
Abstract
There is still limited information on the diversity of viruses co-circulating in humans and animals. Here, we report data obtained from a large field collection of enteric samples taken from humans, pigs, rodents and other mammal hosts in Vietnam between 2012 and 2016. Each of 2100 stool or rectal swab samples was subjected to virally-enriched agnostic metagenomic sequencing; the short read sequence data are accessible from the European Nucleotide Archive (ENA). We link the sequence data to metadata on host type and demography and geographic location, distinguishing hospital patients, members of a cohort identified as a high risk of zoonotic infections (e.g. abattoir workers, rat traders) and animals. These data are suitable for further studies of virus diversity and virus discovery in humans and animals from Vietnam and to identify viruses found in multiple hosts that are potentially zoonotic.
Measurement(s) | viral metagenome |
Technology Type(s) | DNA Sequencing |
Factor Type(s) | related animal |
Sample Characteristic - Organism | Homo sapiens • Rodentia • Chiroptera • Simiiformes • Sus • Carnivora • Capra hircus |
Sample Characteristic - Environment | fecal environment |
Sample Characteristic - Location | Viet Nam |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9889709
Similar content being viewed by others


Background & Summary
Viruses of zoonotic origin are a significant public health concern worldwide having given rise to a number of high profile health emergencies in recent years, including SARS and MERS coronaviruses, H5N1 influenza A and ebolavirus1. However, detailed information on the epidemiology and evolution of viruses co-circulating in human and animal populations in the absence of significant outbreaks or epidemics is generally lacking. To help overcome this deficiency, the Vietnam Initiative on Zoonotic InfectIONS (VIZIONS) project ran between 2011 and 2016 with the overall aim of improving understanding of the epidemiology and evolution of zoonotic virus infections (particularly emerging infections) in Vietnam in south-east Asia.
VIZIONS was a Wellcome Trust-funded collaboration between the Oxford University Clinical Research Unit (OUCRU) in Ho Chi Minh City, Vietnam, the Sanger Institute and the University of Edinburgh in the UK, and Metabiota Inc. in the US, working closely with seven Vietnam hospitals, Hanoi Medical University, and Vietnam regional medical centres and sub-departments for animal health1.
VIZIONS supported studies in hospitals, communities and animal populations throughout Vietnam covering mainly enteric, respiratory, CNS infections and jaundice and generating data on clinical symptoms and outcomes, diagnostics, epidemiology and risk factors, virus identification, and virus genome sequences2,3. The data reported here comprise metagenomic sequence data and linked metadata for 2100 enteric samples from humans and animals and come from two major components of the VIZIONS project, one hospital-based and one community-based.
The first component was a large hospital-based surveillance study: a retrospective case series of hospitalised patients presenting with enteric disease believed to result from an infection3. The main aims of the study were to obtain high resolution data on all viruses present in these patients; to identify viruses of possible zoonotic origin causing severe disease (resulting in hospitalisation); and to estimate the disease burden due to viruses of zoonotic origin. This component contributed two additional sub-studies: a small study of only patients who tested positive for rotavirus A infection using a PCR test; and another small study of patients with undiagnosed enteric disease at four hospital sites.
The second component was a high-risk sentinel cohort: a prospective longitudinal cohort of people who have frequent occupational contact with animals and are thus perceived to be at high risk of developing zoonotic infections2. This component contributed two sub-studies: the high-risk sentinel cohort itself; and a parallel study of non-human mammals with which the human subjects were in close contact. The main aims of the study were to characterise the diversity of viruses circulating in high-risk human populations and in the animals with which they were in contact (including those that were asymptomatic or caused only mild illness that would not be picked up in a hospital-based study), estimate the incidence of virus infections in the human cohort, and identify possible instances of cross-species transmission. Subjects were selected based on high levels of exposure to animals (mainly on farms, in wet markets or slaughterhouses) and were sampled routinely at regular intervals over a three-year period and during episodes of diarrhoea.
A metagenomic sequencing protocol using random primers and a reverse transcription step was used to identify viruses in faecal samples and rectal swabs from these studies. This method was considered to be less biased towards common pathogens and to yield more information than a battery of diagnostic PCRs targeting specific, known pathogens. Metagenomic sequence data would allow in-depth characterisation of virus diversity in the study populations. Subsequent phylogenetic analysis would provide insights into the evolutionary and epidemiological dynamics of the identified viruses, including their spatial and temporal spread through human and animal populations and the inference of any zoonotic transmission events.
The great majority of the data reported here were obtained from Dong Thap, a province in the south of Vietnam that is regarded as a potential hot-spot for the emergence of novel zoonotic viruses. Dong Thap province is in the Mekong Delta region (Fig. 1). Due to its mostly flat terrain, the Mekong Delta is one of the most important agricultural centres in Vietnam, and it has high human and livestock population densities. Dong Thap has an area of 3,379 km2 and is inhabited by just under 1.7 million people, approximately 4.7 million ducks, chickens and other poultry, 233,000 pigs, and 26,000 cattle and buffalo3. Additionally, there is a vibrant rat trade, with many tonnes of live rats sold for human consumption each year2. In addition, a small number of samples are included from other sites in Vietnam (see below).

Map indicating approximate geographical locations of subjects and sampling points within Dong Thap province. Inset shows position of Dong Thap province within Vietnam.
Overall, sequence data and metadata are available for 2100 samples collected in Vietnam between November 2012 and May 2016. The numbers of samples from each of the five sub-studies are shown in Table 1.
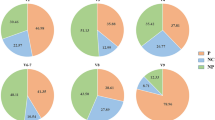
1258 samples were obtained from human subjects (Fig. 2). 707 samples were collected from patients admitted to hospital with symptoms of enteric disease. The great majority of these (95%) were from Dong Thap General Hospital (Table 1). 551 samples were obtained from a high-risk sentinel cohort in Dong Thap province, most of whom were sampled on multiple occasions. These subjects are divided into four risk categories (Table 2). 842 samples were obtained from a variety of non-human animals in Dong Thap province in parallel with sampling the high-risk cohort (Table 3). Information on host species, sampling date, location and other attributes (see below) is available for all samples.

Age and gender distribution of participants in the high-risk and hospital based study. Age (years) and genders (male (blue), female (green)) were recorded for each participant in the hospital based study on the day of sampling. The age of participants in the high-risk cohort was recoded on the day of sampling. The genders of participants in the high-risk cohort were not disclosed.
Metagenomic sequencing yielded from 3670 to 59.5 M read pairs (Illlumina 2 × 250 nt reads) per sample, with a mean of 3.38 M, a median of 2.66 M and a mode of 3.1 M. 67.7% of samples were from stool and 32.3% were from rectal swabs. Stool samples had higher modal read counts (Fig. 3).

Distribution of log10(read count per sample) grouped by sample collection method and host type. For each sample, the total number of reads produced by metagenomic sequencing were calculated. Samples were split into four categories based on sample collection method (faecal sample or rectal swab) and host type (human or non-human animal).
The sequence data and linked metadata have been used in a series of published research papers on viruses in Vietnam:
-
Complete genome characterization of two wild-type measles viruses from Vietnamese infants4;
-
Genome sequences of a novel Vietnamese bat bunyavirus5;
-
Whole-genome deep sequencing of rotaviruses from human and porcine stool samples with evidence of a putative zoonotic infection6;
-
Characterization of Posa and Posa-like virus genomes in faecal samples from humans, pigs, rats, and bats collected7;
-
Genetic diversity and cross-species transmission of kobuviruses in Vietnam8;
-
Virus diversity in enteric bat samples from Vietnam9;
-
Identification of coronavirus genomes from bats and rodents10.
We anticipate that the data may be used for further studies of virus diversity and virus discovery in humans and animals from Vietnam and to identify viruses found in both kinds of host that are potentially zoonotic.
Methods
Methodological summaries are provided below for each of the 5 sub-studies listed in Table 1. These are followed by a summary of the metagenomic sequencing methodology – this was common to all 5 sub-studies.
Hospital patients – enteric disease study
Study design and recruitment
This study took place in Dong Thap General Hospital between November 2012 and May 2015. The study method are described in full elsewhere1. All hospital admissions during the study period with suspected enteric disease were considered for recruitment and up to six patients per day were enrolled into the study.
Patient inclusion and exclusion criteria
Patients were included in the study if they satisfied the following criteria: a clinical diagnosis of acute diarrhoeal disease (defined as a minimum of three loose stools within 24 hours, or one bloody loose stool) believed to be caused by an infection; provision of written informed consent; and requiring admission to the hospital as decided by the attending physician. Patients were excluded from the study if they had been hospitalised with enteric disease within the previous six months, if they had previously been enrolled to the study, if the diarrhoea was deemed likely to be due to prior antibiotic treatment, or if they had unrelated medical complications.
Data collection
Upon enrolment, study staff collected a stool sample (on the day of admission, before any antimicrobial treatment) and a variety of demographic and other epidemiologically relevant information. The patient’s residential address was recorded and mapped with GPS software. Stool samples were subjected to a range of diagnostic tests at the microbiology departments of the participating hospitals and at the central study laboratory at OUCRU: these data are incomplete and are not reported here. All 602 samples collected during this study were sent for metagenomic sequencing (see below).
Hospital patients – rotavirus study
Faecal samples from a subset of hospital patients admitted to Dong Thap General Hospital with enteric disease tested positive by PCR test for the presence of rotavirus A (full details are provided elsewhere6). Between March 2014 and May 2016, 49 such patients were purposefully sampled in order to provide rotavirus positive samples as a partial validation of the metagenomic sequencing. These patients were additional to the routine sample collection from enteric disease patients described above. In all other respects these samples were treated in an identical manner to those from the enteric disease study.
Hospital patients – disease of unknown origin (DUO) study
Between November 2012 and June 2014, stool samples from 56 hospital patients with an enteric disease of unknown origin (DUO) were sent for metagenomic sequencing. The samples were collected from Dak Lak Provincial Hospital (23), Khanh Hoa Provincial Hospital (8), Hue Provincial Hospital (8) and Dong Thap General Hospital (19). Inclusion and exclusion criteria were as for the main enteric disease study, with the additional criterion that no pathogen was identified by diagnostic screening at the hospital. In all other respects these samples were treated in an identical manner to those from the enteric disease study at Dong Thap General Hospital.
High-risk sentinel cohort study
Study design and recruitment
This study took place in Dong Thap Province between March 2013 and May 2016. The study design, methods and baseline characteristics of the cohort are described in detail elsewhere11,12. Four categories of subjects with residential or occupational exposure to animals were recruited into the cohort:
-
Animal farmers and their relatives, representing people with typical residential exposures to a variety of livestock species. This included keepers of both domestic and exotic animals, but excluded keepers of cold blooded animals. Poultry, pig and cattle farmers were originally selected at random from the animal farm census. Exotic animal farmers were actively sought as a group of special interest with the aim of maximising diversity of animals sampled. Up to four family members, including children, were enrolled from 61 farms.
-
Pig and poultry slaughterers, representing people with more intense occupational exposures (particularly to blood). These were recruited from the three major abattoirs and slaughter points in Dong Thap Province.
-
Animal health workers, representing people with intense occupational exposures to sick animals. These were recruited through the Dong Thap Sub-Department of Animal Health and District Veterinary Stations.
-
Rat traders and sellers from three marketplaces. These were actively sought as a group of special interest.
Inclusion and exclusion criteria
Potential participants were included in the cohort study if they were long-term members of one of the above four risk groups; gave informed consent to sampling at regular intervals for three years; and resided within 40 km of Dong Thap General Hospital for clinical presentation in the event of illness.
Data collection
Included individuals were followed up for three years. Cohort members were requested to provide a rectal swab at enrolment, yearly thereafter, and when they reported an episode of enteric illness. In the third year of the study, stool samples were collected instead of rectal swabs, as it had become apparent that human rectal swabs generally did not have sufficient concentrations of viral nucleic acid for reliable metagenomic sequencing. Detailed demographic, socio-economic and behavioural data relating to food preparation and consumption were collected from each participant; these data are not reported here. A total of 551 samples were sent for metagenomic sequencing. These comprised:
-
rectal swabs taken during baseline data collection (March–October 2013);
-
rectal swabs from episodes of enteric illness (April–September 2013);
-
faecal samples from the final data collection period (March–May 2016).
Non-human animal study
The high-risk cohort study also included the sampling of associated mammals and birds. This part of the study is described in detail elsewhere10. A subset of 842 these samples was selected for metagenomic sequencing. This subset comprised all available rodent and bat samples, and a representative selection from other mammals (Table 3). Bat and monkey species were not recorded during sampling, therefore this was predicted by querying the raw reads from bat and monkey samples against mitochondrial DNA databases containing Chiroptera (NCBI:txid9397, February 2017) and Simiiformes (excluding Homo sapiens) (NCBI:txid314293, June 2018) sequences respectively. This was performed using Blastn (e value < 1e-50) and selecting the species with the highest frequency of hits.
Viral metagenomic sequencing
A total of 2100 samples obtained from the 5 sub-studies were sent for metagenomic sequencing at the Sanger Institute, UK. Samples were stored at −80 °C after collection and maintained at this temperature until sample processing (up to a maximum of 12 months). Samples were processed according to a modified Virus Discovery by cDNA Amplified Fragment Length Polymorphism (VIDISCA) method13,14,15. This method enriches samples for viral genetic material, which is otherwise difficult to sequence as it is naturally present at much lower concentrations than host and bacterial nucleic acid. It then converts genetic material from both DNA and RNA viruses to dsDNA to allow for metagenomic library preparation, using a process that is sequence-independent and reduces contamination by host ribosomal RNA.
Detailed procedures were as follows. Samples were suspended in an equal volume of phosphate-buffered saline, and 110 µl were used for nucleic acid extractions. First, viral nucleic acid was enriched for by the removal of cellular debris, mitochondria and bacteria with a centrifugation step (10 minutes at 10,000 × g), and subsequent digestion of any residual DNA with DNase (20 units TURBO DNase, Ambion) - viral nucleic acid was presumed to be protected from this treatment by encapsidation within virions. Protected viral nucleic acid was then extracted using the Boom extraction method16. Viral RNA was converted to a cDNA intermediate using reverse transcriptase (Superscript II, Invitrogen) and a mixture of hexamer primers, designed to avoid binding to ribosomal RNA17. Subsequently, cDNA and viral ssDNA were subjected to second strand synthesis with 5 units Klenow fragment (3′-5′ exonuclease defective, New England Biolabs), and the resulting dsDNA was purified by phenol/chloroform extraction and ethanol precipitation.
Library preparation and sequencing
Isolated viral dsDNA was used to prepare metagenomic sequence libraries for deep sequencing, as previously described6. Library preparation followed standard Illumina protocols, with up to 96 samples being prepared at a time for multiplex sequencing. Nucleic acids were sheared to 400–500 nucleotides (nt) in length. Distinct 8 nt indexing barcodes were added to each sample’s nucleic acid before pooling. Each pool was sequenced divided over two lanes. Sequencing was done on Illumina HiSeq. 2500 machines, yielding up to several million 250 nt paired-end reads per sample. Adapters were trimmed off with Biobambam218. For ethical reasons, human-derived sequences were identified by aligning reads to the GRCh38 reference genome (without Epstein-Barr virus) with BWA-backtrack19, and then removed. The resulting sequence data were deposited in the European Nucleotide Archive (ENA) in two batches, the first by Wellcome Sanger Institute as study PRJEB650520 and the second by the University of Edinburgh as study PRJEB2668721. These sequencing studies are linked by umbrella study PRJEB2788122, where sequence data for each sample is available for download in fastq or cram format.
Ethics
Informed consent was obtained from all human subjects. All sampling and testing procedures were carried out in accordance with guidelines that had previously been reviewed and approved by the relevant institutional ethics committees for human and veterinary medicine. This includes the Ethics Board of Dong Thap Hospital and the review board of sub-Department of Animal Health (for work carried out in Dong Thap); the Ethics Board Hospital of Tropical Diseases and the review board of the sub-Department of Animal Health (for work carried out in Dong Thap province) and the Hanoi Medical University and the review board of the sub-Department of Animal Health in Ha Noi (for work carried out in Ha Noi). In addition, all methods were approved by the Oxford Tropical Research Ethics Committee (OxTREC) (No. 157-12) in the United Kingdom.
Data Records
The sample metadata are available in xslx format via Edinburgh DataShare23. The metagenomic sequence data are freely available through the ENA via studies PRJEB650520 and PRJEB2668721 (linked under study PRJEB2788122). Data linkage is via ENA sample accession in Column T of the metadata file.
The metadata file contains the following information:
Column A (Batch) – Processing group identifier: samples with the same identifier were processed in the laboratory at approximately the same time. This may be useful information for users wanting to take batch effects into account.
Column B (Run) – Illumina HiSeq run identifier: samples with the same identifier were multiplexed and sequenced together at the Sanger Institute. This may be useful information for users wanting to take batch effects into account.
Column C (Sample ID) – Unique identifier for each sample and its corresponding sequence data.
Column D (Subject ID) – Identifier for each individual study subject (hospital patient, cohort member or animal). Study subjects may have had multiple samples taken, hence this identifier is not unique to a sample. It consists of a study code (04VZ for the hospital study, 05VZ for the cohort study), a location code (10 for Dong Thap hospital, or 75-nn for cohort study sites), and an individual identifier.
Column E (Study component) – VIZIONS study component under which the sample was collected.
Column F (Sampling date) – Sample collection date in format YYYY-MM-DD.
Column G (Sampling occasion) – Occasion on which the sample was collected. For patients in the hospital study, this is “hospitalisation (enteric disease)”. For high-risk cohort members, this is “study baseline”, “clinical episode”, or “study end”. For bats and rats, this is “routine sampling”. For other (farm) animals, this is “study baseline”.
Column H (Host category) – Host category of the study subject: “human”, “swine”, “cat”, “dog”, “goat”, “monkey”, “rat”, or “bat”.
Column I (Host species) – Species of the study subject (scientific name), if known.
Column J (Risk category) – Risk category of the high-risk cohort member: “abattoir worker (pig slaughterer)”, “animal health worker”, “farm household”, “rat trader”, “poultry slaughterer”.
Column K (Age of host) – Age of the study subject at time of sampling, in years.
Column L (Sex/gender of host) – Sex or gender of the study subject.
Column M (Sample type) – Sample type: “faeces” or “rectal swab”.
Column N (Province) – Province of the subject’s residence (for patients in the hospital study) or of the sampling site (for cohort members and animals).
Column O (District) – District of the subject’s residence (for patients in the hospital study) or of the sampling site (for cohort members and animals).
Column P (Latitude) – Latitude (degrees North) of the subject’s residence (for patients in the hospital study) or of the sampling site (for cohort members and animals). For samples collected in the community, latitude is reported to only 2 decimal places to preserve subjects’ anonymity.
Column Q (Longitude) – Longitude (degrees East) of the subject’s residence (for patients in the hospital study) or of the sampling site (for cohort members and animals). For samples collected in the community, longitude is reported to only 2 decimal places to preserve subjects’ anonymity.
Column R (Study accession) – ENA accession number for the study.
Column S (Secondary study accession) – ENA secondary accession number for the study.
Column T (Sample accession) – ENA accession number for the sample.
Column U (Secondary sample accession) – ENA secondary accession number for the sample.
Column V (Read count) - Number of raw reads produced upon sequencing the sample.
Technical Validation
RNA was quantified prior to sequencing using the Qubit RNA HS Assay Kit. Sequence data had adapters trimmed off and human-derived reads filtered out (as described in the Methods section). The average read quality (based on Phred score24,25) was determined for all samples using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) (Fig. 4). Of the 14.4 billion reads generated, 99.59% had an average quality score of 20 or above. Users of the sequence data are responsible for performing their own quality checks.

Average read quality grouped by sample collection method and host type. The average read quality for each read was calculated using FastQC. Reads were split into four categories based on collection method (faecal sample or rectal swab) and host type (human or non-human animal) of the sample they belong to.
Metadata were curated manually and all entries cross-checked by a data manager.
Usage Notes
The metadata associated with the sequence data is located on Edinburgh DataVault23. As the VIZIONS metadata include anonymised information on human subjects it cannot be made openly available. Researchers are required formally to request access to the database by contacting the Corresponding Author. Researchers will be required to complete a Data Release Request form that covers the purpose of the study, funding, ethical approvals, publication and intellectual property (Supplementary File 1).
References
Rabaa, M. A. et al. The Vietnam Initiative on Zoonotic Infections (VIZIONS): A Strategic Approach to Studying Emerging Zoonotic Infectious Diseases. Ecohealth 12, 726–735, https://doi.org/10.1007/s10393-015-1061-0 (2015).
Nguyen Tri Khiem, L. Q. C. Ho Van Chien. Rats, mice and people: rodent biology and management. 543–547 (ACIAR, 2002).
GSO. Nien Giam Thong Ke (Statistical Yearbook of Vietnam) (2015).
Oude Munnink, B. B., Phan, M. V., Kellam, P., Cotten, M. & Consortium, V. Complete Genome Characterization of Two Wild-Type Measles Viruses from Vietnamese Infants during the 2014 Outbreak. Genome Announc 4, https://doi.org/10.1128/genomeA.00250-16 (2016).
Oude Munnink, B. B. et al. Genome Sequences of a Novel Vietnamese Bat Bunyavirus. Genome Announc 4, https://doi.org/10.1128/genomeA.01366-16 (2016).
Phan, M. V. T. et al. Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection. Virus Evolution 2, vew027–vew027, https://doi.org/10.1093/ve/vew027 (2016).
Oude Munnink, B. B. et al. Characterization of Posa and Posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol 3, vex022, https://doi.org/10.1093/ve/vex022 (2017).
Lu, L. et al. Genetic diversity and cross-species transmission of kobuviruses in Vietnam. Virus Evol 4, vey002, https://doi.org/10.1093/ve/vey002 (2018).
O’Toole, A., Bogaart, C., Lu, L., Ashworth, J. & Woolhouse, M. E. A68 Bats as a source of zoonotic spillover: Investigating viruses in enteric bat samples from Viet Nam. Virus Evolution 4, vey010.067–vey010.067, https://doi.org/10.1093/ve/vey010.067 (2018).
Carrique-Mas, J. J. et al. The baseline characteristics and interim analyses of the high-risk sentinel cohort of the Vietnam Initiative on Zoonotic InfectiONS (VIZIONS). Scientific reports 5, 17965, https://doi.org/10.1038/srep17965 (2015).
Phan, M. V. T. et al. Identification and characterization of Coronaviridae genomes from Vietnamese bats and rats based on conserved protein domains. Virus Evolution 4, https://doi.org/10.1093/ve/vey035 (2019).
Saylors, K. et al. Mobilising community-based research on zoonotic infections: A case study of longitudinal cohorts in Vietnam. Gateways: International Journal of Community Research and Engagement 8, 23–43, https://doi.org/10.5130/ijcre.v8i1.3922 (2015).
de Vries, M. et al. Performance of VIDISCA-454 in feces-suspensions and serum. Viruses 4, 1328–1334, https://doi.org/10.3390/v4081328 (2012).
de Vries, M. et al. A sensitive assay for virus discovery in respiratory clinical samples. PLoS One 6, e16118, https://doi.org/10.1371/journal.pone.0016118 (2011).
Cotten, M. et al. Full genome virus detection in fecal samples using sensitive nucleic acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS One 9, e93269, https://doi.org/10.1371/journal.pone.0093269 (2014).
Boom, R. et al. Rapid and simple method for purification of nucleic acids. J Clin Microbiol 28, 495–503 (1990).
Endoh, D. et al. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res 33, e65, https://doi.org/10.1093/nar/gni064 (2005).
Tischler, G. & Leonard, S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code for Biology and Medicine 9, 13–13, https://doi.org/10.1186/1751-0473-9-13 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
European Nucleotide Archive, https://identifiers.org/ena.embl:PRJEB6505 (2014).
European Nucleotide Archive, https://identifiers.org/ena.embl:PRJEB26687 (2018).
European Nucleotide Archive, https://identifiers.org/ena.embl:PRJEB27881 (2018).
Ashworth, J. L., Bogaardt, C. & Woolhouse, M. E. J. Sample descriptors linked to metagenomic sequencing data from human and animal enteric samples from Vietnam. Edinburgh DataVault. https://doi.org/10.7488/b130ef3f-d3f2-4c8a-956e-e69f974b08ad (2018).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8, 175–185, https://doi.org/10.1101/gr.8.3.175 (1998).
Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8, 186–194 (1998).
Acknowledgements
The VIZIONS project was supported through a Wellcome Trust research grant ref. 093724. We gratefully acknowledge the contributions made to this study by the following Vietnam institutions: the Preventive Medicine Centre Dong Thap Province; the Sub-Department of Animal Health Dong Thap Province; the Preventive Medicine Centre Dak Lak Province; the Regional Animal Health Laboratory, Buon Ma Thuot; the Sub-Department of Animal Health Dak Lak Province; Hanoi Medical University, Ha Noi; Ba Vi District Health Centre, Ha Noi; and Ba Vi District Veterinary Station, Ha Noi. We acknowledge the roles played by the entire VIZIONS consortium in obtaining these data. At the Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam: Bach Tuan Kiet, Stephen Baker, Alessandra Berto, Maciej F. Boni, Juliet E. Bryant, Bui Duc Phu, James I Campbell, Juan Carrique-Mas, Dang Manh Hung, Dang Thao Huong, Dang Tram Oanh, Jeremy N. Day, Dinh Van Tan, H. Rogier van Doorn, Duong An Han, Jeremy Farrar, Hau Thi Thu Trang, Ho Dang Trung Nghia, Hoang Bao Long, Hoang Van Duong, Huynh Thi Kim Thu, Lam Chi Cuong, Le Manh Hung, Le Thanh Phuong, Le Thi Phuc, Le Thi Phuong, Le Xuan Luat, Luu Thi Thu Ha, Ly Van Chuong, Mai Thi Phuoc Loan, Behzad Nadjm, Ngo Thanh Bao, Ngo Thi Hoa, Ngo Tri Tue, Nguyen Canh Tu, Nguyen Dac Thuan, Nguyen Dong, Nguyen Khac Chuyen, Nguyen Ngoc An, Nguyen Ngoc Vinh, Nguyen Quoc Hung, Nguyen Thanh Dung, Nguyen Thanh Minh, Nguyen Thi Binh, Nguyen Thi Hong Tham, Nguyen Thi Hong Tien, Nguyen Thi Kim Chuc, Nguyen Thi Le Ngoc, Nguyen Thi Lien Ha, Nguyen Thi Nam Lien, Nguyen Thi Ngoc Diep, Nguyen Thi Nhung, Nguyen Thi Song Chau, Nguyen Thi Yen Chi, Nguyen Thieu Trinh, Nguyen Thu Van, Nguyen Van Cuong, Nguyen Van Hung, Nguyen Van Kinh, Nguyen Van Minh Hoang, Nguyen Van My, Nguyen Van Thang, Nguyen Van Thanh, Nguyen Van Vinh Chau, Nguyen Van Xang, Pham Ha My, Pham Hong Anh, Pham Thi Minh Khoa, Pham Thi Thanh Tam, Pham Van Lao, Pham Van Minh, Phan Van Be Bay, Phan Vu Tra My, Maia A Rabaa, Motiur Rahman, Corinne Thompson, Guy Thwaites, Ta Thi Dieu Ngan, Tran Do Hoang Nhu, Tran Hoang Minh Chau, Tran Khanh Toan, Tran My Phuc, Tran Thi Kim Hong, Tran Thi Ngoc Dung, Tran Thi Thanh Thanh, Tran Thi Thuy Minh, Tran Thua Nguyen, Tran Tinh Hien, Trinh Quang Tri, Vo Be Hien, Vo Nhut Tai, Vo Quoc Cuong, Voong Vinh Phat, Vu Thi Lan Huong, Vu Thi Ty Hang, Heiman Wertheim. At the Sanger Institute, Hinxton, Cambridge, UK: Matthew Cotten, Bas Oude Munnink, Paul Kellam, My Vu Tra Phan. At the University of Edinburgh, Edinburgh, UK: Jordan Ashworth, Carlijn Bogaardt, Liam Brierley, Margo Chase-Topping, Al Ivens, Ha Minh Lam, Lu Lu, Sema Nickbakhsh, Dung Nyugen, Aine O’Toole, Andrew Rambaut, Mark Woolhouse. At Metabiota Inc., San Francisco, USA: Kimberly Dodd, Jason Euren, Ashley Lucas, Nancy Ortiz, Len Pennacchio, Edward Rubin, Karen E. Saylors, Tran Minh Hai, Nathan D. Wolfe. At the University of Oxford, UK: Peter Simmonds. At the University of Amsterdam, The Netherlands: Lia van der Hoek. We particularly note the contribution of Matthew Cotten, My Vu Tra Phan and Paul Kellam who led the metagenomic sequencing to which these data are linked.
Author information
Authors and Affiliations
Contributions
Mark Woolhouse, Carlijn Bogaardt and Jordan Ashworth wrote the manuscript. Jordan Ashworth and Carlijn Bogaardt prepared and deposited the metadata file. Ngo Tri Tue, Steve Baker and Guy Thwaites led the field data collection. Tran My Phuc managed the field data.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Woolhouse, M., Ashworth, J., Bogaardt, C. et al. Sample descriptors linked to metagenomic sequencing data from human and animal enteric samples from Vietnam. Sci Data 6, 202 (2019). https://doi.org/10.1038/s41597-019-0215-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0215-2
