
Abstract
Kidney fibrosis represents an urgent unmet clinical need due to the lack of effective therapies and an inadequate understanding of the molecular pathogenesis. We have generated a comprehensive and combined multi-omics dataset (proteomics, mRNA and small RNA transcriptomics) of fibrotic kidneys that is searchable through a user-friendly web application: http://hbcreports.med.harvard.edu/fmm/. Two commonly used mouse models were utilized: a reversible chemical-induced injury model (folic acid (FA) induced nephropathy) and an irreversible surgically-induced fibrosis model (unilateral ureteral obstruction (UUO)). mRNA and small RNA sequencing, as well as 10-plex tandem mass tag (TMT) proteomics were performed with kidney samples from different time points over the course of fibrosis development. The bioinformatics workflow used to process, technically validate, and combine the single omics data will be described. In summary, we present temporal multi-omics data from fibrotic mouse kidneys that are accessible through an interrogation tool (Mouse Kidney Fibromics browser) to provide a searchable transcriptome and proteome for kidney fibrosis researchers.
Design Type(s) | transcription profiling design • proteomic profiling design • stimulus or stress design |
Measurement Type(s) | transcription profiling assay • protein expression profiling assay |
Technology Type(s) | RNA sequencing • mass spectrometry |
Factor Type(s) | experimental condition • temporal_instant • biological replicate |
Sample Characteristic(s) | Mus musculus • kidney |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
More than 10 percent of adults in developed countries present with some degree of chronic kidney disease (CKD). One of the hallmarks of CKD is the development of fibrosis and subsequent renal failure. Mechanisms and pathways underlying the development of kidney fibrosis are still not widely understood and therefore treatment strategies are limited. Mouse models are frequently used to gain more insights into the fibrosis development and to evaluate potential drug candidates.
Two well-established mouse models for kidney fibrosis are folic acid (FA) induced nephropathy and unilateral ureteral obstruction (UUO)1. Both models display human relevant pathological tubulointerstitial fibrosis shortly after induction of injury, are easy to perform and have good reproducibility. Transcriptomics and proteomics studies for both models have been published before, mostly as a basis for follow-up experiments focusing on one selected gene/protein2,3,4,5,6,7. However, data for mRNA, miRNA and protein expression, including a combination of all three omics datasets, have not been generated in parallel for these models; therefore, we aimed to generate this comprehensive data to (1) characterize the UUO and FA model in depth, (2) allow integration of the three layers of omics data, and (3) provide a tool for hypothesis generation as well as testing.
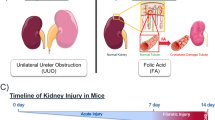
In the FA model, mice were sacrificed before the treatment (day 0) and 1, 2, 7, and 14 days after a single injection (250 mg/kg i.p.) of folic acid (Fig. 1). For the UUO model, mice were sacrificed before obstruction (day 0) and 3, 7, and 14 days after the ureter of the left kidney was obstructed via ligation (Fig. 1). For both studies, kidneys were removed at each time point for total RNA isolation and protein sample preparation. Total RNA was used for mRNA and small RNA sequencing on the Illumina platform. The transcriptomics data (mRNAs and miRNAs) from the FA model were previously published by our group2,3, but here we re-analysed the existing FA sequencing data in parallel with the newly generated UUO sequencing data, applying the same algorithms for consistency and inclusion. Proteomics in matching kidney samples from both models were measured by liquid chromatography/mass spectrometry (LC/MS) using 10-plex TMT. Temporal profiles were generated for each mRNA, miRNA and protein dataset in both models using day 0 as reference point. To combine all three sets, mRNAs and corresponding proteins were matched according to their annotations, whereas mRNAs and miRNAs were paired based on targets predicted using TargetScan.

Schematic of study design, data generation and processing. Overview of how the kidney fibrosis models were set up including flow charts for mRNA-seq, proteomics and small RNA-seq profiling in the kidneys.
For the UUO mRNA-seq data, an average of 30 million reads were sequenced, with 97% mapping to the transcriptome, less than 1% mapping to rRNA genes, and with 88% of the aligned reads mapping to exonic regions. For the UUO small RNA-seq data, an average of 20 million reads were sequenced, with 50% mapping to approximately 700 annotated miRNA genes.
In both proteomics datasets over 8,000 proteins were quantified, thus yielding overall a unique and comprehensive dataset of gene, protein and miRNA expression in the fibrotic mouse kidney.
Finally, all datasets can be viewed and interrogated by an online tool, the Mouse Kidney Fibromics browser: http://hbcreports.med.harvard.edu/fmm/.
Methods
Animal studies
Male BALC/c mice were obtained from Charles River Laboratories (USA). All experimental protocols concerning the use of laboratory animals were performed according to the NIH guidelines for the care and use of laboratory animals, and approved by the Institutional Animal Care and Use Committees (IACUC) of Harvard Medical School. Mice were housed in groups of three on a 12h light/dark cycle with access to food and water ad libitum. At the age of 8–10 weeks they entered the experiment.
Folic acid (FA) model
Folic acid was prepared at 25 mg/ml in 0.3 M sodium bicarbonate. Mice received a single dose of 250 mg/kg via intraperitoneal injection. Before the injection and 2, 7, and 14 days later, mice were sacrificed; kidneys were removed and immediately snap frozen in liquid nitrogen.
Unilateral ureter obstruction (UUO) model
Before surgery, mice were anaesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg of body weight), the left kidney was exposed via a flank incision and 3.0 silk suture thread was used to tie off the ureter at the lower pole. Before the obstruction and 3, 7, and 14 days later, mice were sacrificed; kidneys were removed and immediately snap frozen in liquid nitrogen.
More details about the in vivo experiments can be found elsewhere8.
mRNA-seq: RNA extraction, library preparation and sequencing
Folic acid (FA) model
mRNA sequencing in kidneys from the FA model was published before4 (GSE65267)9,10. In brief, quantity and quality of isolated RNA were assayed on an Agilent 2200 TapeStation instrument and by SYBR qRT-PCR assay. 10 ng total RNA was used to prepare libraries with the IntegenX Apollo 324 system and NuGEN SPIA reagents. Libraries were multiplexed in groups of three per lane of a flow cell, and 50 cycles, paired-end sequencing was performed on an Illumina HiSeq2000 instrument.
Unilateral ureter obstruction (UUO) model
Total RNA was isolated using Qiagen’s RNeasy Mini Kit. Quality and quantity of the RNA was assessed photometrically and with the Bioanalyzer (Agilent). 330 ng total RNA were transcribed utilizing Illumina’s TruSeq Stranded mRNA Library Prep Kit. Libraries were pooled and sequenced on Illumina’s NextSeq500 as single end, 75 bp reads. Sequencing service was performed by the Molecular Biology Core Facilities at Dana-Farber Cancer Institute. Data were deposited to the Gene Expression Ombibus (GEO) database: GSE1183399,11.
Small RNA-seq: RNA extraction, library preparation and sequencing
Folic acid (FA) model
small RNA sequencing in kidneys from the FA model was published before3 (GSE61328)9,12. In brief, total RNA was isolated using the miRNeasy Mini Kit (Qiagen). 1 µg total RNA was used to prepare small libraries utilizing the TruSeq Small RNA Sample Preparation Kit (Illumina) according to manufacturer instructions. All samples were multiplexed into a single lane of a flow cell on the HiSeq2000 platform to produce 50 cycles, single-end reads.
Unilateral ureter obstruction (UUO) model
Total RNA was isolated using Qiagen’s miRNeasy Mini Kit. Quality and quantity of the RNA was assessed photometrically and with the Bioanalyzer (Agilent). 1 µg total RNA was transcribed utilizing Illumina’s TruSeq Small RNA Library Prep Kit. Libraries were pooled and sequenced on Illumina’s NextSeq500 as single end, 75 bp reads. Sequencing service was performed by the Molecular Biology Core Facilities at Dana-Farber Cancer Institute. Data were deposited on GEO database: GSE1183409,13.
Proteomics: protein sample preparation, TMT labelling and LC-MS3 measurement
Kidney samples (from both FA and UUO models) were mechanically homogenized in lysis buffer (8 M urea, 1% SDS, Roche complete protease inhibitors and phosphatase inhibitors, 50 mM Tris pH 8.5). Approximately one third of a kidney was used for sample preparation. Protein concentration was determined using the BCA assay (Pierce, Rockford, IL).
The homogenate was reduced with 5 mM DTT and alkylated with 15 mM iodoacetamide (Sigma, St. Louis, MO). 0.15 mg of protein was precipitated using chloroform:methanol. Pellets were washed twice with cold methanol and re-solubilized in 8 M urea with 20 mM EPPS, pH 8.5. After diluting the samples to 4 M urea using 20 mM EPPS, they were digested with Lys-C (Wako Chemicals, Richmond, VA) overnight at room temperature. On the next day, samples were further diluted to 1.5 M urea using 20 mM EPPS and digested for 6h at 37 °C using Trypsin (Promega, Madison, WI). 60 µg of each sample were then brought to 10% (v/v) acetonitrile and labeled with 2:1 (TMT:Peptide) by mass of TMT-10 reagent (Pierce). The reaction was quenched with hydroxylamine (0.5% final volume). Afterwards, samples were acidified by adding formic acid to 2% final volume, combined, and desalted using a C18 Sep-Pak (Waters, Milford, MA). The now combined sample was fractionated using basic pH reversed phase chromatography using a 1200 HPLC (Agilent; Santa Clara, CA) equipped with a UV-DAD detector and fraction collection system. Then, the resulting 12 fractions were desalted using the C18 StageTip procedure14. Each fraction was loaded onto a 100 µm id, 35 cm long column packed with 1.8 µm beads (Sepax, Newark DE) and separated using a 3 h gradient from 8–27% buffer B (99% acetonitrile and 1% formic acid) and buffer A (96% water, 3% acetonitrile and 1% formic acid) on an Easy 1000 nano-LC (Thermo-Fisher Scientific, San Jose, CA). All MS analyses were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo-Fisher Scientific, San Jose, CA) applying a multi-notch MS3 method15,16. The FA proteomics was performed as a service at the Thermo Fisher Center for multiplexed Proteomics at the Harvard Medical School.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via PRIDE17 partner repository with the dataset identifiers PXD01145318 (FA) and PXD01086119 (UUO).
Bioinformatic analysis
mRNA-seq data
All samples were processed using an RNA-seq pipeline implemented in the bcbio-nextgen project (https://bcbio-nextgen.readthedocs.org). Raw reads were examined for quality issues using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to ensure library generation and sequencing were suitable for further analysis. Adapter sequences, other contaminant sequences (such as polyA tails and low quality sequences with PHRED quality scores less than five) were trimmed from reads using atropos20. Trimmed reads were aligned to UCSC build mm10 of the Mus musculus genome, augmented with transcript information from Ensembl release GRCm38.84 using STAR21. Alignments were checked for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity and other quality checks using a combination of FastQC, Qualimap22, MultiQC23 and custom code within the bcbio-nextgen pipeline. Counts of reads aligning to known genes were generated by featureCounts24. In parallel, Transcripts Per Million (TPM) measurements per isoform were generated by quasialignment using Salmon25. Normalization at the gene level was called with DESeq224,25,26, preferring to use counts per gene estimated from the Salmon quasialignments by tximport11,24,25,26,27. The DEGreport Bioconductor package was used for QC and clustering analysis (https://bioconductor.org/packages/release/bioc/html/DEGreport.html). A Quality metrics report for UOO and FA sequencing data can be found on https://github.com/hbc/MouseKidneyFibrOmics/tree/master/reports.
Small RNA-seq data
All samples were processed using a small RNA-seq pipeline implemented in the bcbio-nextgen project (https://bcbio-nextgen.readthedocs.org/en/latest/). Quality control of the raw reads was performed as above for the RNA-seq data using FastQC and atropos. In the following, we focused on miRNA analysis but the small RNA seq dataset includes also t-RNAs and pi-RNAs.Trimmed reads were aligned to miRBase v2128 to the specific species with seqbuster29. In addition, the trimmed reads were aligned to the Mus musculus genome (version mm10) using STAR21,29. The aligned reads were analyzed with seqcluster30 to characterize the whole small RNA transcriptome and classify reads into rRNA, miRNA, repeats, genes, tRNAs and others from the UCSCannotation31. Finally, aligned reads were analyzed using miRDeep232, an algorithm that assesses the fit of sequenced RNAs to a biological model of miRNA generation and correct folding. Alignments were checked for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity and other quality checks using a combination of FastQC, MultiQC23 and custom code within the bcbio-nextgen pipeline.
Data were loaded into R using the bcbioSmallRna R package (https://github.com/lpantano/bcbioSmallRna) and isomiRs Bioconductor package33,34 to get normalized expression values13.
Proteomics data
Raw data were converted to mzXML and searched via Sequest35 version 28 against a concatenated Uniprot36 database downloaded 02/04/2014. Variable modifications of oxidized methionine and over-labelling of TMT on serine, threonine and tyrosine were considered37. Mass tolerance parameters for peptide identification were ±25 ppm for precursor ions and ±0.9 Da for fragment ions. To distinguish forward and reverse hits, linear discriminant analysis38 was used and reverse hits were filtered to an FDR of 1% at the protein level. Using rules of parsimony shared peptides were collapsed into the minimally sufficient number of proteins (Table 1). Quantitation filters of >200 sum reporter ion S:N and >0.7 isolation specificity were incorporated. All abundance values were normalized with edgeR using TMM method39 and transformed the abundance values to log2 scale. UNIPROT ids were mapped to ensembl gene ids using the GRCm38.84 release to combine the proteomic data with the gene and miRNA expression data.
Data processing
We used the normalized abundance values for each data type to populate the Rshiny app. To pair miRNA with genes, we used TargetScanHuman database40. Only pairs described in the database and pairs with the abundance correlation along time was lower than −0.7 were kept as valid miRNA-Gene pairs. This information is shown in the Rshiny app, at the bottom of the page, where the user can inspect filtered targets of miRNAs.
Data Records
A list of all datasets per biological replicate is summarized in an experimental study table (Online-only Table 1).
GitHub as an easily accessible and widely used platform was used for our codes and QC reports. All raw data and processed data were uploaded to GEO and PRIDE and the code was finally archived at Zenodo, too41.
The raw sequencing data have been deposited in the GEO database with ID number GSE11834142.
The raw protein data have been deposited in the PRIDE database with ID numbers PXD01145318 and PXD01086118,19.
Gene expression estimates using salmon and tximport for the UUO model can be found in uuo_mrna.csv11.
Gene expression estimates using salmon and tximport for the FA model can be found in fa_mrna.csv10.
miRNA data resulting from seqbuster and isomiRs for the UUO model can be found in uuo_mirna.csv10,13.
miRNA data resulting from seqbuster and isomiRs for the FA model can be found in fa_mrna.csv12.
Protein data resulting from limma for the UUO model can be found in uuo_protein.csv43.
Protein data resulting from limma for the FA model can be found in fa_protein.csv43,44.
Technical Validation
To assess the quality of the mRNA and small RNA sequencing libraries, basic quality metrics were summarized per sample in Tables 2 and 3 as well as in the supplementary information (Supplementary Tables 1 and 2). The full quality metrics report for UOO and FA sequencing data can be found on https://github.com/hbc/MouseKidneyFibrOmics/tree/master/reports. All mRNA-seq samples had a mapping rate >90%, ribosomal content <1% and exonic mapping rate >80%, showing a good enrichment of reads on coding genes. Small RNA-seq samples had 3′ adapter in more than 80% of the reads, a read size distribution of 22 after adapter removal. After removal of reads shorter than 18 nts, more than 50% of the reads mapped to miRNA sequences.
To further assess the quality of the data, we performed principal component analysis of the normalized gene and protein expression values to determine if the biological replicates show consistency and group by time after injury. Figure 2 shows strong clustering of the replicates and separation among time points for UUO miRNA (a), UUO mRNA (b), UUO protein (c) and FA protein (d). FA mRNA and miRNA data are shown in the supplementary information (Supplementary Fig. 1). The separation among time points is maximal in the protein datasets, while the mRNA and miRNA datasets show how day 7 and day 14 (for mRNA) and day 3 and day 7 (for miRNA) are more closely related. This difference can be explained by the fact that miRNA changes happen before mRNA changes, and the miRNA profile of day 3 could be impacting the day 7 mRNA profile. The same might be true for day 7 of the miRNA profile and day 14 of the mRNA profile.

Principal component analysis (PCA) of all UUO datasets and FA proteins. The normalized expression abundance of mRNAs, proteins and miRNA was used. Each color represents a time point in the dataset. (a) miRNA expression in kidneys from the UUO model shows day 3 and 7 being in the same cluster, while the normal and the latest time points are distinct. (b) The greatest variation in gene expression in the UUO model is observed along the first principal component (PC) between normal and injured samples, with the second PC separating injury times. (c) A similar pattern is observed using UUO protein expression, with higher consistency within sample groups allowing for better discrimination between time points. (d) Protein expression in the FA model shows different clusters for each time point, PC1 separating normal from the injured samples, and PC2 separating early injury from later time points.
Additionally, we have looked at the expression of known housekeeping genes and well-known fibrosis and injury markers (Fig. 3 and Supplementary Fig. 2) to evaluate the validity of the animal and omics experiments. The selection of these genes is purely based on literature and did not result from any statistical analysis. The coefficient of variation average for housekeeping genes is 3.3%, indicating stable expression in all kidney samples. Classical fibrosis markers α-smooth muscle actin (Acta2), collagen (Col1a1) and fibronectin (Fn1) are continuously increased over time for the irreversible UUO model whereas there is a decrease towards normal in the reversible FA model. Kidney injury markers clusterin (Clu), kidney injury molecule 1 (Kim-1 alias Havcr) and lipocalin-2 (Ngal alias Lcn2) are strongly increased early on without further significant increases over time in the UUO model. In the FA model, similar to fibrosis markers, injury markers indicate recovery at the later time points. Thus, fibrosis and injury marker profiles correspond to previously published data. miR-192, a kidney-enriched miRNA involved in regulation of the sodium transport, is decreased in de-differentiated fibrotic kidneys45 while miR-21 increased according to its role in fibrosis46,47.

Expression profiles of fibrosis and injury markers and housekeeping genes. UUO mRNA (dotted line), UUO protein (dashed line), FA protein (standard line), and UUO miRNA (standard line). (a) The fibrosis markers α-smooth muscle actin (Acta2), collagen (Col1a1) and fibronectin (Fn1) show increasing expression over time for UUO mRNA, UUO protein and FA protein datasets. (b) Kidneyinjury markers clusterin (Clu), kidney injury molecule 1 (Kim-1 alias Havcr) and lipocalin-2 (Ngal alias Lcn2) show increased expression early on without further significant increases over time. (c) Nine commonly used housekeeping genes show no change of expression in all the datasets. (d) miRNAs miR-192 and -21 are involved in kidney pathogenesis and show expression changes over time for the UUO model.
Usage Notes
Analyses of parts of this dataset have been published before in separate publications4,5 using the FA mRNA and miRNA data to identify new biomarkers of fibrosis and to find miRNAs involved in the pathophysiology, respectively. This bigger dataset here with mRNA, miRNA and protein data from two kidney fibrosis models could be utilized, among other possibilities, to (1) identify and validate new target genes or miRNAs for kidney fibrosis; (2) develop a more comprehensive understanding of the pathophysiology; (3) identify novel gene-miRNA regulatory networks related to kidney fibrosis; and (4) discover novel transcripts (genes and miRNAs) in fibrotic kidneys. The various ways to use and re-use proteomics data have been reviewed and well-stated elsewhere48,49. Furthermore, a large number of algorithms for differential gene expression analysis are available through the BioConductor project website to re-analyse or further investigate this dataset. In addition, we have developed a searchable web-tool for simple and quick inquiries related to this dataset: http://hbcreports.med.harvard.edu/fmm/.
Kidney fibrosis as well as any other fibrotic disease are complex and involve complementary changes in gene and protein expression as the disease initiates and progresses; thereby, affecting various signalling pathways50. Restricting data generation and analysis to a single omic dataset shows only one facet of this complex pathophysiology. Therefore, the value of using multi omics data lies in the generation of more representative multi-layered networks which can uncover causative changes51. Comparable approaches have been made for other fibrotic diseases either in one study52 or retrospectively by reviewing individually generated and published single omics datasets53. Similarly, individual omics datasets generated by others using human kidney disease samples54 or human fibrotic diseases could be combined and integrated with our dataset to explore translational aspects or “universal” fibrotic patterns. A plethora of different integration algorithms are available and could be used for this purpose55.
One main limitation in the data reported here is that bulk omics datasets do not distinguish among different kidney cell types and infiltrated immune cells; however, this bulk data could be useful for power calculations for designing future single-cell and omics studies.
Code Availability
The code used to process the data and perform the quality control and visualization analysis can be found at: https://github.com/hbc/MouseKidneyFibrOmics and at Zenodo41.
References
Yang, H.-C., Zuo, Y. & Fogo, A. B. Models of chronic kidney disease. Drug Discov. Today Dis. Models 7, 13–19 (2010).
Craciun, F. L. et al. RNA Sequencing Identifies Novel Translational Biomarkers of Kidney Fibrosis. J. Am. Soc. Nephrol. 27, 1702–1713 (2016).
Pellegrini, K. L. et al. Application of small RNA sequencing to identify microRNAs in acute kidney injury and fibrosis. Toxicol. Appl. Pharmacol. 312, 42–52 (2016).
Husi, H. et al. A combinatorial approach of Proteomics and Systems Biology in unravelling the mechanisms of acute kidney injury (AKI): involvement of NMDA receptor GRIN1 in murine AKI. BMC Syst. Biol. 7, 110 (2013).
Arvaniti, E. et al. Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci. Rep. 6, 26235 (2016).
Chen, W.-Y. et al. Upregulation of Interleukin-33 in obstructive renal injury. Biochem. Biophys. Res. Commun. 473, 1026–1032 (2016).
Furini, G. et al. Proteomic Profiling Reveals the Transglutaminase-2 Externalization Pathway in Kidneys after Unilateral Ureteric Obstruction. J. Am. Soc. Nephrol. 29, 880–905 (2018).
Craciun, F. L. et al. Pharmacological and genetic depletion of fibrinogen protects from kidney fibrosis. Am. J. Physiol. Renal Physiol. 307, F471–84 (2014).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Craciun, F. L., Vaidya, V. S. & Hutchinson, J. N. Next Generation Sequencing identifies Cdh11 and Mrc1 as novel translational biomarkers of kidney fibrosis. Gene Expression Omnibus, http://identifiers.org/GEO:GSE65267 (2015).
Pantano, L. & Pavkovic, M. Multi Omics analysis of fibrotic kidneys in two mouse models [RNA-Seq]. Gene Expression Omnibus, http://identifiers.org/GEO:GSE118339 (2018).
Pellegrini, K. L. & Vaidya, V. S. Next generation sequencing of miRNAs in folic acid-induced kidney damage. Gene Expression Omnibus, http://identifiers.org/GEO:GSE61328 (2015).
Pantano, L. & Pavkovic, M. Multi Omics analysis of fibrotic kidneys in two mouse models [miRNA-Seq]. Gene Expression Omnibus, http://identifiers.org/GEO:GSE118340 (2018).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using Stage Tips. Nat. Protoc. 2, 1896–1906 (2007).
Ting, L., Rad, R., Gygi, S. P. & Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011).
McAlister, G. C. et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–56 (2016).
Berberich, M. Kidney proteomics in the folic acid (FA) induced nephropathy mouse model. PRIDE, http://identifiers.org/pride.project:PXD011453 (2018).
Berberich, M. Kidney proteomics in the unilateral ureter obstruction (UUO) mouse model. PRIDE, http://identifiers.org/pride.project:PXD010861 (2018).
Didion, J. P., Martin, M. & Collins, F. S. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
García-Alcalde, F. et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679 (2012).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Griffiths-Jones S., Grocock R. J., van Dongen S., Bateman A. & Enright A.J. miRBase: a database of microRNA sequences, targets and nomenclature. Nucleic Acids Res 34, D140–D144 (2006).
Pantano, L., Estivill, X. & Martí, E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 38, e34 (2010).
Pantano, L., Estivill, X. & Martí, E. A non-biased framework for the annotation and classification of the non-miRNA small RNA transcriptome. Bioinformatics 27, 3202–3203 (2011).
Mangan, M. E., Williams, J. M., Kuhn, R. M. & Lathe, W. C. The UCSC Genome Browser: What Every Molecular Biologist Should Know. In Current Protocols in Molecular Biology 107, 19.9.1-36 (2014).
Mackowiak, S. D. Identification of Novel and Known miRNAs in Deep-Sequencing Data with miRDeep2. In Current Protocols in Bioinformatics 36, 12.10.1-15 (2011).
Ramos, M. et al. Software for the Integration of Multiomics Experiments in Bioconductor. Cancer Res. 77, e39–e42 (2017).
Pantano, L., Escaramis, G. & Argyropoulos, C. Characterization of miRNAs and isomiRs, clustering and differential expression. Bioconductor, https://doi.org/10.18129/B9.bioc.isomiRs (2016).
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699 (2018).
Böhm, G. et al. Low-pH Solid-Phase Amino Labeling of Complex Peptide Digests with TMTs Improves Peptide Identification Rates for Multiplexed Global Phosphopeptide Analysis. J. Proteome Res. 14, 2500–2510 (2015).
Huttlin, E. L. et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Agarwal, V., Bell, G. W., Nam, J.-W. & Bartel, D. P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 (2015).
Pantano, L. hbc/MouseKidneyFibrOmics: v1.0. Zenodo, https://doi.org/10.5281/zenodo.2592516 (2019).
Pantano, L. & Pavkovic, M. Multi Omics analysis of fibrotic kidneys in two mouse models. Gene Expression Omnibus, http://identifiers.org/GEO:GSE118341 (2018).
Pantano, L. & Pavkovic, M. Multi Omics analysis of fibrotic kidneys in two mouse models (Folic acid (FA) model MS dataset). Gene Expression Omnibus, http://identifiers.org/GEO:GSE126181 (2018).
Pantano, L. & Pavkovic, M. Multi Omics analysis of fibrotic kidneys in two mouse models (Unilateral ureter obstruction (UUO) model MS dataset). Gene Expression Omnibus, http://identifiers.org/GEO:GSE126182 (2018).
Krupa, A. et al. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 21, 438–447 (2010).
Chau, B. N. et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4, 121ra18 (2012).
Loboda, A., Sobczak, M., Jozkowicz, A. & Dulak, J. TGF-β1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediators Inflamm. 2016, 8319283 (2016).
Martens, L. & Vizcaíno, J. A. A Golden Age for Working with Public Proteomics Data. Trends Biochem. Sci. 42, 333–341 (2017).
Vaudel, M. et al. Exploring the potential of public proteomics data. Proteomics 16, 214–225 (2016).
Humphreys, B. D. Mechanisms of Renal Fibrosis. Annual Review of Physiology 80, 309–326 (2018).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biology 18, 83 (2017).
Santolini, M. et al. A personalized, multiomics approach identifies genes involved in cardiac hypertrophy and heart failure. NPJ Syst Biol Appl 4, 12 (2018).
Yu, G., Ibarra, G. H. & Kaminski, N. Fibrosis: Lessons from OMICS analyses of the human lung. Matrix Biol. 68–69, 422–434 (2018).
Papadopoulos, T. et al. Omics databases on kidney disease: where they can be found and how to benefit from them. Clin. Kidney J. 9, 343–352 (2016).
Huang, S., Chaudhary, K. & Garmire, L. X. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 8, 84 (2017).
Acknowledgements
We thank Matt Berberich for data management support. M.P. is a recipient of a research fellowship from the Deutsche Forschungsgemeinschaft. R.A.E. was supported by NIH Grants P50-GM107618 and U54-HL127365. Work in the Vaidya laboratory was supported by Outstanding New Environmental Sciences award from NIH/NIEHS (ES017543), Innovation in Regulatory Science Award from Burroughs Wellcome Fund (BWF-1012518) and a collaborative research agreement with Biogen (A24378). We acknowledge the support of the Molecular Biology Core Facilities at Dana-Farber Cancer Institute for sequencing and the Thermo Fisher Center for Multiplexed Proteomics at the Harvard Medical School for the proteomics service on the FA samples. Work by L.P. and S.H.S. at the Harvard Chan Bioinformatics Core was funded by the Harvard Medical School Tools and Technology Committee and Harvard Catalyst|The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.P., L.P., V.S.V., conception and design, pipeline development, analysis of data, drafting and revising the article; M.P., L.P., C.V.G., S.B., S.A.B., R.A.E., J.V.S. and V.S.V. data analysis, interpretation, and manuscript revision.
Corresponding author
Ethics declarations
Competing Interests
M.P. is a full time employee of Bayer Healthcare, R.A.E. and V.S.V. are full time employees of Pfizer Inc., J.V.S. is a full time employee of Cobalt Biomedicine.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
ISA-Tab metadata file
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Pavkovic, M., Pantano, L., Gerlach, C.V. et al. Multi omics analysis of fibrotic kidneys in two mouse models. Sci Data 6, 92 (2019). https://doi.org/10.1038/s41597-019-0095-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0095-5
This article is cited by
-
WNT-dependent interaction between inflammatory fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic kidney disease
Nature Communications (2024)
-
Construction of a novel rabbit model of ureteral calculi implanted with flowable resin
BMC Urology (2022)
-
A novel renal perivascular mesenchymal cell subset gives rise to fibroblasts distinct from classic myofibroblasts
Scientific Reports (2022)
-
The Nax (SCN7A) channel: an atypical regulator of tissue homeostasis and disease
Cellular and Molecular Life Sciences (2021)
-
TGF-β1 is a regulator of the pyruvate dehydrogenase complex in fibroblasts
Scientific Reports (2020)
