
Abstract
The London Planetree (Platanus acerifolia) are present throughout the world. The tree is considered a greening plant and is commonly planted in streets, parks, and courtyards. The Sycamore lace bug (Corythucha ciliata) is a serious pest of this tree. To determine the molecular mechanism behind the interaction between the London Planetree and the Sycamore lace bug, we generated a comprehensive RNA-seq dataset (630,835,762 clean reads) for P. acerifolia by sequencing both infected and non-infected leaves of C. ciliata using the Illumina Hiseq 4000 system. We assembled the transcriptomes using the Trinity De Novo assembly followed by annotation. In total, 121,136 unigenes were obtained, and 80,559 unigenes were successfully annotated. From the 121,136 unigenes, we identified 3,010,256 SNPs, 39,097 microsatellites locus, and 1,916 transcription factors. The transcriptomic dataset we present are the first reports of transcriptome information in Platanus species and will be incredibly useful in future studies with P. acerifolia and other Platanus species, especially in the areas of genomics, molecular biology, physiology, and population genetics.
Design Type(s) | transcription profiling design • sequence assembly objective • sequence annotation objective |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | experimental condition • temporal_interval |
Sample Characteristic(s) | Platanus × hispanica • leaf |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Transcriptional sequencing technology is used in biological research for the gene expression profile investigation, the biological molecular evolution, and molecular marker acquisition1,2,3,4. The technology is particularly convenient for non-model organisms, for which there is no genome data available5,6. Abundant transcriptome data of some garden trees are reported as the demand for continuous development of urban landscaping7,8,9.
The London Planetree (Platanus acerifolia) is a hybrid cross between the American sycamore (P. occidentalis) and the Oriental Planetree (P. orientalis)10. P. acerifolia is a woody arbor plant with a large crown that grows rapidly, provides dense shade, and is tolerant to urban pollution11. This species is commonly grown around the world and is known as “the king of street trees12”. Despite its widespread use, there is a lack of research regarding the molecular biology of the tree, and there are no publicly available genome or transcriptome resources for the species or the genus. For this reason, research on genetic diversity and work on genetic engineering using molecular biotechnology is limited.
A particularly harmful pest to P. acerifolia is the sycamore lace bug (Corythucha ciliata), which is native to North America but was introduced to Europe in the 1960s13. The bug was first found in Hunan province in China in 2002 and has since spread to Hubei, Shanghai, Shandong, Henan, and Beijing, where heavy infestations have been reported14,15. The sycamore lace bug specifically damages Platanus trees, causing chlorotic or bronzed foliage and premature senescence of leaves16. Currently, transcriptome resources are not available for the genus Platanus, even though such data would deepen our understanding of the interaction mechanism between P. acerifolia and C. ciliata and promote related research between in the two other Platanus species.
The objectives of our study were to determine the leaf transcriptome dataset of this tree. The leaf transcriptome of P. acerifolia was sequenced on the Illumina HiSeq 4000 platform, and 637,324,886 raw reads were generated. After filtering reads of low quality, the 630,835,762 clean reads were assembled de novo and led to 121,136 unigenes. A total of 76,203, 52,758, 48,527, 8,849, 57,997, and 34,193 unigenes were annotated with a significant Blastx against non-redundant (Nr), SwissProt, Protein family (Pfam), Clusters of Orthologous Groups (COG), gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, respectively. After transcriptome sequences, molecular marker and transcription factor were mined. A total of 3,010,256 single nucleotide polymorphisms (SNPs) were identified in all samples, and 39,097 microsatellites (simple sequence repeats, SSR) were identified cross the 121,136 unigenes. In addition, 1,916 transcription factors were identified. This data descriptor provides an opportunity to identify the functional genes and molecular marker for P. acerifolia. This comprehensive P. acerifolia transcriptomic information can be utilized to promote the insect defense mechanisms in P. acerifolia.
Methods
Material treatment
Leaf samples of P. acerifolia were collected from mature trees that were in the courtyard of Beijing Academy of Agriculture and Forestry Sciences (Beijing, China) during July 2017 (Table 1). Only healthy leaves were selected. The leaves, including the petiole, were detached from the tree and placed in a glass tube with 10 mL sterile water. The glass tubes were sealed with absorbent cotton and placed in a 2 L glass beaker. Each leaf was inserted into 100 C. ciliata, which were raised according to previous research16. The experiments were performed in a growth chamber (25 ± 2 °C, 50–70% RH, 16:8 L:D). The insects on the leaves were treated for 24 h, 48 h and removed with a soft brush. Control leaves (control) were grown as the others but without C. ciliate infestation. After treatment, each plant leaf sample was collected for RNA extraction. Each treatment was performed in three biological replicates.
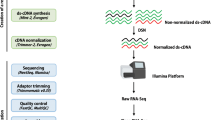
RNA isolation, cDNA library, and illumina sequencing
Total RNA was extracted using the TRIzol reagent (Invitrogen, CA, USA). The integrity and the purity of total RNA were verified using an Agilent Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent Technologies, CA, USA) with a minimum RNA integration number of 7. Approximately 10 μg of the total RNA representing a specific adipose type was subjected to isolate Poly (A) mRNA with poly-T oligo-attached magnetic beads (Invitrogen, CA, USA). After purification, the poly(A)− or the poly(A)+ RNA fractions were fragmented into small pieces using divalent cations under elevated temperatures. The cleaved RNA fragments were reverse-transcribed to create the final cDNA library in accordance with the protocol for the mRNA-Seq sample preparation kit (Illumina, San Diego, USA). The average insert size for the paired-end libraries was 300 bp (±50 bp). The paired-end sequencing was performed on an Illumina Hiseq 4000 following the vendor’s recommended protocol.
De Novo assembly, unigene annotation, and functional classification
Fastp17 was used to remove the readings that contained adaptor contamination, low quality bases, and undetermined bases. The sequence quality was verified via FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), including the Q20, the Q30, and the GC-content of the clean data. The downstream analyses were based on high-quality clean data. De Novo assembly of the transcriptome was performed with Trinity 2.4.018. Next, TransRate19 and BUSCO20 were used to assess De Novo transcriptome assembly quality. The assembled unigenes were aligned against the Nr protein (http://www.ncbi.nlm.nih.gov/), Pfam, COG, and the SwissProt (http://www.expasy.ch/sprot/) databases using BLASTx21 with an E-value threshold of <0.00001. The gene ontology (GO) annotations were obtained using Blast2GO22 (http://www.blast2go.com/b2ghome). Metabolic pathway analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/)23.
SNPs, SSRs, and transcription factor identification
SAMtools package24 was used to detect potential SNPs. SNPs were filtered based on the following criteria: (1) the number of reads to cover a candidate SNP above 8; (2) remove low quality where base calls with low Phred quality below 25; (3) frequency of mutated bases among all reads covering the position above 30%. For all unigenes, SSRs were identified using MISA25 (http://pgrc.ipk-gatersleben.de/misa/misa.html) according to default parameters, and the primer for each SSR was designed by Primer3 (http://primer3.sourceforge.net/releases.php)26. The transcription factor families were identified using the Plant Transcription Factor Database PlantTFDB 4.0 (http://planttfdb.cbi.pku.edu.cn/prediction.php)27.
Data Records
The annotation, molecular markers, and transcription factor output files were provided in Figshare28. Raw FASTQ files for the RNA-Seq were deposited to the NCBI SRA database under SRA accession number SRP15664029. The final assembled unigenes sequences were deposited at NCBI GenBank (GGXZ00000000.2)30.
Technical Validation
High throughput sequencing generated 46,890,842–57,342,752 pairs of raw reads per sample29, and the Q20 scores (the average quality value) were greater than 97%. The GC content of clean reads was similar, ranging from 46.14% to 47.36% (Online-only Table 1). The total length of the combined reads for the 12 samples that represented the different stages of damage was 202,095,905 bp and 121,136 unigenes28; the average length was 1015.15 bp with an N50 of 1579 bp and an E90N50 of 1762 bp (Table 2).
All 121,136 unigenes found in P. acerifolia leaves were functionally annotated using six public databases (Table 3). Of unigenes, 62.91% (76,203) were annotated to the NR database, 43.55% (52,758) were annotated to proteins in the Swiss-Prot database, 40.06% (48,527) were annotated to proteins in the Pfam database, 7.31% (8,849) were annotated to the COG database, 47.88% (57,997) were annotated to the GO database, and 28.23% (34,193) were annotated to the nucleotide sequences in the KEGG database. In total, 66.5% of unigenes (80,559) were annotated to a database.
The similarity analysis of the NR database demonstrated that there were 39,436 unigenes with significant homology (E-values < 1e−30) to other sequences in the Nr database and 36,767 unigenes with E-values between 1e−5 and 1e−30. The NR annotation species distribution analysis showed that 22,670 unigenes had higher homology with nelumbo_nucifera, which accounted for 29.94% of the total (Fig. 1)28. In addition, Swiss-Prot and Pfam annotation results were deposited in Swiss-prot_annotation.xls and Pfam_annotation.xls, respectively28.

Species distribution of the NR annotation.
After COG based annotation, a total of 8,849 unigenes were assigned to 24 functional categories (Fig. 2)28. For COG annotation, the two largest COG categories were “Translation, ribosomal structure, and biogenesis” (803, 16.85%) and “Posttranslational modification, protein turnover, chaperones” (550, 11.54%). The following abundant groups were “General function prediction only” (457, 9.59%), “Energy production and conversion” (324, 6.80%), “Carbohydrate transport and metabolism” (300, 6.30%), “Signal transduction mechanisms” (265, 5.56%), and “Amino acid transport and metabolism” (262, 5.50%). The two groups involving “Cell motility” (7, 0.147%) and “Nuclear structure” (3, 0.063%) represented the smallest COG classifications. Lastly, 43 unigenes (0.902%) were classified into “Defense mechanisms”.

The COG functional categories.
A total of 57,997 unigenes were annotated in the GO database, 53.14% (29,079) for the biological process, 58.80% (49,763) for the molecular function, and 56.13% (32,553) for the cellular component. The categories “cellular process,” “metabolic process,” and “single-organism process” were most abundant among the biological process GO category. Within the cellular component category, the “cell” and “cell part” terms were most abundant. For the molecular function, the unigenes were chiefly related to “binding” and “catalytic activity” (Fig. 3)28.

GO classification of the Platanus acerifolia unigenes.
We mapped the unigenes to the reference authoritative pathway in KEGG for further functional classification and annotation. In total, 34,193 unigenes were distributed among 130 KEGG pathways, and 11,229 (32.84%) were related to metabolic pathways. The largest number of unigenes involved were in the “Carbohydrate metabolism” (2741) category, followed by the “Amino acid metabolism” (1771) category, whereas “Glycan biosynthesis and metabolism” (309) was the smallest group (Fig. 4 and kegg_annotation.xls)28.

KEGG pathway distribution of the Platanus acerifolia unigenes.
We screened the P. acerifolia unigene dataset to determine potential SNPs and SSRs for future populations and genetics analysis. Among unigenes sequences, we detected 28,144 unigenes containing SSRs and 6,053 unigenes containing more than one SSR. According to the repeat motif, the SSR loci can be divided into six categories: mono-nucleotide repeats (21,895), di-nucleotide repeats (11,388), tri-nucleotide repeats (5,353), tetra-nucleotide repeats (373), penta-nucleotide repeats (55), and hexa-nucleotide repeats (33) (Fig. 5, ssr_repeats.xls, ssr_analysis_details.xls)28.

Percentage of different SSR motifs.
A total of 3,010,256 SNPs were obtained from the twelve leaves samples. Among these SNPs, 1,503,269 and 1,506,987 SNPs were obtained from the CK and insect treated samples, respectively. And, 1,005,449 SNPs were homo-type, 2,004,807 were hete-type (snp_homo_hete_statistics.xls, snp_detail.xls)28. Among them, 1,349,858 were putative transitions, and 791,734 were putative transversions. The transition-type SNPs include four classes (A/G, C/T, G/A, and T/C) and the transversion-type SNPs include eight classes (A/C, A/T, C/A, C/G, G/C, G/T, T/A, and T/G). (snp_transition_tranversion_statistics.xls, snp_detail.xls)28.
In order to promote functional gene research in P. acerifolia, we identified a series of transcription factors, which included 35 gene families. Among them, MYB_superfamily had as many as 311 unigenes, and C2C2 and AP2/ERF had 168 and 166 unigenes, respectively. NAC had 132 unigenes, bHLH had 122 unigenes, and both WRKY had 107 unigenes (Fig. 6, Transcription_Factor_annotation.xls)28.

Transcription factor family of the Platanus acerifolia unigenes.
The comprehensive datasets we present are the first reports of transcriptome information in Platanus species and will facilitate the identification of insect defense-related genes in the future. The annotated unigenes are a significant improvement on the sequence information available for P. acerifolia and other closely related species. The identified SNPs and SSR locus resources will be of help in population genetic structure, gene flow studies, and parentage analysis for P. acerifolia. The reported transcription factors in this dataset will be useful resources to further explore the physiological and biochemical mechanisms of growth development and stress response in P. acerifolia and other Platanus species.
Code Availability
The following software were used for data analysis:
1. Fastp was used for preprocessing for FastQ files. https://github.com/OpenGene/fastp.
2. FastQC was used for quality control. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Trinity 2.4.0 was used to de novo transcriptome assembly. https://github.com/trinityrnaseq/trinityrnaseq.
4. Transrate v1.0.3 and BUSCO v3 were used for assessing assembly quality. http://hibberdlab.com/transrate/ and https://busco.ezlab.org/.
5. Blast2GO was used for GO annotation. http://www.blast2go.com/b2ghome.
6. KEGG database was used for metabolic pathway annotation. http://www.genome.jp/kegg/.
7. SAMtools was used for detecting SNPs. https://samtools.github.io/bcftools/howtos/variant-calling.html.
8. MISA was used to identify SSRs. http://pgrc.ipk-gatersleben.de/misa/misa.html.
9. PlantTFDB 4.0 database was used for predicting transcription factor. http://planttfdb.cbi.pku.edu.cn/prediction.php.
References
Simon, S. A. et al. Short-read sequencing technologies for transcriptional analyses. Annual Review of Plant Biology 60, 305 (2009).
Dunwell, J. M., Moya-LeaN, M. A. & Herrera, R. Transcriptome analysis and crop improvement (a review). Biological Research 34, 153–164 (2001).
Zhang, Q. L. et al. Characterization of ladybird Henosepilachna vigintioctopunctata transcriptomes across various life stages. Scientific Data 5, 180093 (2018).
D’Esposito, D. et al. Transcriptome characterisation and simple sequence repeat marker discovery in the seagrass Posidonia oceanica. Scientific Data 3, 160115 (2016).
Li, F., Cao, D., Liu, Y., Yang, T. & Wang, G. Transcriptome Sequencing of Lima Bean (Phaseolus lunatus) to Identify Putative Positive Selection in Phaseolus and Legumes. International Journal of Molecular Sciences 16, 15172–15187 (2015).
Yin, Z. et al. Transcriptome sequencing and molecular markers discovery in the gonads ofPortunus sanguinolentus. Scientific Data 5, 180131 (2018).
Klocko, A. L. et al. Floral Transcriptome of Eucalyptus grandis. Plant & Animal Genome XXI, 0808 (2013).
Kurth, F. et al. Large scale transcriptome analysis reveals interplay between development of forest trees and a beneficial mycorrhiza helper bacterium. Bmc Genomics 16, 658 (2015).
Padovan, A. et al. Transcriptome sequencing of two phenotypic mosaic eucalyptus trees reveals large scale transcriptome re-modelling. Plos One 10, e0123226 (2015).
Santini, A. The hybrid plane (Platanus × Acerifolia L.) Platanaceae family. Sherwood - Foreste ed Alberi Oggi 7, 37–41(2001).
Weinberger, K. R., Kinney, P. L. & Lovasi, G. S. A review of spatial variation of allergenic tree pollen within cities. Arboriculture & Urban Forestry 41, 57–68 (2015).
Liu, G., Huang, J., Chen, L. & Bao, M. Plant regeneration from excised hypocotyl explants of Platanus acerifolia willd. Vitro Cellular & Developmental Biology Plant 38, 558–563 (2002).
Li, F. et al. Sequencing and Characterization of the Invasive Sycamore Lace Bug Corythucha ciliata (Hemiptera: Tingidae) Transcriptome. Plos One 11, e0160609 (2016).
Fu, N. et al. Analysis of Corythucha ciliata CcilCSP1 Structure and Prediction of Its Binding to Host-Plant Volatiles. Scientia Silvae Sinicae 53, 109–117(2017).
Li, F. Q. et al. Understanding the mechanisms of dormancy in an invasive alien Sycamore lace bug, Corythucha ciliata through transcript and metabolite profiling. Scientific Reports 7, 2631 (2017).
Li, F. et al. Identification of an Alarm Pheromone-Binding Chemosensory Protein From the Invasive Sycamore Lace Bug Corythucha ciliata (Say). Frontiers in Physiology 9, 354 (2018).
Chen, S., Zhou, Y., Chen, Y. & Jia, G. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644 (2011).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Research 26, 1134 (2016).
Simao, F. A., Waterhouse, R. M., Panagiotis, I., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25, 3389–3402 (1997).
Ana, C. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Wixon, J. & Kell, D. The Kyoto encyclopedia of genes and genomes–KEGG. Yeast 17, 48–55 (2000).
Li, H., Handsaker, B., Wysoker, A., Fennell, T. & Ruan, J. The Sequence Alignment-Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Sharma, P. C., Atul, G. & Günter, K. Mining microsatellites in eukaryotic genomes. Trends in Biotechnology 25, 490–498 (2007).
Rozen, S. & Skaletsky, H. Primer3 on the WWW for General Users and for Biologist Programmers. Humana Press, Totowa, NJ, 365–386 (2000).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Research 45, D1040–D1045 (2017).
Li, F. et al. The annotation, molecular markers, and transcription factor of Platanus acerifolia transcriptome. figshare, https://doi.org/10.6084/m9.figshare.7866407.v2 (2019).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP156640 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GGXZ00000000.2 (2019).
Acknowledgements
This study was supported by the National Key R&D Program of China (Grant No. 2016YFC1201200), Beijing Outstanding Talents Cultivation Youth Backbone Project (2017000020060G116), and the International Cooperation Projects of Beijing Academy of Agriculture and Forestry Sciences (GJHZ2018-07).
Author information
Authors and Affiliations
Contributions
Experiments were conceived and designed by C.L. and performed by F.L., M.G. and M.J. Data were analyzed by F.L. and A.G. Paper was written by L.F. and C.W. Language in the manuscript was improved by A.G.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Li, F., Wu, C., Gao, M. et al. Transcriptome sequencing, molecular markers, and transcription factor discovery of Platanus acerifolia in the presence of Corythucha ciliata. Sci Data 6, 128 (2019). https://doi.org/10.1038/s41597-019-0111-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0111-9
This article is cited by
-
Systematic identification and evolutionary analysis of cytochrome P450 genes in Platanus acerifolia induced by Corythucha ciliata damage
Brazilian Journal of Botany (2023)
