
Abstract
Spatholobus suberectus Dunn (S. suberectus), which belongs to the Leguminosae, is an important medicinal plant in China. Owing to its long growth cycle and increased use in human medicine, wild resources of S. suberectus have decreased rapidly and may be on the verge of extinction. De novo assembly of the whole S. suberectus genome provides us a critical potential resource towards biosynthesis of the main bioactive components and seed development regulation mechanism of this plant. Utilizing several sequencing technologies such as Illumina HiSeq X Ten, single-molecule real-time sequencing, 10x Genomics, as well as new assembly techniques such as FALCON and chromatin interaction mapping (Hi-C), we assembled a chromosome-scale genome about 798 Mb in size. In total, 748 Mb (93.73%) of the contig sequences were anchored onto nine chromosomes with the longest scaffold being 103.57 Mb. Further annotation analyses predicted 31,634 protein-coding genes, of which 93.9% have been functionally annotated. All data generated in this study is available in public databases.
Design Type(s) | sequence assembly objective • sequence annotation objective |
Measurement Type(s) | whole genome sequencing assay |
Technology Type(s) | DNA sequencing |
Factor Type(s) | growth condition |
Sample Characteristic(s) | Spatholobus suberectus • leaf |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

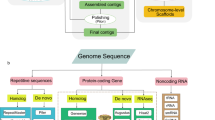
Background & Summary
Spatholobus suberectus Dunn is widely used as a food supplement in tea, wine, and soup as well as being one of the most important Chinese medicinal plants (Fig. 1a) for treatment of various diseases such as blood stasis syndrome, abnormal menstruation, and rheumatism1. It is mainly distributed in Fujian, Guangdong, Yunnan Province, and the Guangxi Zhuang Autonomous Region of China2. The vine stem of S. suberectus, called “chicken blood vines” in China due to an outflow of red juice outflow when the vine stem is injured (Fig. 1b), is the critical medicinal component3. Pharmacological and clinical studies have demonstrated that S. suberectus exhibits various functions against oxidation4, viruses5, bacteria6, cancer7, and platelets8. The crud drug of S. suberectus is therefore used in many patented Chinese medicines, and the market demand for the wild resource is increasing rapidly. But unlike other Leguminosae plants, the seed setting rate of S. suberectus is low (Fig. 1c), and most of the fruit falls off before seed maturation, which results in a low natural reproductive capacity. The growth cycle of S. suberectus is very long and the crud drug must grow for more than seven years before it can be used as medicine. These factors have combined to decrease the wild resources of S. suberectus in China to the verge of extinction.

Morphological character of S. suberectus. (a) A picture of S. suberectus plant. (b) The vine stem of S. suberectus is called “chicken blood vines”. (c) The pod of S. suberectus has only one seed.
To investigate biosynthesis of the main bioactive components and seed development mechanism needed for future S. suberectus production we generated a high-quality draft version of the S. suberectus genome. Whole-genome sequencing of several species in Leguminosae plants have been performed, for instance, Lotus japonicus9, Glycine max10, Medicago truncatula11, Glycyrrhiza uralensis12, Cicer arietinum13, and Cajanus cajan14, however, there are few reports of Subtribe Erythrininae Benth, containing nine genera of Leguminosae2. As one of the members of this subtribe, genomic information of S. suberectus can fill this gap.
The genome size of S. suberectus, a diploid (2n = 18) species, was estimated to be 793 Mb using 17-mer frequency distribution analysis with SOAPdenovo. In this study, we combined sequences generated on the Illumina, PacBio, and 10X Genomics GemCode platform as well as the new assembly technique FALCON to generate the first draft genome assembly of S. suberectus. The assembled genome is about 798 Mb with scaffold and contig N50 sizes of 6.9 Mb and 2.1 Mb, respectively. The S. suberectus assembly was further refined using 233.19 Gb Hi-C data: 748 Mb (93.73%) of the contig sequences were anchored onto nine chromosomes, the scaffold N50 was improved to be 86.99 Mb, and the longest scaffold was 103.57 Mb.
Almost half of the S. suberectus genome (47.82%) was occupied by repetitive elements, the largest amount of which was long terminal repeat retrotransposons (17.32%). Combined with homology-based predictions, de novo predictions and transcriptome-based predictions, 31,634 protein-coding genes with an average transcript size of 1,097.55 bp were predicted in the genome. In total, 93.9% (29,688) of protein-coding genes were successfully functionally annotated.
Methods
Plant materials and DNA extraction
S. suberectus samples from Nanning, Guangxi Zhuang Autonomous Region, China (22°51′28″N, 108°22′2″E) were selected for genome sequencing. The samples were kept at the Guangxi Botanical Garden of Medicinal Plants for breeding and research purposes. Total genomic DNA was isolated from fresh young leaves of 8-year-old S. suberectus using the Plant DNA Kit (TIANGEN) according to the manufacturer’s instructions.
Library construction and sequencing
The DNA was sheared by a Covaris® M220 focused-ultrasonicatorTM (Covaris, Woburn, Massachusetts, USA). The sheared DNA, with fragment sizes of 250 bp and 450 bp, was processed using the TrueSeq DNA PCR-Free LT Library Kit protocol. PCR products were purified (AMPure XP system) and library quality was assessed on an Agilent Bioanalyzer 2100 system. These PCR-Free libraries were sequenced with a HiSeq X Ten instrument as 150 bp paired-end reads. In total, 77.73 Gb of raw sequence data were generated (Table 1).
Sheared DNA (40 μg) was purified and concentrated with AMPure PB beads (PacBio) and further used for SMRTbell preparation according to the manufacturer’s protocol (Pacific Biosciences; 20-kb template preparation using BluePippin (Sage Science) size selection system with a 15-kb cut-off). The libraries were then sequenced with a PacBio sequel instrument (Pacific Biosciences, Menlo 31 Park, CA, USA). A total of 11 SMRT Cells were used to yield 79.79-fold genome coverage of sequence data (Table 1), consisting of 63.27 Gb sequence data with an N50 read length of 14,288 bp (Table 2).
The linked read sequencing libraries were constructed on a 10X Genomics GemCode platform15. Sample indexing and partition barcoded libraries were prepared using the Chromium Genome Reagent Kit (10x Genomics) according to the manufacturer’s instructions. The barcode sequencing library was first quantified by Qubit2.0, insert size was checked using an Agilent2100, and finally quantified by qPCR. The 123.09 Gb library was sequenced with 150 bp paired-end reads on an Illumina HiSeq X Ten platform (Table 1).
For the Hi-C library, chromatin was fixed in place with formaldehyde in the nucleus. Fixed chromatin was digested with DpnII restriction endonuclease, 5′ overhangs were filled in with biotinylated nucleotides, and free blunt ends were ligated. After ligation, cross-links were reversed, and the DNA was purified from protein. Purified DNA was treated to remove biotin that was not internal to the ligated fragments. The DNA was then sheared to a mean fragment size of 350 bp, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adaptors. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeq platform to produce 233.19 Gb Hi-C sequence data (Table 1). The quality of Hi-C sequencing was evaluated using HiCUP16. The effect rate (%) = Unique di-tigs/Total Reads Processed = 4,356,614/10,000,000 = 43.57% (Table 3). Typically, 35.91% of the alignable read pairs represent interchromosomal interactions. Eleven percent represents intrachromosomal interactions between fragments less than 10 kb apart and 53.09% are intrachromosomal read pairs that are more than 10 kb apart (Table 3).
Estimation of the S. suberectus genome size
Quality-filtered reads from the Illumina platform were subjected to 17-mer frequency distribution analysis with SOAPdenovo17. K-mer 17 was selected to estimate the genome size and heterozygosity of S. suberectus (Fig. 2). We plotted the distribution of k-mer depth against frequency with a main peak occurring at the depth of 40 (Fig. 2). Based on the total number of k-mers (32,476,446,092), the S. suberectus genome size was calculated to be approximately 793.39 Mb, using the following formula: genome size = k-mer_Number/Peak_Depth and Revised Gsize = Genome size × (1-Error Rate). The heterozygosity of the S. suberectus genome is 0.74%.

Estimation of S. suberectus genome size by K-mer analysis.
Genome assembly
De novo assembly of the 63.27 Gb PacBio single-molecule long reads from SMRT Sequencing was performed using FALCON (https://github.com/PacificBiosciences/FALCON/)18. In order to get enough corrected reads, the longest 60 subreads were first selected as seed reads to do error correction. Then error-corrected reads were aligned to each other and assembled into genomic contigs using FALCON with parameters length_cutoff_pr = 5000, max_diff = 120, max_cov = 130. The draft assembly was polished using the quiver algorithm. Pilon was used to perform error correction of p-contigs with 98.02X coverage of short paired-end reads generated from Illumina HiSeq Platforms19. The assembly consisted of 1,954 contigs, with a contig N50 length of 2.06 Mb (total length = 794 Mb) (Table 4).
We used BWA-MEM20 to align the 10X Genomics data to the assembly using default settings. Scaffolding was performed by fragScaff (in vitro, long-range sequence information for de novo genome assembly via transposase contiguity) with the barcoded sequencing reads.
The assembly consisted of 1,146 scaffolds, with the scaffold N50 length improving to 6.9 Mb (total length = 798 Mb) and contig N50 of 2.1 Mb (Table 5). The genome assembly size is similar to the estimated genome size by k-mer analysis.
The input de novo assembly, shotgun reads, and Dovetail Hi-C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies21. Shotgun and Dovetail Hi-C library sequences were aligned to the draft input assembly using a modified SNAP read mapper (http://snap.cs.berkeley.edu). The separations of Dovetail Hi-C read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, score prospective joins, and make joins above a threshold. After scaffolding, shotgun sequences were used to close gaps between contigs.
The S. suberectus assembly was further refined using 233.19 Gb Hi-C data (Table 1): 748 Mb (93.73%) of the contig sequences were anchored onto nine chromosomes (Fig. 3). The scaffold N50 was finally improved to be 86.99 Mb and the longest scaffold was 103.57 Mb.

Diagrammatic sketch of the annotation pipeline.
Indentification of repetitive elements in S. suberectus
Tandem Repeat Finder22 was employed to identify tandem repeats in the S. suberectus genome. RepeatMasker (http://www.repeatmasker.org) and RepeatProteinMasker23 were used against Repbase24 to identify known transposable element repeats. In addition, RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html), RepeatScout (http://www.repeatmasker.org/)25, PILER (http://www.drive5.com/piler/)26, and LTR_Finder (http://tlife.fudan.edu.cn/ltr_finder)27 were utilized to identify de novo evolved repeats (Fig. 3).
The combined results show that almost half of the S. suberectus genome (47.82%) was occupied by repetitive elements (Fig. 4b–e). Among these, long terminal repeat (LTR) retrotransponsons represent the largest amount of repetitive elements, reaching 17.32% of the genome, fewer than soybean (42%)10 and chickpea (46%)28, but are similar to Lotus japonicus (18%)9. LTR/Copia repeats were the most abundant, making up 10.06% of the genome (Fig. 4d), followed by LTR/Gypsy elements (6.61%; Fig. 4e).

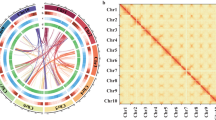
Circos Plot Showing the Genomic Features of S. suberectus. Concentric circles, from outermost to innermost, show (a) gene density (blue), (b) tandem repeats density (green), (c) transposon element density (purple), (d) LTR-Copia density (yellow), (e) LTR-Gypsy density (red) and intra-genome collinear blocks connected by curved lines. All distributions are drawn in a window size of 300 kb, chromosomes_ scale = 5,000,000 bp.
Gene annotation
Genes in the S. suberectus genome were annotated using multiple methods, including homology-based predictions, de novo predictions and transcriptome-based predictions (Fig. 3). For de novo predictions, Augustus29, GENSCAN30, GlimmerHMM31, geneid32 and SNAP33 analysis were performed on the repeat-masked genome, with parameters trained from Arabidopsis thaliana. Predicted protein sequences from Nelumbo nucifera (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/plant/Nelumbo_nucifera/latest_assembly_versions/GCF_000365185.1_Chinese_Lotus_1.1 version 1.1), Arabidopsis thaliana (ftp://ftp.ensemblgenomes.org/pub/plants/release-32/gff3/arabidopsis_thaliana/, version 10.32), Glycine max (ftp://ftp.ensemblgenomes.org/pub/plants/release-32/fasta/glycine_max/dna/, version 1.0), Petunia axillaris (ftp://ftp.solgenomics.net/genomes/Petunia_axillaris/, version 1.6.2), Solanum lycopersicum (ftp://ftp.ensemblgenomes.org/pub/plants/release-32/fasta/solanum_lycopersicum/, release-32), and Oryza sativa (ftp://ftp.ensemblgenomes.org/pub/plants/release-32/fasta/oryza_sativa/, version 1.0) were used for homology-based predictions. First, query sequences were subjected to tblastn analysis with an Expect (E)-value cutoff of 1e-5. BLAST hits corresponding to reference proteins were concatenated by Solar software, and low-quality records were removed. The genomic sequence of each reference protein was extended upstream and downstream by 2,000 bp to represent a protein-coding region. Gene structures contained in each protein region were predicted using GeneWise software34. For transcriptome-based predictions, RNA from five organs (root, petiole, leaves, flowers, and stems) was isolated and RNA-seq data were used for gene annotation, processed by TopHat and Cufflinks35. RNA-seq data were also assembled by Trinity36. PASA37 software (http://pasapipeline.github.io/) was then used to generate a full transcriptome-based genome annotation. The homology, de novo, and transcriptomic gene sets were merged to form a comprehensive and non-redundant reference gene set using EVidenceModeler38 software. Next, PASA37 was used to generate UTRs as suggested by the RNA-seq data.
Our analysis indicates that 31,634 protein-coding genes with an average transcript size of 1,097.55 bp were predicted in the genome (Fig. 4a).
Functional annotation of the protein-coding genes was carried out using blastp (E-value cut-off 1e-05) against SwissProt39 and NR databases. Protein domains were annotated by searching against InterPro40 and Pfam database41, using InterProScan and HMMER (http://hmmer.janelia.org), respectively. The GO terms for genes were obtained from the corresponding InterPro or Pfam entry. The pathways in which the genes might be involved were assigned by BLAST against the KEGG database42 with the E-value cut-off of 1e-05.
Overall, 79% (24,976), 70.8% (22,394), and 82.5% (26,082) of genes showed enrichment in InterPro, KEGG, and GO respectively. In total, 93.9% (29,688) of protein-coding genes were successfully annotated for conserved functional motifs or functional terms.
Non-coding RNA annotation
Annotation of tRNA was performed using tRNAscan-SE43 software with default parameters. rRNA annotation was based on homology with rRNAs from several diverse higher plant species (not shown), using blastn with ‘E-value = 1e-5’. miRNA and snRNA genes were predicted by INFERNAL software44 using the Rfam database45.
The final results included 820 miRNA, 672 tRNA, 261 rRNA, and 550 snRNA with average lengths of 117.33, 75.32, 305.41 and 115.50 bp respectively.
Data Records
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession QUWT0000000046. The version described in this paper is version QUWT01000000. Raw read files are available at NCBI Sequence Read Archive47. All the annotation tables containing results of an analysis of the draft genome are available at figshare48.
Technical Validation
Evaluation of the completeness of the S. suberectus genome assembly
To estimate the quality of genome assembly, short reads were mapped back to the consensus genome using BWA49 and an overall 97.29% mapping rate was found, suggesting that our assembly results contained comprehensive genomic information. Gene region completeness was evaluated by RNA-Seq data (Table S1): of the 53,538 transcripts assembled by Trinity36, 99.62% could be mapped to our genome assembly, and 95.94% were considered as complete (more than 90% of the transcript could be aligned to one continuous scaffold).
The completeness of gene regions was further assessed using CEGMA (conserved core eukaryotic gene mapping approach)50: 240 of 248 (96.77%) conserved core eukaryotic genes from CEGMA were captured in our assembly, and 206 (83.06%) of these were complete (Table S2). Furthermore, we performed BUSCO (Benchmarking Universal Single-Copy)51 analysis based on a benchmark of 956 conserved plant genes, of which 96% had complete gene coverage (including 18% duplicated ones), 1% were fragmented and only 2.6% were missing (Table S3). These data largely support a high quality S. suberectus genome assembly, which can be used for further investigation.
Code Availability
The execution of this work involved many software tools, whose versions, settings and parameters are described below.
(1) SOAPdenovo: version 3.0, default parameters; (2) FALCON: version 3.1, length_cutoff_pr = 5000, max_diff = 120, max_cov = 130; (3) HiCUP: version 0.5.10, (4) HiRise: Dovetail Genomics LLC, Santa Cruz, CA, USA; (5) BWA: version 0.7.8, default parameters; (6) Tandem Repeat Finder: version 409, default parameters; (7) RepeatMasker: version 4.0.5, default parameters; (8) Repbase: version 15.02; (9) RepeatModeler: version 1.0.11, default parameters; (10) RepeatScout: version 1.0.5, default parameters; (11) PILER: version 1.06, default parameters; (12) LTR_FINDER: version 1.0.7, default parameters; (13) Augustus: version 3.0.2, default parameters; (14) GENSCAN: version 1.0, default parameters; (15) geneid: version 1.4, default parameters; (16) GlimmerHMM: version 3.0.2, default parameters; (17) SNAP: version 11-29-2013; (18) BLAST: version 2.2.26, default parameters; (19) GeneWise: version 2.2.0, default parameters; (20) TopHat: version 2.0.8, default parameters; (21) Cufflinks: version 2.1.1, default parameters; (22) Trinity: version 2.4.0, default parameters; (23) PASA: version 2.3.3, default parameters; (24) EVidenceModeler: version 1.1.1, default parameters; (25) InterPro: version 5.16, default parameters; (26) Pfam database: version 03-30-2016; (27) InterProScan: version 4.8, default parameters; (28) NR database: version 08-10-2015; (29) KEGG database: version 08-31-2015; (30) SwissProt database: version 05-24-2016; (31) HMMER: version 3.1b1, default parameters; (32) tRNAscan-SE: version 1.3.1, default parameters; (33) BUSCO: version 3.0.2, Embryophyta Version odb9; (34) CEGMA: version 2.5.
References
Lee, M. H., Lin, Y. P., Hsu, F. L., Zhan, G. R. & Yen, K. Y. Bioactive constituents of Spatholobus suberectus in regulating tyrosinase-related proteins and mRNA in HEMn cells. Phytochemistry 67, 1262–1270 (2006).
Wu, Z. Y., Raven, P. H. & Hong, D. Y. Flora of China. (Beijing: Science Press & St. Louis: Missouri Botanical Garden Press, 2010).
Cui, Y. J., Liu, P. & Chen, R. Y. Studies on the active constituents in vine stem of Spatholobus suberectus. Chin. J. Chin. Mater. Med 30, 121–123 (2005).
Fu, Y. F. et al. Immunomodulatory and antioxidant effects of total flavonoids of Spatholobus suberectus Dunn on PCV2 infected mice. Sci. Rep 7, 8676 (2017).
Chen, S. R. et al. In Vitro Study on Anti-Hepatitis C Virus Activity of Spatholobus suberectus Dunn. Molecules 21, 1367 (2016).
Cho, H. et al. Spatholobus suberectus Dunn. constituents inhibit sortase A and Staphylococcus aureus cell clumping to fibrinogen. Arch. Pharm. Res. 40, 518–523 (2017).
Peng, F., Meng, C., Zhou, Q., Chen, J. & Xiong, L. Cytotoxic Evaluation against Breast Cancer Cells of Isoliquiritigenin Analogues from Spatholobus suberectus and Their Synthetic Derivatives. J. Nat. Prod. 79, 248–251 (2016).
Lee, B. J. et al. Antiplatelet effects of Spatholobus suberectus via inhibition of the glycoprotein IIb/IIIa receptor. J. Ethnopharmacol. 134, 460–467 (2011).
Sato, S. et al. Genome Structure of the Legume, Lotus japonicus. DNA Res. 15, 227–239 (2008).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).
Mochida, K. et al. Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 89, 181–194 (2017).
Gupta, S. et al. Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. DNA Res. 24, 10 (2016).
Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89 (2012).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C. data. F1000Res. 4, 1310 (2015).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18–18 (2012).
Chin, C. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at, https://arxiv.org/abs/1303.3997 (2013).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 4, Unit 4.10 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–158 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–268 (2007).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res 32, W309–312 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinf. 18, Unit 4.3 (2007).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res 10, 547–548 (2000).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Bairoch, A. M. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol 396, 59–70 (2007).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 28, 263–266 (2004).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Qin, S. S. et al. Draft genome of Spatholobus suberectus. GenBank, https://identifiers.org/ncbi/insdc:QUWT00000000 (2019).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP157950 (2019).
Qin, S. S. et al. Draft genome of Spatholobus suberectus. figshare, https://doi.org/10.6084/m9.figshare.c.4414709.v1 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. (Oxford University Press, 2009).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
We thank Huizhen Lv for offering photos of S. suberectus. This study was supported by the Guangxi science and technology research project (AB16450012), the National Natural Science Foundation of China (81503179, 81473309), the National Public Welfare Special Project of China “Quality Guarantee system of Chinese herbal medicines” (201507002), the China Agriculture Research System (CARS-21), the Guangxi science and technology research project (AA18242040).
Author information
Authors and Affiliations
Contributions
J.H.M. and Z.Y.Z. designed the project. S.S.Q. and L.Q.W. analyzed data and wrote the paper. S.S.Q., K.H.W. and Y.L. performed experiments. S.S.Q., Z.J.S., X.L.Z., S.W. and Q.H.W. contributed samples, materials, or data. M.J.L., K.J.Z., Y.Y.H. and S.Y.W. helped with the data analysis and examined the results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ISA-Tab metadata file
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Qin, S., Wu, L., Wei, K. et al. A draft genome for Spatholobus suberectus. Sci Data 6, 113 (2019). https://doi.org/10.1038/s41597-019-0110-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0110-x
This article is cited by
-
Differential selection of yield and quality traits has shaped genomic signatures of cowpea domestication and improvement
Nature Genetics (2024)
-
Applications of some advanced sequencing, analytical, and computational approaches in medicinal plant research: a review
Molecular Biology Reports (2024)
-
Comparative cytogenomics reveals genome reshuffling and centromere repositioning in the legume tribe Phaseoleae
Chromosome Research (2022)
-
Oligo-FISH barcode in beans: a new chromosome identification system
Theoretical and Applied Genetics (2021)
-
Antimutagenic components in Spatholobus suberectus Dunn against N-methyl-N-nitrosourea
Genes and Environment (2019)
