
Abstract
Despite the use of Hymenolepis diminuta as a model organism in experimental parasitology, a full genome description has not yet been published. Here we present a hybrid de novo genome assembly based on complementary sequencing technologies and methods. The combination of Illumina paired-end, Illumina mate-pair and Oxford Nanopore Technology reads greatly improved the assembly of the H. diminuta genome. Our results indicate that the hybrid sequencing approach is the method of choice for obtaining high-quality data. The final genome assembly is 177 Mbp with contig N50 size of 75 kbp and a scaffold N50 size of 2.3 Mbp. We obtained one of the most complete cestode genome assemblies and annotated 15,169 potential protein-coding genes. The obtained data may help explain cestode gene function and better clarify the evolution of its gene families, and thus the adaptive features evolved during millennia of co-evolution with their hosts.
Measurement(s) | whole genome sequencing assay • transcription profiling assay • sequence_assembly • sequence annotation • mitochondrial DNA |
Technology Type(s) | DNA sequencing • RNA sequencing • genome assembly • bioinformatics analysis |
Sample Characteristic - Organism | Hymenolepis diminuta |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.10283018
Similar content being viewed by others
Background & Summary
The study of the genomics and transcriptomics of parasite model species has led to advances in the basic aspects of parasite biology, as well as new trends in human and veterinary medicine. Modern genomic tools, especially those based on a combination of multiple methods, allow detailed analyses of genome structure.
Our study used hybrid genome sequencing to examine the genome of the tapeworm Hymenolepis diminuta by three technologies: Illumina sequencing pair-end, Illumina mate-pair and MinION Oxford Nanopore DNA sequencing. H. diminuta is a well-described representative of the class Cestoda, the large group of parasitic flatworms that includes members known to be serious pathogens of vertebrate animals and humans1,2.
H. diminuta was chosen for the present study since it is commonly used in studies of new therapeutics, biochemical processes, immune responses and other host-parasite interrelationships during cestodiasis3,4,5,6,7,8 and is considered the most important model species in experimental cestodology. Hymenolepis diminuta has a worldwide distribution as an intestinal parasite of rodents (primarily rats) and humans9, and the tapeworms of the genus Hymenolepis are considered to be among the most frequent causative agents of the human cestodiasis1.
Despite its importance as a model organism, the genome of H. diminuta is available only as a draft genome acquired as part of the 50 Helminth Genomes project initiative10. In addition, Gauci et al.11 using the example of Echinococcus granulosus, highlight the possible limitations of published draft genomes of selected tapeworm species, one being the fact that they were sequenced using only Illumina short reads technology. Therefore, the ultimate goal of our study was to improve the accuracy of the draft genome of H. diminuta, by integrating data from three complementary approaches; this approach may significantly enhance the scientific value of the achieved datasets for future studies.
The combination of the recent progress in sequencing technologies and unlimited access to genomic data has fueled rapid development in the biomedical sciences, including parasitology. Most recently, the International Helminth Genomes Consortium released draft genomes (both published and unpublished) to present lineage-specific trends rather than individual species-specific differences10. This dataset of helminths genomes provides a number of new details important in studies of parasitic worms; however, there is an urgent need to continue helminth genome sequencing and improve the available genomes. An examination of the number and organization of EG95 E. granulosus vaccine-related encoding genes based on two available E. granulosus draft genomes published by Tsai et al.12 and Zheng et al.13 indicates that the genome sequence data available for E. granulosus offers limited potential for practical use11; in part, it was not possible to map any of the EG95 gene family members previously characterized by cloning and sequencing genomic DNA fragments. These results have revealed limitations in available genomic data and highlights deficiencies present in current genomic resources, and thus, reinforced the need to supplement available datasets with new sequencing results.
This can be achieved by simultaneous use of available sequencing technologies, providing both short and long reads. In recent years, such a hybrid approach has proven to be useful in improving quality of genome assemblies and improving discovery of gene family expansions. For instance, a hybrid approach was introduced for de novo human genome sequence14, one of the best described genomes. When assembling clownfish genome using high-coverage Illumina short reads and low-coverage Nanopore long reads, Tan et al.15 observed substantial improvement in the genome statistics when compared with Illumina-only assembly. They suggest that development and improvement of Nanopore technology will shift toward the use of high-coverage long read-only assembly, followed by multiple iterations of genome polishing using Illumina reads. Genome improvements due to the use of hybrid sequencing have been applied to characterize the genetic polymorphism in Wuchereria bancrofti populations, and provide, among others, a list of genetic markers useful for monitoring changes in parasite genetic diversity16.
The present paper provides the first results of hybrid de novo whole-genome sequencing of H. diminuta combined with RNAseq analysis. Our assembly appears to be more complete than that available in WormBase ParaSite17 and offers improved genome statistics. In this respect our results suggest that the procedure yielded one of the most comprehensive tapeworm genome assemblies available. In addition, our results are supported with RNA-seq analyses, which allow a better overview of the entire structure of the H. diminuta genome.
Here we confirm that the hybrid sequencing approach is the optimal method for obtaining the high quality data resulting in determination of a complete genome sequence. This cost-effective approach combining Illumina paired-end, mate-paired, and MinION Nanopore long reads allowed the retrieval of one of the most comprehensive tapeworm (or any parasitic worm) genome available, complimented by RNA sequencing data. These may result in better understanding of the biology of the parasite, its genetic diversity, adaptation to parasitic way of life and may allow new treatments and/or diagnostic tools to be identified in the near future.
Methods
Experimental animals
Approximately three month old male Lewis rats (Rattus norvegicus domesticus) were used as definitive hosts for adult H. diminuta. The rats were kept in plastic cages in the laboratory animal facilities of the Medical University of Warsaw, Poland. Food and water were provided ad libitum. This study was approved by the 3rd Local Ethical Committee for Scientific Experiments on Animals in Warsaw, Poland (Permit Number 51/2012, 30th of May 2012).
Cultivation of H. diminuta adult cestodes
Six-week-old H. diminuta cysticercoids were removed from dissected Tenebrio molitor beetles under a microscope (100× magnification). Ten three-month-old rats were infected by voluntary oral uptake of six cysticercoids of H. diminuta per rat. Smears of their fecal samples were examined under a microscope (magnification 400×) five to six weeks from the initial infection, to verify the presence of adult parasites by their eggs. The rats were euthanized with 100 mg/kg intraperitoneal thiopental anaesthesia (Biochemie GmbH, Austria). The small intestines were removed immediately, adult parasites were isolated and washed up to 5× with 100 mM PBS with antibiotics added (1% penicillin) to remove debris.
DNA isolation
Briefly after recovery from host intestine, DNA was isolated from tapeworm fragments containing only scolex and immature proglottids. Genomic DNA was isolated using a Genomic Midi AX isolation kit with ion-exchange membranes (A&A Biotechnology, Gdynia, Poland) according to the manufacturer’s instructions. The integrity of the genomic DNA molecules was checked using agarose gel electrophoresis. The obtained DNA extracts were used immediately or stored at −20 °C until use.
RNA isolation and sequencing
A total of three adult H. diminuta tapeworms were homogenized in RLT buffer and total RNA was isolated from the homogenate using RNeasy Midi Kit (Qiagen, Germany). The sequencing library was prepared from 1 μg total RNA using TruSeq RNA Sample Preparation v2 Kit (Illumina, San Diego, CA, USA) according to manufacturer’s instructions; the library was paired-end sequenced (2 × 100 bp) on the Illumina HiSeq 1500 platform.
WGS library preparation and sequencing
For whole genome sequencing (WGS) 2.5 μg of high quality genomic DNA was used. Prior to the library preparation DNA was fragmented using Covaris M220 (Covaris, Inc, Woburn, MA, USA) and size selection was performed using BluePippin (Sage Science, Inc, Beverly, MA, USA) for the average insert size 600 bp. The library was prepared using NEBNext Ultra® II DNA Library Prep Kit (New England BioLabs, Inc, Ipswich, MA, USA) according to manufacturer’s instruction.
For mate-pair whole genome sequencing (MP-WGS) two different libraries, with (4 μg input DNA) and without (1 μg input DNA) size selection, were prepared. Libraries were constructed using Nextera Mate Pair Library Preparation Kit (Illumina) according to manufacturer’s instruction. Size selection was performed using BluePippin (Sage Science) for fragments ranging from 5000 to 10000 bp (average size 8000 bp). The mean fragment size for the library without size selection was 2000 bp.
The WGS library was paired-end sequenced on a HiSeq. 1500 (Illumina) (S59, S66, S70, S13, S41, S34, S47: 2 × 100 bp, Table 1) and on an MiSeq (Illumina) (S36: 2 × 300 bp, S3: 2 × 250 bp, Table 1). S1 was single-read sequenced (1 × 500 bp) on an MiSeq (Illumina) (Table 1). The MP-WGS library was paired-end sequenced (2 × 100 bp) on a HiSeq 1500 (Illumina).
For Oxford Nanopore sequencing (ONT) high molecular DNA was isolated from tapeworm using phenol-chloroform extraction. Briefly, 200 mg of tapeworm tissue sample was washed twice with PBS buffer to remove excess rat stool material. After washing, the sample was submerged in 900 μl of TE buffer. The sample was lysed by the addition of 90 μl of 10% SDS, 10 μl of Proteinase K (20 mg/ml) and incubated at 37 °C for one hour until all cells were lysed. Following this, 200 μl of 5 M NaCl was added to the cleared lysate, which was subjected to phenol:chloroform:isoamyl alcohol extraction until no protein debris was visible in the interphase. After protein removal, the DNA was precipitated with isopropanol (0.7 volume added) and centrifuged for 10 minutes at 14000 rpm and washed with 70% ethanol. The DNA pellet was dried for a short time at room temperature and re-suspended in 100 μl of low-TE buffer (10 mM Tris and 0.1 mM EDTA pH = 8.0) containing RNase (50 μg/ml). DNA quality and integrity were checked using electrophoresis in standard 1% agarose gel and by PFGE using Biorad CHEF-II instrument. DNA quantity was measured with Qubit 3.0 fluorimeter and Broad Range chemistry (Thermo Scientific, Life Technologies).
The Oxford nanopore library was constructed by 1D ligation using two strategies. In the first, 8 μg of DNA was sheared into 20 kbp fragments using Covaris g-Tube and 5 μg of sheared template was taken for 1D library construction using SQK-LSK108 kit (Oxford Nanopore Technologies). Approximately 1 μg of library was loaded into R9.4 flowcell system and sequenced on a MinION instrument for 24 hours. In the second approach, 20 μg of DNA was sheared into 20 kbp fragments followed by size selection on BluePippin instrument (Sage Science). Fragments above 10 kbp were recovered using PAC 30 kb cassette. 5 μg of recovered DNA was taken for 1D library construction using SQK-LSK108 kit and 1.5 μg of final library was loaded into R9.4.1 flowcell and sequenced on MinION sequencer.
De novo genome assembly
A hybrid assembly approach was employed, with several types of reads used in the assembly. Firstly, the datasets created from high quality of DNA reads from Illumina paired-end and Illumina mate-pair sequencing were assembled using tools based on de Bruijn graph, ABySS18 and dnaasm19. The software versions are reported in Table 2. Secondly, the set of contigs (results of assembly) were combined based on the Oxford Nanopore long reads using two different tools: LINKS20 and dnaasm-link21. This step was developed in an iterative way: firstly, results obtained from only short DNA reads were linked, where distance parameter in LINKS tool was set to 6 kbp. The obtained results were linked with those obtained for distance values of 7 kbp, then 8 kbp, 19 kbp, 20 kbp and 30 kbp. However, as the LINKS application requires a very large amount of RAM, the procedure was performed using dnaasm-link running on C++ instead of Perl. In addition, dnaasm-link has a module to fill the gaps between contigs using sub-sequences from long DNA reads.
Functional annotation
The annotation pipeline was run using newly-obtained transcriptomic and genomic data from H. diminuta. During the first step, the RNA-seq data were mapped to the assembled genomic scaffolds using the STAR aligner. Obtained BAM file and genomic scaffolds were analyzed with BRAKER2 software with the Augustus tool to acquire the protein-expressing coding sequences. In the next step, BRAKER2 (amino acid sequences) and Trinity (transcriptomic sequences) outputs were used to obtain detailed genomic annotations using using MAKER2 pipeline (with -est2genome = 1; -prot2genome = 1).
All de novo assembled transcripts were searched against UniProt/SwissProt22 database using BLASTx and BLASTp with an e-value < 10−5. Open reading frames (ORFs) were predicted using Transdecoder. The remaining functional annotation was obtained using g:Profiler and Trinotate pipeline, which uses several software packages: Hmmer, a protein domain identification (Pfam) tool, Rnammer to predicts ribosomal RNA and SignalP to predicts signal peptide sites.
Mitochondrial genome
Mitochondrial DNA was obtained and sequenced with Illumina technology as described above. The mtDNA was bioinformatically obtained from de novo assembly from the PET1 dataset (S59, S66, S70, S13, S41, S34 and S36 sets of reads). The mitogenome was analyzed and and characterized using CLC Main Workbench and MacVector software. The organization of mitochondrial genome is given in the ‘Technical validation’ section, where it is also compared with NC_002767.
Data Records
Data supporting the results of this article has been deposited at European Nucleotide Archive (EMBL). The study titled ‘Hybrid sequencing of Hymenolepis diminuta genome’ got Access Number ERP11343723, the project identifier is PRJEB30942. Raw Illumina and Nanopore reads have been given the indexes ERS3052629–ERS3052634, the assembly output is deposited under name ‘H.diminuta_WMSil1’ and identifier GCA_90217791524, mitochondrial genome under name ‘Hymenolepis diminuta strain WMSil1 genome assembly, organelle: mitochondrion’, LR53642925. Annotation is included. Supporting data, also including script parameters, are available at figshare26.
Technical Validation
Paired-end reads
Firstly, the quality of input data was checked using FastQC tool. The results confirmed the high quality of DNA reads – the reports were collected by the MultiQC tool and are available online at https://doi.org/10.6084/m9.figshare.8798111.v1. Following this, the basic statistics of the paired-end tags were studied using the BBmap package (Table 1).
Further analysis used two data sets: PET1 and PET2. PET1 is a set with coverage 150× created from S59, S66, S70, S13, S41, S34 and S36, while PET2 was created from S3 with S47, and has 370× coverage.
Mate-pairs reads
MP-WGS sequencing identified two sets of reads: MP1 and MP2. Both data sets consist of 100 bp DNA reads; however, MP1 was found to include 69, 558, 283 raw pairs of reads while MP2 had 54, 688, 723. Read quality, determined by FastQC, showed problems with adapter content, over-represented sequences and per sequence GC content, which is typical for this type of sequencing. To overcome this issue, the NxTrim27 tool was used to filter only correctly paired reads based on the adapter location (Table 3). The resultant sets of DNA reads were again checked by FastQC, and no such problems with DNA reads were observed.
After rejecting improperly paired DNA reads, the insert size value of the remaining mate pairs were examined using BBmap package. The results are presented in Fig. 1.

MP dataset after NxTrim trimming insert size histogram. The graphs on the left and right present the histograms for MP1 and MP2 datasets, respectively.
ONT reads
Sequencing of nanopore library without size selection (ONT1) yielded 546,222 reads and 3.5 GB of sequence data with a mean read length of 6.37 kbp. Size selected library (ONT2) sequencing yielded 156,168 reads and 1.6 GB of sequence data with the mean read length of 10.1 kbp. Nanopore sequencing yielded totally 702,390 reads and 5.1 GB of data.
Raw nanopore data was base-called using Albacore (Oxford Nanopore Technologies, Oxford, UK). After quality filtering for quality and residual adapter removal using NanoFilt and Porechop. Long nanopore read data statistics, generated using NanoPlot, are presented in Figs. 2 and 3.

Raw ONT dataset length histogram. The graphs on the left and right present the histograms for ONT1 and ONT2 datasets, respectively.

Raw ONT dataset quality diagrams. The graphs on the left and right present the diagrams for ONT1 and ONT2 datasets, respectively.
The error rate was checked using the BBmap package by mapping long DNA reads to contigs produced only from paired-end tags. The obtained results indicated a 25% error rate from nanopore DNA reads; therefore, the raw DNA reads were corrected using a Canu28 correcting module, resulting in the error rate falling to 11%.
De novo assembly results
A hybrid assembly approach was employed, where short paired-end reads PET1 and PET2 datasets (depicted in Table 1) and mate-pair reads MP1 and MP2 (given in Table 3) and ONT1 and ONT2 long reads were used together.
The present study investigates the effect of applying reads from third-generation sequencers on de novo assembly results. In a typical de novo project, sequencing and assembly are performed iteratively until the results are of good enough quality and funds still remain. During each iteration, sufficient funds need to remain available for the next sequencing process, because the assembly costs are lower. From this point of view, two approaches can be used when performing a new experiment: (1) use the sequencing technology previously used in the project, or (2) complement results with sequencing technology not used previously in the project. Our results indicate that option (2) is a better choice, as adding results from new sequencing technology gives better statistics than additional reads obtained by the previously used technology.
As depicted in Table 4 we observed a significant improvement in assembly results between column 2 and column 3, when mate-pair reads were added, and between column 4 and column 5, when Nanopore reads were added. The improvements in assembly between column 2 and column 1, between column 4 and column 3, and between column 6 and column 5 were less pronounced since they were obtained using the same sequencing methods. In particular, using N50 statistics as a measure of quality, we observed 21% better results (from 69.7 kbp to 84.2 kbp) when using PET1 + PET2 reads instead of PET1 reads (the sequencing coverage increases from 150× into 520×). Adding the mate-pair MP1 dataset (sequencing coverage 77×) into PET1 + PET2 (dataset has 597× coverage instead of 520×) improved N50 by 1000% (from 84.2 kbp to 842.2 kbp). The next mate-pair dataset, MP2 (sequencing coverage 61×, therefore all reads cover genome 658×) improved N50 by 0.2% (from 842.2 kbp to 844.2 kbp). Using the Nanopore dataset (ONT1, coverage 19×, mean read length 6.4 kbp) improved N50 by 206% (from 844.2 kbp to 1.7 Mbp), and the next Nanopore dataset ONT2 (coverage 8×, mean read length 10.1 kbp) improved N50 by 134%. A similar effect was observed when using a number of scaffolds.
In addition, our proposed approach is cost- and time-effective, and limited basically by the access to diverse sequencing technologies.
De novo transcriptome assembly
The Trimmomatic tool was used to trim out adaptors and low-quality fragments (Phred < 30) from the raw data. Reads shorter than 90 bp were removed from the dataset. Processed sequences were de novo assembled with Trinity with default parameters (k-mer = 25). This allowed to obtain a reference transcriptome comprising 28,282 transcripts. To confirm compatibility of RNA-Seq and DNA-Seq datasets, whole-transcriptiome mapping was performed to genomic scaffolds using BBMap, obtaining 85.65% (24,223/28,282) uniquely aligned transcripts.
We used the BUSCO tool on the transciptome, yielding 784 complete, 668 complete and single-copy, 116 complete and duplicated, 40 fragmented and 154 missing BUSCOs. This result is better than the results of the scaffold analysis (Table 4).
Genome characteristics
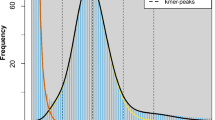
Firstly, the k-mers distribution of the genome was studied using Jellyfish and GenomeScope tools. Jellyfish was used to obtain 51-mer count histogram in a subset of 7 GB of the short DNA reads, which was used to estimate genome size, heterozygosity and repeat content with the aid of GenomeScope. The size of the test genome was found to be approximately 185 Mbp (value close to the 177 Mbp resultant assembling size, see Table 5) with low heterozygosity (below 0.05%) and 15.4% repeat content (Fig. 4).

Results obtained by GenomeScope application. Shortcuts on the diagram: len – inferred total genome length, uniq – percent of the genome that is unique (not repetitive), het – overall rate of heterozygosity, kcov – mean k-mer coverage for heterozygous bases, err – error rate of the reads, dup – average rate of read duplications, k – k-mer size, observed – the observed k-mer profile, full model – estimated GenomeScope model, unique sequence – line representing unique sequences (k-mers below the line are treated as unique), errors – line representing sequencing errors (k-mers below the line are treated as incorrect), k-mer peaks – increased number of k-mers compared to the number of k-mers with lower and higher coverage.
We tried to confirm the high repeat content value by launching the RepeatMasker29,30 tool with Repbase31 database (databases Dfam_Consensus-20170127 and RepBase-20181026). Several families of repeat elements covering only 0.72% of the genome were identified. However, in the presented genome assembly 9.127 Mbp of the 177.074 Mbp (5.2%) is known as ‘N’ signs. In addition, we estimate that approximately 8 Mbp (4.5%) of the genome has not been assembled. Most of the ‘N’ signs and unassembled sequences may consist of repetitive sequences, which may be a response to the high value of the predicted repeat content.
Genome assembly results comparison to results available at WormBase ParaSite
The H. diminuta genome has previously been studied and the genome draft is available10. However, our sequencing effort23 resulted in approximately 45× better N50 statistics (2.3 Mbp versus 51.0 kbp; in presented study 13× fewer scaffolds were obtained: 719 scaffolds in comparison with 9867, with the longest being almost 7 Mbp compared to 356 kbp in the previous work (Table 5). Our results were also evaluated using the Circoletto tool; example results are presented in Fig. 5.

Results obtained by the Circoletto application. The presented diagram compares the HMN_01_pilon sequence (subsequence from 18 Mbp to 24 Mbp indices) from the Hymenolepis microstoma genome (from WormBase ParaSite) to two scaffolds from the presented study: scaffold26 and scaffold28. Colors mean identity level: blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75, red > 0.75.
Mitochondrial genome characteristics
Our results indicate that the complete mitogenome of H. diminuta WMS-il1 strain consists of 13,829 bp, and includes 36 genes: two rRNA genes (l-rRNA, s-rRNA), 22 tRNA genes (Ala, Arg, Asn, Asp, Cys, Gln, Glu, Gly, His, Ile, Leu-1, Leu-2, Met, Lys, Phe, Pro, Ser-1, Ser-2, Thr, Trp, Tyr, Val), and 12 protein-coding genes (atp6, cox1, cox2, cox3, cytb, nad1, nad2, nad3, nad4, nad4L, nad5, nad6). All identified genes are oriented in the same direction (Fig. 6).

The organization of mitochondrial genome of Hymenolepis diminuta (WMS-il1 strain). All genes are transcribed in the same direction. The two leucine tRNA genes are designated by tRNA-LeuCUN and tRNA-LeuUUR, respectively, and two serine tRNA genes by tRNA-SerUCN and tRNA-SerAGN, respectively. Gene scaling is only approximate.
The rrnL gene (967 bp) is separated from the rrnS gene (709 bp) by the tRNA-Cys gene. The length of the tRNA genes vary from 59 bp (tRNA-Ser) to 72 bp (tRNA-His). The 12 protein-coding genes encoded a total number of 3,363 amino acids. The total length of all protein-coding genes was found to be 10,089 bp. The length of the individual protein-coding genes varied from 261 bp (nad4L gene) to 1599 bp (cox1 gene). Except for the nad4 gene, all the protein-coding genes use the ATG start codon, whereas the nad4 gene uses ATT as a start codon. The majority of identified protein-coding genes are terminated with the TAG termination codon; the only exceptions are cox2 and nad6 genes, which are terminated with the TAA codon.
In the mitogenome of H. diminuta (WMS-il1 strain) two non-coding regions were found: the larger between ND5 and tRNA-Gly genes, and the shorter is between the tRNA-Tyr and tRNA-Ser genes. The nucleotide composition of the obtained mitogenome is A = 25.4%, T = 45.6%, G = 19.3% and C = 9.6%.
Our mitogenome analysis of the H. diminuta WMS-il1 strain mitogenome was performed using data from the Illumina next-generation sequencing. All 36 genes previously found in mitogenomes of other cestode species were identified32,33,34. The length, structure and composition of the coding regions are also similar to these previously described in tapeworms, including NCBI H. diminuta reference sequence NC_002767.135. No differences were observed in the gene sequence encoding tRNA. However, both rRNA-coding genes differed with regard to two bases when compared to the reference sequence. Interestingly, the protein-coding regions showed substantial variability and only ND3 was identical as these described in reference sequence. These differences are shown in Table 6.
Gene prediction
Gene prediction was performed with genomic scaffolds according to the protein sequences of H. diminuta (PRJEB507) and other closely-related organisms: H. nana (PRJEB508), H. microstoma (PRJEB124) and Echinococcus multilocularis (PRJEB122), downloaded from WormBase ParaSite database36 Version: WBPS12 (WS267). This step was processed again by MAKER2 software (with -est2genome = 0; -prot2genome = 0). The annotation files (GFF3) obtained from each species were combined and both results were compared using custom script in the R environment ver. 3.5.0. Next, CDS annotations not confirmed in either pathway which were shorter than 150 nt (as suggested by NCBI) were removed from the final GFF3 file using Genome Annotation Generator (GAG)37 with -rcs 150 option. The general statistics of GFF file modifications using GAG are presented in Table 7.
A total of 15,169 potential protein-coding genes were predicted in the assembled H. diminuta genome and functionally annotated, which encodes 19,651 mRNAs. For extracting CDS sequences, the gffread (https://github.com/gpertea/gffread) script was applied. In total 16,983 (86.42%) homologs were identified in H. diminuta with a median sequence identity of 98.91%, 15,144 (77.06%) homologs in H. microstoma with a median sequence identity of 80.36%, 14,668 (74.74%) homologs in H. nana with a median sequence identity of 78.04%, and 14,132 (71.91%) homologs in E. multilocularis with a median sequence identity of 60.00% (Fig. 7(a,b)) by searching WormBase ParaSite database using BLASTp38 and CDS sequences as query.

The results of bidirectional BLAST of predicted protein coding genes (proteins) against four reference proteomes. (a) The distribution of the de novo assembled protein coding sequences across four closely related cestode species. (b) The Venn diagram of 15,169 predicted proteins. The four included cestode species shared a core set of 5,416 proteins, a total of 8,543 proteins were included with reference to the H. diminuta proteome and 1,152 were unique for this tapeworm across all analyzed species.
Annotation results
Our sequencing and annotation results enriched de novo assembly reference of the H.diminuta genome available from WormBase ParaSite. A considerable body of the annotation created only by in silico prediction is incomplete, and requires re-annotation. Our acquisition of RNA-seq data offers a significant improvement in the finalization of the annotation processes, as even mRNA sequences from related organisms (H. microstoma, H. nana, E. multilocularis) do not always form the best basis for exon–intron structure prediction. By using transcriptome evidence from the same species (H. diminuta) it was possible to confirm intronic donor-acceptor sites according to the alignment of cDNA and genomic DNA. Our improved annotation allowed the splice site to be corrected according to de novo assembly transcriptome aligned to H. diminuta genome. Software applied in this study allowed us to add UTR regions to previously-annotated genes (Fig. 8a and Suppl. A). Our data includes some fixes of the reference CDS regions (Fig. 8b and Suppl. B); in addition, the H. diminuta genome was supplemented with genes that have not yet been annotated in the reference genome (Fig. 8c and Suppl. C). In some cases, two gene annotations, predicted by Sanger Institute (annotated on two separated scaffolds, blue -HDID_scaffold0000291 and orange -HDID_scaffold0000029 bars on the Fig. 8d and Suppl. D) were merged into individual complete protein-coding gene.

The schematic diagram showing the types of improvements in the annotation of the H. diminuta genome. (a) Additions to the UTR annotations; (b) improvement of the CDS regions; (c) new gene annotations; (d) merging of two reference annotations. More detailed diagram, including examples of improvements, is presented in the Supplementary Figure (A–D).
Change history
10 February 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41597-020-0394-x
References
Sun, T. Parasitic disorders: Pathology, diagnosis, and management. (Williams & Wilkins, 1999).
Garcia, L. S. Diagnostic medical parasitology. (American Society for Microbiology Press, 2006).
Kapczuk, P. et al. Selected molecular mechanisms involved in the parasite–host system Hymenolepis diminuta–rattus norvegicus. Int. J. Mol. Sci. 19, 2435 (2018).
Skrzycki, M. et al. Hymenolepis diminuta: experimental studies on the antioxidant system with short and long term infection periods in the rats. Exp. Parasitol. 129, 158–163 (2011).
Stradowski, M. Effects of inbreeding in Hymenolepis diminuta [Cestoda]. Acta Parasitol. 3, 146–149 (1994).
Čadkova, Z. et al. Is the tapeworm able to affect tissue Pb-concentrations in white rat? Parasitology 141, 826–836 (2014).
Sulima, A. et al. Comparative Proteomic Analysis of Hymenolepis diminuta Cysticercoid and Adult Stages. Front Microbiol. 8, 2672, https://doi.org/10.3389/fmicb.2017.02672 (2018).
Bień, J. et al. Mass spectrometry analysis of the excretory-secretory (ES) products of the model cestode Hymenolepis diminuta reveals their immunogenic properties and the presence of new es proteins in cestodes. Acta Parasitol. 61, 429–442 (2019).
Burt, M. D. B. Aspects of the life-history and systematics of Hymenolepis diminuta. In Biology of the Tapeworm Hymenolepis diminuta (ed. Arai, H. P.), 1–57 (London and New York: Academic Press, 1980).
International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 51, 163–174 (2019).
Gauci, C. G., Rojas, C. A. A., Chow, C. & Lightowlers, M. W. Limitations of the Echinococcus granulosus genome sequence assemblies for analysis of the gene family encoding the eg95 vaccine antigen. Parasitology 145, 807–813 (2018).
Tsai, I. J. et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature 496, 57 (2013).
Zheng, H. et al. The genome of the hydatid tapeworm Echinococcus granulosus. Nat. Genet. 45, 1168 (2013).
Mostovoy, Y. et al. A hybrid approach for de novo human genome sequence assembly and phasing. Nat. Methods 13, 587 (2016).
Tan, M. H. et al. Finding nemo: hybrid assembly with oxford nanopore and illumina reads greatly improves the clownfish (Amphiprion ocellaris) genome assembly. GigaScience 7, gix137 (2018).
Small, S. T. et al. Human Migration and the Spread of the Nematode Parasite Wuchereria bancrofti. Mol. Biol. Evol., pii: msz116, https://doi.org/10.1093/molbev/msz116 (2018).
Howe, K. L., Bolt, B. J., Shafie, M., Kersey, P. & Berriman, M. Wormbase parasite — a comprehensive resource for helminth genomics. Mol. Biol. Parasitol. 215, 2–10 (2017).
D Jackman, S. et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 27, gr.214346.116 (2017).
Kuśmirek, W. & Nowak, R. De novo assembly of bacterial genomes with repetitive DNA regions by dnaasm application. BMC Bioinformatics 19(273), 1–10 (2018).
Warren, R. et al. LINKS: Scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 4, 35 (2015).
Kuśmirek, W., Franus, W. & Nowak, R. Linking de novo assembly results with long DNA reads by dnaasm-link application Biomed Res. Int 2019, 1–10 (2019).
Consortium, U. Uniprot: a hub for protein information. Nucleic Acids Research 43, D204–D212 (2014).
Hybrid sequencing of Hymenolepsis diminuta genome. European Nucleotide Archive, https://identifiers.org/ena.embl:ERP113437 (2019).
NCBI Assembly, https://identifiers.org/ncbi/insdc.gca:GCA_902177915.1 (2019).
Nowak, M. R. Hymenolepis diminuta strain WMS-il1 genome assembly, organelle: mitochondrion. GenBank, https://identifiers.org/ncbi/insdc:LR536429.1 (2019).
Nowak, R. et al. Hybrid de novo whole-genome assembly and annotation of the model tapeworm Hymenolepis diminuta genome. figshare, https://doi.org/10.6084/m9.figshare.c.4485695 (2019).
O’Connell, J. et al. NxTrim: Optimized trimming of Illumina mate pair reads. Bioinformatics 31, 2035–2037 (2015).
Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, gr.215087.116 (2017).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4–10 (2004).
Tarailo-Graovac, M. et al. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4–10 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Li, W. X. et al. The complete mitochondrial dna of three monozoic tapeworms in the caryophyllidea: a mitogenomic perspective on the phylogeny of eucestodes. Parasites Vectors 10, 314 (2017).
Li, W. X. et al. Comparative mitogenomics supports synonymy of the Genera ligula and Digramma (Cestoda: Diphyllobothriidae). Parasites Vectors 11, 324 (2018).
Guo, A. et al. Mitochondrial genome of paruterina candelabraria (Cestoda: Paruterinidae), with implications for the relationships between the genera cladotaenia and paruterina. Acta Trop. 189, 1–5 (2019).
von Nickisch-Rosenegk, M., Brown, W. M. & Boore, J. L. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that platyhelminths are eutrochozoans. Mol. Biol. Evol. 18, 721–730 (2001).
Howe, K. et al. Wormbase: annotating many nematode genomes. In Worm, vol. 1, 15–21 (Taylor & Francis, 2012).
Geib, S. M. et al. Genome annotation generator: a simple tool for generating and correcting wgs annotation tables for ncbi submission. GigaScience 7, giy018 (2018).
Camacho, C. et al. Blast+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Bushnell, B. Bbmap: a fast, accurate, splice-aware aligner. Tech. Rep., Lawrence Berkeley National Lab. (LBNL), Berkeley, CA (United States) (2014).
Andrews, S. Fastqc a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2014).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. Nanopack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–70 (2011).
Vurture, G. et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Simão, F., Waterhouse, R., Ioannidis, P., V Kriventseva, E. & M Zdobnov, E. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs Bioinformatics 31, 3210–3212 (2015).
Darzentas, N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26, 2620–2621 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from rna-seq data. Nat. Biotechnol. 29, 644 (2011).
Dobin, A. et al. Star: ultrafast universal rna-seq aligner. Bioinformatics 29, 15–21 (2013).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. Braker1: unsupervised rna-seq-based genome annotation with genemark-et and augustus. Bioinformatics 32, 767–769 (2015).
Stanke, M. et al. Augustus: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439 (2006).
Holt, C. & Yandell, M. Maker2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from rna-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494 (2013).
Reimand, J. et al. g: Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Research 44, W83–W89 (2016).
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Reports 18, 762–776 (2017).
Wheeler, T. J. & Eddy, S. R. Nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Punta, M. et al. The pfam protein families database. Nucleic Acids Research 40, D290–D301 (2011).
Lagesen, K. et al. Rnammer: consistent and rapid annotation of ribosomal rna genes. Nucleic Acids Research 35, 3100–3108 (2007).
Nielsen, H. Predicting secretory proteins with SignalP. Protein Function Prediction, 59–73 (Springer, 2017).
Acknowledgements
Financial support for this study was provided by the National-Science-Center-Poland (Grant Number 2014/13/B/NZ6/00881). We thank Dr Jan Gawor and Dr Robert Gromadka from the DNA Sequencing and Oligonucleotide Synthesis Laboratory, IBB Polish Academy of Science for their technical assistance during High molecular genomic DNA isolation, long read library construction and MinION sequencing.
Author information
Authors and Affiliations
Contributions
D.M. conceived and managed the project. D.M., R.S., R.P. and V.V.T. obtained funding and designed the study. R.S. and A.S. provided H. diminuta material. A.S.-K., R.S., A.S. and K.B. isolated DNA and RNA. R.P., M.R. managed DNA and RNA sequencing. R.N., W.K., J.J., Ł.P. carried out genome assembly, gene prediction, and functional genome annotation. J.J., Ł.P. and R.S. carried out comparative genomics. R.N., D.M., J.J. and Ł.P. drafted the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Nowak, R.M., Jastrzębski, J.P., Kuśmirek, W. et al. Hybrid de novo whole-genome assembly and annotation of the model tapeworm Hymenolepis diminuta. Sci Data 6, 302 (2019). https://doi.org/10.1038/s41597-019-0311-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0311-3
This article is cited by
-
Cestodes in the genomic era
Parasitology Research (2022)
-
Comparative genomic analysis of Echinococcus multilocularis with other tapeworms
Biologia (2022)
-
Complete genome sequence of lovastatin producer Aspergillus terreus ATCC 20542 and evaluation of genomic diversity among A. terreus strains
Applied Microbiology and Biotechnology (2021)
