommer, D. D. et al. Minimus: A fast, lightweight genome assembler. BMC Bioinform. 8, 1–11 (2007).
Graham, E. D., Heidelberg, J. F. & Tully, B. J. Binsanity: Unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ 5, e3035 (2017).
Parks, D. H. et al. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Eren, A. M. et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015).
Delmont, T. O. & Eren, A. M. Identifying contamination with advanced visualization and analysis practices: Metagenomic approaches for eukaryotic genome assemblies. PeerJ 4, e1839 (2016).
Campbell, J. H. et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA. 110, 5540–5545 (2013).
Chen, L.-X. et al. Accurate and complete genomes from metagenomes. Genome Res. 30, 315–333 (2020).
Hyatt, D. et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, 501–504 (2005).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Edgar, R. C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 1–19 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Lavrinienko, A. et al. Two hundred and fifty-four metagenome-assembled bacterial genomes from the bank vole gut microbiota. figshare https://doi.org/10.6084/m9.figshare.c.4910601 (2020).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Eisenhofer, R. et al. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Ecol. Evol. 27, 105–117 (2019).
Acknowledgements
This work was made possible by funding from the Academy of Finland (nos 287153, 324602 to PCW) and through a collaboration with the EMP500 (http://www.earthmicrobiome.org/emp500/). We thank the CSC Finland for access to national computing facilities and BGI for provision of sequencing services. In addition to providing free access to scripts for phylogenomics analysis of MAGs, we are grateful to Elaina Graham who kindly provided advice and access to scripts to more efficiently process contigs into MAGs using BINSANITY.
Author information
Authors and Affiliations
Contributions
P.C.W., A.L., T.M., T.A.M., R.K. and L.R.T. conceived the study. A.L., E.T., T.M. conducted fieldwork, with A.L. completing the laboratory processing of samples. P.C.W. completed the analyses and led the writing of the paper, with all authors reviewing draft versions of the manuscript. P.C.W., T.M., T.A.M., L.R.T. and R.K. provided funding and access to equipment and other resources to perform the analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Lavrinienko, A., Tukalenko, E., Mousseau, T.A. et al. Two hundred and fifty-four metagenome-assembled bacterial genomes from the bank vole gut microbiota. Sci Data 7, 312 (2020). https://doi.org/10.1038/s41597-020-00656-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00656-2
This article is cited by
-
The multi-kingdom microbiome of the goat gastrointestinal tract
Microbiome (2023)
-
Trait biases in microbial reference genomes
Scientific Data (2023)
-
The microbiome of the buffalo digestive tract
Nature Communications (2022)
-
Bifidobacterium castoris strains isolated from wild mice show evidence of frequent host switching and diverse carbohydrate metabolism potential
ISME Communications (2022)
