
Abstract
Metagenomic and metatranscriptomic time-series data covering a 52-day period in the fall of 2016 provide an inventory of bacterial and archaeal community genes, transcripts, and taxonomy during an intense dinoflagellate bloom in Monterey Bay, CA, USA. The dataset comprises 84 metagenomes (0.8 terabases), 82 metatranscriptomes (1.1 terabases), and 88 16S rRNA amplicon libraries from samples collected on 41 dates. The dataset also includes 88 18S rRNA amplicon libraries, characterizing the taxonomy of the eukaryotic community during the bloom. Accompanying the sequence data are chemical and biological measurements associated with each sample. These datasets will facilitate studies of the structure and function of marine bacterial communities during episodic phytoplankton blooms.
Design Type(s) | transcription profiling design • sequence assembly objective • biodiversity assessment objective |
Measurement Type(s) | transcription profiling assay • marine metagenome • microbial community |
Technology Type(s) | RNA sequencing • DNA sequencing • amplicon sequencing |
Factor Type(s) | assay protocol • temporal_instant |
Sample Characteristic(s) | marine metagenome • Monterey Bay • ocean biome |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
In pelagic marine ecosystems, a major proportion of primary production is transformed by heterotrophic microbes on the scale of hours to days1,2,3. Much of this rapidly-processed primary production is made available in the form of dissolved organic carbon (DOC), released from phytoplankton by direct excretion or through trophic interactions. Bacterial uptake of DOC produces living biomass and regenerates inorganic nutrients1.
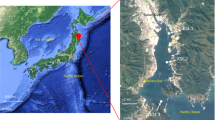
Monterey Bay is a coastal ecosystem with high primary production driven by frequent upwelling of nutrient-rich waters4,5. Intense phytoplankton blooms can develop6, and these vary dynamically in terms of taxonomic composition. In 2016, the fall phytoplankton bloom (Fig. 1) was dominated by an unusually intense bloom of the dinoflagellate Akashiwo sanguinea7. A. sanguinea cell abundances reached 4.9 × 106 cells L−1, and chlorophyll a concentrations reached 57 µg L−1 (at ~6 m depth) over the period spanning mid-September to mid-November. Here we present metagenomic, metatranscriptomic, and iTag data on the bacterial and archaeal communities during a 52-day period spanning this unusual plankton bloom in Monterey Bay (Table 1). iTag data on the eukaryotic microbial communities provides contextual information on community dynamics of the bloom-forming phytoplankton and grazer communities.

MODIS satellite image on September 26, 2016 of the phytoplankton bloom occurring in Monterey Bay and extending into the Pacific. The red dot represents the sampling station M0, located at 36.835 N, 121.901 W.
Methods
Sampling protocol
From September 26 through November 16, 2016, microbial cells were collected at Monterey Bay station M0 for sequence analysis. A moored autonomous robotic instrument, the Environmental Sample Processor (ESP)8, filtered up to 1 L of seawater sequentially through a 5.0 µm pore-size polyvinylidene fluoride filter to capture primarily eukaryotic microbes, which was stacked on top of a 0.22 µm pore-size polyvinylidene fluoride filter to capture primarily bacteria and archaea (Table 1). The samples were collected between 5 and 7 m depth at approximately 10 a.m. PST. Samples were collected daily except during October 7 – November 1 when the ESP was offline for repair. ESP filters were preserved with RNAlater at the completion of sample collection and stored in the instrument until retrieval. While the ESP was offline, grab samples were collected by Niskin bottle at the M0 mooring site 2–3 times per week, with time of sampling, depth of sampling, and filters the same as for the ESP samples except that filters were flash frozen in liquid nitrogen.
Environmental data (temperature, salinity, chlorophyll a fluorescence, light transmission, and dissolved O2 concentrations) were collected by a CTD instrument mounted with the ESP9. Additional environmental data were obtained from grab samples collected at the M0 mooring 2–3 times per week [total dimethylsulfoniopropionate concentration (DMSPt), dissolved DMSP concentration (DMSPd), DMSPd consumption rate, chlorophyll a, and cell counts by flow cytometry and microscopy]10,11 (Online-only Table 1).
DNA/RNA extraction
Total community nucleic acids for metagenome, metatranscriptome, and 16S iTag sequencing were obtained from the same 0.22 µm filter (0.22–5.0 μm size fraction) using the ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research, Irvine CA). At extraction start, internal standards were added to the lysis buffer tube (see Usage Notes), and the filter was cut into small pieces under sterile conditions to facilitate extraction. RNA was treated according to the manufacturer’s instructions with in-column DNase I treatment. After elution, RNA was treated with Turbo DNase (Invitrogen, Carlsbad CA) and concentrated using Zymo RNA Clean and Concentrator (Zymo Research). Except for a few cases of low nucleic acid yields, duplicate filters were sequenced for each sample date.
DNA for 18S rRNA gene sequencing was extracted from the 5.0 μm filters using the DNeasy Plant Mini Kit (Qiagen, Venlo NL) with modifications. Filters were cut into pieces and added into a prepared lysis tube containing ~200 µl of 1:1 mixed 0.1 and 0.5 mm zirconia/silica beads (Biospec Products, Bartlesville, OK) and 400 μl Buffer AP1. Internal standards (see Usage Notes) were added just prior to extraction. Three freeze-thaw cycles were performed using liquid nitrogen and a 65 °C water bath. Following freeze-thaw, bead beating was performed for 10 min, followed by centrifugation at 8,000 rpm for 10 min to remove foam. Following centrifugation, 45 μl of proteinase K (>600 mAU/ml, Qiagen) was added to each tube and incubated at 55 °C for 90 min with gentle rotation. Filters were then removed and the tubes incubated at 55 °C for 1 h. The DNeasy kit protocol was resumed at the RNase A addition step. Final DNA was eluted in 75 μl of diluted (1:10) TE buffer.
Metagenome sequencing and analysis
Sequence data were generated at the Department of Energy (DOE) Joint Genome Institute (JGI) using Illumina technology. Libraries were constructed and sequenced using the HiSeq-2000 1TB platform (2 × 151 bp). For assembly, reads were trimmed and screened, and those with no mate pair were removed using BFC (v r181)12. Remaining reads were assembled using SPAdes (v 3.11.1)13. The read set was mapped to the final assembly and coverage information generated using BBMap (v 37.78)14 with default parameters. Assembled metagenomes were processed through the DOE JGI Metagenome Annotation Pipeline (MAP) and loaded into the Integrated Microbial Genomes and Microbiomes (IMG/M) platform15,16.
Metatranscriptome sequencing and analysis
Sequence data were generated at the DOE JGI using Illumina technology. Libraries were constructed and sequenced using the HiSeq-2500 1TB platform (2 × 151 bp). Metatranscriptome reads were assembled using MEGAHIT (v 1.1.2)17. Cleaned reads were mapped to the assembly using BBMap.
16S and 18S iTag sequencing and analysis
Sequence data were generated at the DOE JGI using Illumina technology. Primers 515FB18 (5′-GTGYCAGCMGCCGCGGTAA) and 806RB19 (5′-GGACTACNVGGGTWTCTAAT) were used for 16S rRNA gene amplification, and primers 565F (5′-CCAGCASCYGCGGTAATTCC) and 948R (5′-ACTTTCGTTCTTGATYRA) were used for 18S rRNA gene amplification20. Libraries were constructed and sequenced using the Illumina MiSeq platform (2 × 301 bp). Contaminant reads were removed using the kmer filter in BBDuk, and filtered reads were processed by the JGI iTagger (v 2.2) pipeline (https://bitbucket.org/berkeleylab/jgi_itagger).
To generate an overview of microbial community composition during the bloom (Figs 2 and 3), the 16S and 18S rRNA amplicon libraries (raw reads) were primer-trimmed using Cutadapt (v 1.18)21 and analyzed using QIIME2 (v 2018.6)22. The DADA223 plugin in QIIME2 was used to generate exact sequence variants (ESVs), which were classified using the QIIME2 naive Bayes classifier trained on 99% Operational Taxonomic Units (OTUs) from the SILVA rRNA database (v 132)24 after trimming to the primer region. Taxonomic bar plots were generated using QIIME2.

Relative abundance of bacterial and archaeal taxa at Monterey Bay station M0 during the fall of 2016. Samples were collected at ~6 m, and 16S rRNA genes were amplified from community DNA in the 0.22 to 5.0 µm size range. Taxonomic groups were defined based on exact sequence variants using DADA2 in QIIME 2 (https://qiime2.org) and assigned taxonomy with the naive Bayes q2-feature-classifier trained using the 515F/806R region from 99% operational taxonomic units from the SILVA 132 16S rRNA database. Assignments of the 30 most abundant taxa are given at the family level.

Relative abundance of eukaryotic taxa at Monterey Bay station M0 during the fall of 2016. Samples were collected at ~6 m, and 18S rRNA genes were amplified from community DNA in the >5.0 µm size range. Taxonomic groups were defined based on exact sequence variants using DADA2 in QIIME 2 (https://qiime2.org) and assigned taxonomy with the naive Bayes q2-feature-classifier trained using the 565F/948R region from 99% operational taxonomic units from the SILVA 132 18S rRNA database.
Data Records
The raw Illumina sequencing reads for metagenomes, metatranscriptomes, and 16S rRNA and 18S rRNA iTags are available from the NCBI Sequence Read Archive under 342 separate project IDs (summarised in Online-only Table 2) which we have gathered under a single BioProject umbrella ID25.
Contigs assembled within each individual metagenome and metatranscriptome are available from the JGI Integrated Microbial Genomes portal (Online-only Table 2).
Chemical and biological data associated with each sample are available at the Biological and Chemical Oceanography Data Management Office (BCO-DMO)9,10. Measured parameters include temperature, salinity, depth, light transmission, concentrations of dissolved oxygen and chlorophyll, concentration and consumption rates of DMSP, and cell counts for heterotrophic bacteria, Synechococcus, Akashiwo, and photosynthetic eukaryotes.
Technical Validation
For metagenomic and metatranscriptomic Illumina data, BBDuk (version 37.95; https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/) was used to remove contaminants, trim reads that contained adapter sequence, and trim reads where quality dropped to zero. BBDuk was used to remove reads that contained four or more ‘N’ bases, had an average quality score across the read <3, or had a minimum length ≤51 bp or 33% of the full read length. Reads mapped with BBMap to masked human, cat, dog and mouse references at >93% identity were separated into a chaff file. Reads aligned to common microbial contaminants were also separated into a chaff file. For metatranscriptomic data, reads containing ribosomal RNA and known JGI spike-in sequences were removed and placed into separate fastq files. The internal DNA and mRNA standards added for quantification purposes at the nucleic acid extraction step (see Usage Notes) were recovered at 0.5–5.0% of sequences as expected.
For 16S rRNA and 18S rRNA, BBDuk was used to remove contaminants and trim reads that contained adapter sequence. This program was also used to remove reads that contained one or more ‘N’ bases, had an average quality score across the read of <10, or had a minimum length ≤51 bp or 33% of the full read length. Reads mapped with BBMap to masked human, cat, dog and mouse references at >93% identity or aligned to common microbial contaminants were separated into a chaff file. The 16S and 18S rRNA reads amplified from the internal DNA standards added for quantification purposes (see Usage Notes) were recovered at their expected level (0.5–5.0% of sequences).
Sequence datasets were checked for consistency with the expected composition of coastal marine microbial communities. Taxonomic assignments of 16S and 18S rRNA ESVs matched those of marine microbes common in coastal areas in general26,27 and in Monterey Bay seawater in particular11 (Figs 2 and 3). Taxonomic assignments of protein-encoding genes from metagenomic datasets were likewise representative of coastal and Monterey Bay microbial communities, and had taxonomic assignments consistent with the iTag datasets.
Usage Notes
Sample processing included the addition of internal standards to allow for calculation of volume-based absolute copy numbers for each gene or transcript type (i.e., counts L−1 rather than % of sequence library)28,29. The DNA standards consisted of genomic DNA from Thermus thermophilus DSM7039 HB829 and Blautia producta strain VPI 4299 (American Type Culture Collection, Manassas, VA). mRNA standards consisted of custom-designed 1006 nt artificial transcripts29. Artificial transcript sequences are available at Addgene Plasmid Repository (https://www.addgene.org; products MTST5 and MTST6). All four standards (two DNA and two mRNA) were added to the 0.22 μm pore size samples at the initiation of nucleic acid extraction. In the case of 18S iTag samples, genomic DNA from Arabidopsis (BioChain Institute, Inc., Newark, CA) and Mus musculus (Millipore Sigma, Burlington MA) was similarly added to the 5.0 μm pore size samples at initiation of extraction. Added amounts of internal standards were estimated at ~1% of final yields of DNA or mRNA based on prior recoveries from similar filters. Actual yields averaged ~2% of reads. The internal standards should be removed from the raw data prior to analysis. Information on how internal standards can be used for volume-based quantification is available elsewhere29,30.
Environmental data collected in association with the nucleic acid samples are given in Online-only Table 1. Available data differ between sampling dates depending on whether sampling was done by the ESP, from Niskin grab samples, or both.
Code Availability
Software versions and parameters used are as follows:
BFC v r181
MEGAHIT v 1.1.2: –k-list 23, 43, 63, 83, 103, 123
SPAdes v 3.11.1: -m 2000, -k 33, 55, 77, 99, 127 –meta
BBDuk v 38.08 for 16S, v 38.06 for 18S
BBMap v 37.78
iTagger v 2.2
For 16S iTags:
Cutadapt v 1.18: –interleaved -g GTGYCAGCMGCCGCGGTAA -G
GGACTACNVGGGTWTCTAAT -m 275 –discard-untrimmed
QIIME2 v 2018.6:
qiime dada2 denoise-paired\
–p-trunc-len-f 210\
–p-trunc-len-r 181
For 18S itags:
Cutadapt v 1.18: –interleaved -g CCAGCASCYGCGGTAATTCC -G
ACTTTCGTTCTTGATYRA -m 275 –discard-untrimmed
QIIME2 v2018.6:
qiime dada2 denoise-paired\
–p-trunc-len-f 259\
–p-trunc-len-r 200
References
Azam, F. et al. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 10, 257–263 (1983).
Moran, M. A. The global ocean microbiome. Science 350, aac8455 (2015).
Williams, P. Microbial contribution to overall marine plankton metabolism-direct measurements of respiration. Ocean Acta 4, 359–364 (1981).
Pennington, J. T. & Chavez, F. P. Seasonal fluctuations of temperature, salinity, nitrate, chlorophyll and primary production at station H3/M1 over 1989–1996 in Monterey Bay, California. Deep Sea Res Part II: Top Stud Oceanogr 47, 947–973 (2000).
Ryan, J. P. et al. Influences of upwelling and downwelling winds on red tide bloom dynamics in Monterey Bay, California. Cont Shelf Res 29, 785–795 (2009).
Schulien, J. A., Peacock, M. B., Hayashi, K., Raimondi, P. & Kudela, R. M. Phytoplankton and microbial abundance and bloom dynamics in the upwelling shadow of Monterey Bay, California, from 2006 to 2013. Mar Ecol Prog Ser 572, 43–56 (2017).
Wells, B. K. et al. State of the California Current 2016–17: Still anything but normal in the north. CalCOFI. Rep 58, 1–55 (2017).
Scholin, C. et al. The quest to develop ecogenomic sensors: A 25-year history of the Environmental Sample Processor (ESP) as a case study. Oceanography 30, 100–113 (2017).
Moran, M. A. Environmental data from CTD during the Fall 2016 ESP deployment in Monterey Bay, CA. Biological and Chemical Oceanography Data Management Office (BCO-DMO), https://doi.org/10.1575/1912/bco-dmo.756376.1 (2019).
Moran, M. A. & Kiene, R. P. Environmental data from Niskin bottle sampling during the Fall 2016 ESP deployment in Monterey Bay, CA. Biological and Chemical Oceanography Data Management Office (BCO-DMO), https://doi.org/10.1575/1912/bco-dmo.756413.1 (2019).
Nowinski, B. et al. Microdiversity and temporal dynamics of marine bacterial dimethylsulfoniopropionate genes. Environ Microbiol 12, 1687–1701 (2019).
Li, H. BFC: correcting Illumina sequencing errors. Bioinformatics 31, 2885–2887 (2015).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol 19, 455–477 (2012).
Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. Lawrence Berkeley National Laboratory, DOE Joint Genome Institute, https://jgi.doe.gov/news-publications/scientific-posters/bb_user-meeting-2014-poster-final/ (2014).
Chen, I.-M. A. et al. IMG/M v. 5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res 47, D666–D677 (2018).
Huntemann, M. et al. The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v. 4). Stand Genomic Sci 11, 17 (2016).
Li, D. et al. MEGAHITv1. 0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18, 1403–1414 (2016).
Apprill, A., McNally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol 75, 129–137 (2015).
Stoeck, T. et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol 19, 21–31 (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Bolyen, E. et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Preprint at, https://doi.org/10.7287/peerj.preprints.27295v2 (2018).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nature Meth 13, 581 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596 (2012).
BioProject, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA533622 (2018).
Gifford, S. M., Sharma, S., Booth, M. & Moran, M. A. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J 7, 281 (2013).
Satinsky, B. M. et al. Microspatial gene expression patterns in the Amazon River Plume. Proc Nat Acad Sci USA 111, 11085–11090 (2014).
Moran, M. A. et al. Sizing up metatranscriptomics. ISME J 7, 237 (2013).
Satinsky, B. M., Gifford, S. M., Crump, B. C. & Moran, M. A. In Meth Enzymol Vol. 531 (ed. DeLong, E. F.) 237–250 (Elsevier, 2013).
Lin, Y., Gifford, S., Ducklow, H., Schofield, O. & Cassar, N. Towards quantitative microbiome community profiling using internal standards. Appl Environ Microbiol 85, e02634–02618 (2019).
Acknowledgements
We thank B. Roman, B. Ussler, J. Figurski, C. Wahl, B. Kieft, S. Gifford, T. Pennington for sampling and protocol assistance, advice, and technical expertise; K. Selph for flow cytometric analysis, L. Ziccarelli for A. sanguinea microscopy analysis, J. Christmann and the crew of the R/V Shana Rae; the Moss Landing Marine Laboratories Small Boat Facility; J. Ryan for processing MODIS satellite images; and S. Sharma and C. Edwardson for bioinformatic assistance. This work was funded by NSF grants OCE-1342694, OCE-1342699, OCE-1342734, the DOE Community Science Program, and partial support from the David and Lucile Packard Foundation through funds allocated to the Monterey Bay Aquarium Research Institute (MBARI). The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
B.N. managed the project, conducted field sampling, prepared RNA and DNA samples, analyzed data, and wrote the manuscript. C.B.S. prepared RNA and DNA samples. C.M.T. conducted field sampling. K.E. conducted field sampling and chemical data analysis. R.M. managed field sampling and instruments and contributed to manuscript writing. C.M.P. managed field sampling and instruments and contributed to manuscript writing. C.A.S. supervised field sampling and contributed to manuscript writing. J.M.B. supervised field sampling and contributed to manuscript writing. M.H. participated in sequencing and analysis. A.C. participated in sequencing and analysis. B.F. participated in sequencing and analysis. B.F. participated in sequencing and analysis. S.R. participated in sequencing and analysis. K.P. participated in sequencing and analysis. N.V. participated in sequencing and analysis. S.M. participated in sequencing and analysis. T.B.K.R. participated in sequencing and analysis. C.D. participated in sequencing and analysis. A.C. participated in sequencing and analysis. N.N.I. participated in sequencing and analysis. N.C.K. supervised sequencing and analysis. T.G.R. coordinated sequencing and analysis. W.B.W. designed the project and contributed to manuscript writing. R.P.K. designed the project, conducted field sampling, supervised chemical data analysis, and contributed to manuscript writing. E.A.E.F. supervised sequencing and analysis. M.A.M. designed and supervised the project and wrote the manuscript with contributions from all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Tables
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Nowinski, B., Smith, C.B., Thomas, C.M. et al. Microbial metagenomes and metatranscriptomes during a coastal phytoplankton bloom. Sci Data 6, 129 (2019). https://doi.org/10.1038/s41597-019-0132-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0132-4
This article is cited by
-
Reverse engineering environmental metatranscriptomes clarifies best practices for eukaryotic assembly
BMC Bioinformatics (2023)
-
Ecological divergence of syntopic marine bacterial species is shaped by gene content and expression
The ISME Journal (2023)
-
Mutation-induced infections of phage-plasmids
Nature Communications (2023)
-
Functional annotation and importance of marine bacterial transporters of plankton exometabolites
ISME Communications (2023)
-
Salinity effects on the microbiome of a Neotropical water strider
Hydrobiologia (2023)
