
Abstract
Glycoprotein 2 (GP2) and uromodulin (UMOD) filaments protect against gastrointestinal and urinary tract infections by acting as decoys for bacterial fimbrial lectin FimH. By combining AlphaFold2 predictions with X-ray crystallography and cryo-EM, we show that these proteins contain a bipartite decoy module whose new fold presents the high-mannose glycan recognized by FimH. The structure rationalizes UMOD mutations associated with kidney diseases and visualizes a key epitope implicated in cast nephropathy.
Similar content being viewed by others

Main
GP2 and UMOD are structurally related homopolymeric glycoproteins1 (Extended Data Fig. 1a) that prevent bacterial pathogen adhesion2,3 and are implicated in multiple pathologies of the intestine and the urinary tract, respectively4,5. Recent studies revealed how the C-terminal zona pellucida (ZP) module of UMOD mediates its polymerization6,7. However, there is no detailed information on the UMOD N-terminal branch region recognized by FimH8, suggested to contain a domain with eight cysteines (D8C) conserved in different vertebrate proteins9, and it is unknown whether the equivalent region of GP2 is also responsible for binding FimH10.
To address these questions, we first expressed in mammalian cells the whole GP2 branch as well as the corresponding region of UMOD and assessed their ability to selectively capture the lectin domain of FimH (FimHL) from an Escherichia coli periplasmic extract. This showed that, as in the case of UMOD, the branch of GP2 is sufficient for interaction with FimHL (Extended Data Fig. 2).
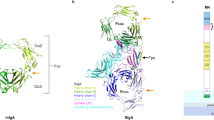
We then obtained crystals of the GP2 branch, but experimental phasing of its 1.9-Å-resolution data was hindered by relatively high diffraction disorder in one direction and low crystal symmetry. However, molecular replacement with models generated by AlphaFold2 (ref. 11) allowed us to solve the structure, which was subsequently used to phase two additional crystal forms diffracting to ~1.4 Å resolution (Extended Data Figs. 3 and 4 and Supplementary Table 1). The electron density maps reveal that the GP2 branch is a protein module (henceforth referred to as ‘decoy module’) that consists of a β-hairpin stabilized by a disulfide bond (Cx48-Cy59), packed against a globular ‘D10C’ domain with a new fold including two 310 helices, nine β-strands (βA–βI) and five intermolecular disulfides (C163-C8157, C285-C9172, C3107-C6145, C4113-C10177, C5138-C7146) (Fig. 1a and Extended Data Fig. 1). Notably, the extent of the latter and its C1-C8, C2-C9 disulfides are not compatible with the original boundaries of the D8C domain9; accordingly, GP2 D10C is secreted comparably with the complete branch, whereas a D8C construct is barely expressed and not secreted (Fig. 1b).

a, Overall structure of the GP2 branch region/decoy module, depicted in cartoon representation with β-strands in blue, 310 helices in cyan and loops in light gray. Disulfides and glycans are shown as yellow and dark gray sticks, respectively, with oxygen atoms in red and nitrogen atoms in blue. b, Reducing western blot comparison of the expression and secretion of GP2 constructs corresponding to the entire branch, D10C or D8C. n = 3. c–g, Details of the GP2 structure rationalize the effect of kidney disease-associated UMOD mutations affecting a set of residues identical between the two proteins (Supplementary Table 2). Selected GP2 D10C domain residues and mutations affecting the corresponding identical residues of UMOD are as follows: GP2 D61, P62, C163→UMOD D172H, P173L/R, C174R (c); GP2 R74, C285, D86, C4113, C10177→UMOD R185C/G/H/L/S, C195F/Y, D196N/Y, C223R/Y, C287F (d); GP2 P62, C163, W92, C8157, V163→UMOD P173L/R, C174R, W202C/S, C267F, V273F/L (e); GP2 C163, R94, C8157→UMOD C174R, R204G/P, C267F (f); GP2 Y164, C10177→UMOD Y274C/H, C287F (g).
The large majority of UMOD pathogenic mutations affect the protein’s branch and, in particular, the residues corresponding to the decoy module of GP2 (ref. 4). Because of 60% sequence identity to UMOD, the crystal structure of the latter immediately explains the effect of many substitutions affecting invariant positions (Fig. 1c–g and Supplementary Table 2). Remarkably, most of these mutations cluster within two structurally important regions of the decoy module, the β-hairpin/D10C domain groove and the disulfide bond-rich region at the opposite end of D10C (Extended Data Fig. 5).
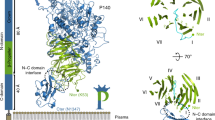
Helical reconstruction of UMOD filaments, together with focused refinement of the protein’s branch, recently yielded a composite map of the full-length molecule (Extended Data Fig. 6); however, this information could only be confidently interpreted at the level of the filament core, due to the lack of a reliable model for the branch residues6. By combining the crystallographic information on GP2 with AlphaFold2 predictions, we could generate a model of the entire UMOD branch (epidermal growth factor (EGF) domains I–III + decoy module) that was fitted into the cryo-EM density and fused with the coordinates of the filament core to describe the complete protein (Fig. 2a and Supplementary Table 3).

a, Complete atomic model of polymeric UMOD, with N-glycans shown as sticks. Elements are colored as in Extended Fig. 1a, with the D10C epitope for BJP in green; additional subunits are gray. b, UMOD cryo-EM map region encompassing the protein’s decoy module. The Asn side chains carrying the two D10C N-glycans and the BJP epitope are indicated. c, Consistent with its location within the structure, the N275 high-mannose glycan can be efficiently cleaved by Endo H only in denaturing conditions. Colored circles indicate the presence of the specified glycans, open circles with a cross indicate their absence. n = 3. d, Recognition of the D10C N275 glycan by the lectin domain of fimbrial adhesin FimH from UPEC UTI89. The cryo-EM map of the UMOD branch + EGF IV is colored gray, the difference map between the densities of the UMOD–FimHL complex and free UMOD is cyan. PNGase F, Peptide:N-glycosidase F.
Inspection of the fitted map revealed that, whereas the complex-type carbohydrate linked to D10C N232 (refs. 8,12) is exposed to the solvent, the high-mannose glycan attached to N275 (refs. 8,12) emerges from the groove between the β-hairpin and D10C, and packs against the EGF III/β-hairpin junction (Fig. 2b). This suggests that the architecture of the decoy module contributes to maintaining the high-mannose structure of the UMOD N275 glycan, which is crucial for capturing FimH2,8. Consistent with this idea, the high-mannose carbohydrate can be fully cleaved by Endoglycosidase H (Endo H) only upon protein denaturation (Fig. 2c). Interestingly, although the GP2 branch also binds FimHL, its D10C domain cannot be glycosylated at the position corresponding to UMOD N275 (R165). However, the presence of a GP2 glycosylation site at N65 (ref. 13)—a residue far away in sequence from R165, but closely located to it within the β-hairpin/D8C groove (Extended Data Fig. 7a)—suggests that this residue may carry a high-mannose glycan equivalent to UMOD N275. In agreement with these considerations, introduction of an N65A mutation in the decoy module of GP2 impairs its interaction with FimHL (Extended Data Fig. 7b) and mass spectrometric analysis of the glycans attached to N65 detects the HexNAc2Hex5 oligomannose structure (Extended Data Fig. 8), indicating that UMOD and GP2 exploit a common molecular strategy to counteract bacterial adhesion.
To gain further insights into this process, which was previously visualized only at low resolution by cryo-electron tomography8, we reconstituted in vitro the complex between UMOD and FimHL from uropathogenic E. coli (UPEC) UTI89 and studied it by single-particle cryo-EM (Extended Data Fig. 9 and Supplementary Table 3). Despite high conformational variability, this yielded a map with a nominal resolution of 7.4 Å, whose comparison with that of free UMOD showed density for a single copy of FimHL bound to the D10C region that presents the N275 glycan (Fig. 2d and Supplementary Table 3). Consistent with our binding studies (Extended Data Fig. 2b), the majority of the UMOD/FimHL interface is clearly made by the decoy module; however, the density of the complex hints at the possibility that the C-terminal region of EGF III may also contribute to the interaction with the lectin.
Finally, our study sheds light on the basis of cast nephropathy, a severe complication of multiple myeloma, by mapping the UMOD epitope recognized by monoclonal light chains/Bence Jones proteins (BJP)14 to the D10C βE/loop/βF region (Extended Data Fig. 1). Rationalizing previous biochemical studies of this medically crucial interaction14, the structure suggests that the epitope adopts a rigid conformation stabilized by its involvement in the C5-C7 and C3-C6 disulfides, close proximity to the N232 glycan and hydrophobic interaction with the C terminus of another subunit within the UMOD filament (Fig. 2a,b).
From a general point of view, this work provides an example of how deep learning techniques can substantially aid the X-ray crystallographic and cryo-EM investigation of challenging biological samples, by providing accurate models that can be used to solve the phase problem and aid the fitting of low-resolution density maps, respectively.
Methods
DNA constructs
Consistent with a cautionary note in UniProt entry P55259 and sequence alignments with homologous sequences from other species, prediction of the signal peptide cleavage propensity of the human GP2 sequence with SignalP15 suggested that M8, rather than M1, corresponds to the protein’s initiator methionine. Moreover, sequence comparisons indicated that GP2 isoform 1 residues V179–R181, which immediately follow the last residue encoded by GP2 exon 2, are not only absent in isoform α (UniProt P55259-3), but also lack counterparts in human UMOD (UniProt P07911). Based on this information, an open reading frame was designed that encoded GP2α residues M8–S181 (corresponding to isoform 1 residues M8–T178 + D182−S184) followed by a 8× His tag. A corresponding gene and an equivalent UMOD construct, as well as GP2 Δ31-59, Δ31-88 and N65A mutant genes, were also synthesized (GenScript) and all constructs were cloned into pLJ6, a mammalian expression vector derived from pHLsec3 (ref. 16).
For expressing the E. coli FimH lectin domain (FimHL; residues F22–T179), synthetic genes encoding non-tagged and C-terminally His-tagged versions of the protein (including its native signal peptide) were cloned into bacterial expression vectors pD451-SR and pD441-SR/CH (ATUM), respectively.
Protein expression and purification
For structural studies, the GP2 branch region was expressed in N-acetylglucosaminyltransferase I-deficient Expi293F GnTI- cells (ThermoFisher Scientific), transiently transfected with 25 kDa linear polyethylenimine (Polysciences) as described17,18. After capture from the conditioned medium by immobilized metal affinity chromatography (IMAC) and partial deglycosylation with Endo H19, recombinant GP2 was purified by size-exclusion chromatography (SEC) using a Superdex 75 Increase 10/300 GL column (GE Healthcare) and concentrated to 7 mg ml−1 in 20 mM Na-HEPES pH 7.5, 150 mM NaCl.
For evaluation of relative protein secretion levels and FimHL binding experiments, branch region constructs and mutants thereof were expressed in HEK293T cells20 grown in DMEM medium supplemented with 4 mM l-Gln, 10% FBS and transiently transfected in 4 mM l-Gln, 2% FBS using 25 kDa branched polyethylenimine (Sigma-Aldrich)19,21.
For in vitro reconstitution of the UMOD–FimHL complex, native human UMOD was purified from a healthy 49-year-old male donor using the diatomaceous earth method22. His-tagged FimHL A27V from UPEC strain UTI89 (ref. 23) was purified by immobilized metal affinity chromatography from the periplasmic extract of E. coli OverExpress C43(DE3) cells (Sigma-Aldrich) grown in mannose-free M9 minimal medium. The eluted protein, which was essentially pure by SDS–PAGE analysis, was then dialyzed against 20 mM Na-HEPES pH 7.5, 150 mM NaCl at 0.7 mg ml−1 concentration. Finally, purified UMOD and FimHL were mixed at a molar ratio of 1:3, incubated for 30 min and dialyzed against 10 mM Na-HEPES pH 7.0 (Extended Data Fig. 9).
For binding experiments, a crude periplasmic extract of E. coli OverExpress C43(DE3) expressing untagged FimHL was used (Extended Data Fig. 2a).
Protein analysis
Proteins separated by SDS–PAGE were detected with SimplyBlue SafeStain (Invitrogen/ThermoFisher Scientific) or transferred to nitrocellulose membranes (GE Healthcare) for immunoblotting with Penta•His BSA-free anti-5His mouse monoclonal (1:1,000; QIAGEN) and horseradish peroxidase-conjugated goat anti-mouse IgG Fc secondary antibody (1:10,000; Life Technologies/ThermoFisher Scientific). Chemiluminescence detection was performed with Western Lightning ECL Plus (PerkinElmer). Protein deglycosylation under denaturing conditions using either Endo H or Peptide:N-glycosidase F (New England Biolabs) was carried out for 1 h at 37 °C, according to the manufacturer’s instructions. Gradient gels (4%–12%) were used for SDS–PAGE, except for the deglycosylation experiment shown in Fig. 2c where a 12% gel was used to maximize the separation between bands.
Protein binding experiments
Purified C-terminally His-tagged UMOD, GP2 and GP2 N65A decoy module proteins in 20 mM Na-HEPES pH 7.5, 150 mM NaCl (binding buffer) were separately incubated with IMAC beads (GE Healthcare) for 1 h at room temperature. E. coli periplasmic extract containing untagged FimHL, adjusted to the binding buffer, was then added and the resulting mixtures were incubated for 2 h at room temperature or overnight at 4 °C. After washing the IMAC beads with binding buffer, bound material was eluted with 20 mM Na-HEPES pH 7.5, 150 mM NaCl, 500 mM imidazole and subjected to SEC as described above. Peak fractions were analyzed by SDS–PAGE, and control SEC runs of the same decoy modules by themselves or a His-tagged version of FimHL were used to determine the elution volumes of the unbound proteins.
Protein crystallization
Crystallization trials of the GP2 branch region, carried out by sitting drop vapor diffusion using a mosquito robot (TTP Labtech), initially yielded triclinic plates that grew in one week at 293K in 25% (v/v) ethylene glycol. After we determined the structure of this crystal form, we obtained two additional forms that also had plate-like morphology but grew at 277K: orthorhombic crystals in 20% (v/v) 1,5-pentanediol, 10% (w/v) PEG 8K, 0.1 M GlyGly/AMPD pH 8.5, 0.5 mM YCl3, 0.5 mM ErCl3, 0.5 mM TbCl3, 0.5 mM YbCl3 (condition E11 of the MORPHEUS II crystallization screen24 (Molecular Dimensions)); and monoclinic crystals in 5% (w/v) PEG 20K, 25% (w/v) 1,1,1-tris(hydroxymethyl) propane, 0.1 M MOPSO/bis-tris pH 6.5, 1% (w/v) NDSB-195, 0.01 M spermine, 0.01 M spermidine, 0.01 M 1,4-diaminobutane, 0.01 M dl-ornithine (MORPHEUS II condition H4). Before data collection at synchrotron, crystals were fished directly from the crystallization drops and flash frozen in liquid nitrogen.
X-ray data collection and reduction
Datasets for the P1, P212121 and C2 crystal forms were collected from single specimens at 100 K at European Synchrotron Radiation Facility beamlines ID23-1 (ref. 25) (λ = 1.0052 Å), ID30B26 (λ = 0.9763 Å) and ID30A-3 (λ = 0.9677 Å), respectively, using MXCuBE3 (ref. 27). All data was processed with XDS28 (Supplementary Table 1), with high-resolution data cutoffs chosen on the basis of statistical indicators CC1/2 and CC*29,30. Although the P1 crystals diffracted reproducibly to better than 3.0 Å resolution, a single specimen yielded data extending well beyond a Bragg spacing of 2.0 Å; unfortunately, probably because of the disorder, the diffraction extent of this particular crystal was severely underestimated by the data collection strategy software, so that we were only able to process the resulting data to 1.9 Å.
Experimental phasing attempts
Despite the workable resolution of its diffraction, the P1 crystal form suffered from disorder parallel to the b*c* planes, that is reflected by relatively high Rmerge and Rmeas values. Although this did not prevent us from ultimately solving the structure by molecular replacement (MR), it precluded multiple attempts to phase the data experimentally by sulfur-single wavelength anomalous dispersion. Parallel attempts to obtain usable derivative data from crystals soaked with Pt or Au compounds also failed, because of the apparent lack of specific binding sites for these heavy atoms. Similarly, no heavy atom bound to the C2 crystal form of the protein despite the fact that this was obtained in the presence of a mixture of different lanthanides and yttrium.
Structure solution by molecular replacement with AlphaFold2 models
AlphaFold2 (AlphaFold Monomer 2.0)11 was used to generate five independent models of residues V29–S181 of GP2α, with relative r.m.s. deviations (r.m.s.d.) of 0.6–1.7 Å. After removal of a low-confidence N-terminal region (residues V29–L44), visual inspection of the models suggested further trimming to residues D61–S181, which clearly belonged to a single globular domain (Extended Data Fig. 3a). The resulting coordinate sets (r.m.s.d. 0.1–0.2 Å), with per-residue pseudo-B factors corresponding to 100-(per-residue confidence (pLDDT11)), were combined into an ensemble that was used to phase the P1 data by MR with Phaser31. Using a search model r.m.s.d. variance of 1 Å, this found a single solution consisting of two molecules per asymmetric unit (LLG 1258, TFZ 31.6), whose correctness was readily confirmed by initial refinement (R 0.31, Rfree 0.36) and positive difference density for the N-acetylglucosamine (GlcNAc) residues attached to GP2 N65, N122 and N134 as well as part of the β-hairpin (Extended Data Fig. 3b,c). After one round of autobuilding in PHENIX32, the structure was completed by alternating manual rebuilding in Coot33 and ISOLDE34 with refinement using phenix.refine35. Protein geometry and carbohydrate structure validation was carried out with MolProbity36 and Privateer37, respectively, and data reduction, refinement and validation statistics calculated using phenix.table_one38 are reported in Supplementary Table 1. Because of a lack of density for the residues making up the loop of the β-hairpin, the final model consists of GP2 residues S41–G49 and H57–S181, as well as five GlcNAc residues attached to N65, N122 (chains A and B) and N134 (chain A only). Using these coordinates as a reference, the top ranked AlphaFold2 model had a Global Distance Test (GDT_TS) score of 94.9 (or 97.2 if only the D10C domain is considered).
An ensemble of the two chains of a partially refined model of the P1 structure was used to phase the P212121 data (with one molecule in the asymmetric unit) by MR (LLG 8167, TFZ 41.7; initial R 0.23, Rfree 0.25); residues D61–S181 of the refined P212121 model were in turn used for MR phasing of the C2 data (LLG 8539, TFZ 82.9; initial R 0.24, Rfree 0.25). As expected on the basis of the P1 MR results, both the orthorhombic and monoclinic structures could, in principle, also have been solved using the initial AlphaFold2 ensemble (P212121: LLG 1325, TFZ 33.5; initial R 0.32, Rfree 0.35; C2: LLG 1232, TFZ 31.9; initial R 0.32, Rfree 0.34). After rebuilding, refinement and validation as described for the P1 crystal form, the final P212121 and C2 models contain amino acids Y42–S181 and L44–S181, respectively, as well as two GlcNac residues attached to N65 and N122; in addition, the orthorhombic model includes two residues belonging to the C-terminal His-tag, whereas the monoclinic one contains the GlcNac attached to N134.
Cryo-EM data collection
Data collection and processing details for full-length native human UMOD have been reported6.
For collecting cryo-EM data from the UMOD–FimHL complex (Supplementary Table 3), prepared as described in the section ‘Protein expression and purification’, the specimen (1.8 mg ml−1) was applied in 3-µl volumes onto glow-discharged Cu R2/2 holey carbon 300 mesh grids (Quantifoil). After blotting for 2 s, grids were plunged into liquid ethane cooled by liquid nitrogen using a Vitrobot Mark IV (ThermoFisher Scientific). Cryo-EM experiments were performed at the Cryo-EM Swedish National Facility, SciLifeLab, Stockholm. Videos were collected using fringe-free imaging and aberration-free image shift with the EPU data acquisition software, on a Titan Krios electron microscope (ThermoFisher Scientific) operated at 300 kV, using a K3 camera equipped with a BioQuantum energy filter (Gatan-Ametek). Videos were taken at ×105,000 nominal magnification in counting mode with a dose rate of 15 e px−1 s−1 and a total dose of 40 e/Å2 distributed over 40 subframes, gain-corrected and then compressed using video compression in RELION39. Motion correction with dose weighting was also performed in RELION40 within the Scipion software suite41.
Cryo-EM data processing
Processing of the cryo-EM data of the UMOD–FimHL complex followed the general workflow used for reconstructing the full-length UMOD filament6. First, contrast transfer function determination was carried out using CTFFIND in RELION. An in-house script designed specifically for filament picking (Cryo-EM-filament-picker)42 was then used to select end-to-end filament coordinates. After two-dimensional classification in cryoSPARC43, selected particle coordinates were transferred back to RELION for three-dimensional (3D) classification, 3D helical refinement, particle subtraction and final non-helical refinement and polishing. Specifically, starting from a total of 13,616 raw micrographs, 3,767,790 particles (helical segments with 70 Å step size) were auto-picked and extracted on the basis of motion correction and contrast transfer function estimation; based on two-dimensional classification quality evaluated with cryoSPARC, a subset of 1,139,808 particles was then selected for further processing. Because FimHL occupancy varied among filaments, segments with higher FimHL occupancy were selected during iterative RELION 3D classification runs. Finally, 225,819 homogeneous particles were subjected to auto-refinement and postprocessing. To improve the local density of the FimHL-binding region, we performed particle subtraction to mask out the UMOD helical core and continued local refinement in RELION. Ultimately, a density representing the UMOD branch–FimHL complex with an overall average resolution of 7.4 Å was obtained by auto-refining the subtracted particles with a UCSF Chimera44-generated mask that only covered the binding region (Extended Data Fig. 9 and Supplementary Table 3).
Cryo-EM map fitting, model refinement and validation
A complete atomic model of full-length UMOD was assembled in several steps. First, five independent models of the whole UMOD branch (residues D25–S191) were generated with AlphaFold2; all these models shared the same domain boundaries, fold and disulfide connectivity, with their overall r.m.s.d. (0.4–4.3 Å) simply reflecting differences in the orientation of EGF I–III (r.m.s.d. 0.2–0.4 Å) relative to the decoy module (r.m.s.d. 0.1–0.2 Å). Second, although the overall r.m.s.d. values between the AlphaFold2 models of the GP2 D10C domain and the corresponding experimental structures (average ~0.5 Å) were not much larger than those between the latter (average 0.1 Å), local differences could be observed at the level of the relatively flexible 310B/βB loop as well as a subset of side chains. To consider these alternatives while fitting the cryo-EM density of the UMOD D10C domain (62% sequence identical to that of GP2), the P212121 and C2 high-resolution structures of GP2 D10C were each used to generate five homology models of UMOD D10C using MODELLER45. The respective models with the best Discrete Optimized Protein Energy (DOPE) scores46 were then used as starting points for exploring different possible conformations by molecular dynamics in YASARA Structure47. Third, the top AlphaFold2 model and P212121/C2-structure derived homology models (r.m.s.d. 0.7/0.8 Å) of D10C were individually rigidly docked with UCSF Chimera into the 3D reconstruction of full-length UMOD (overall nominal resolution 4.7 Å)6, whose masking and postprocessing with RELION was optimized to obtain the best possible density for the D10C-containing region near the center of the map. The resulting map fit correlations of the AlphaFold2 model and the homology models were 0.884 and 0.892/0.896, respectively. Fourth, the placed AlphaFold2 model was locally rebuilt, taking into account—if available—alternative possibilities suggested by the superimposed homology models. At this stage, we also connected the C terminus of D10C to the N terminus of the atomic model of the UMOD filament core (PDB ID 6TQK)6, consisting of the EGF IV domain and the ZP module (Extended Data Fig. 1a); rebuilt the C-terminal end of the ZP-C domain interacting with D10C6; and built the glycan chains attached to N232 and N275. The resulting coordinates were then subjected to global real-space and group ADP refinement in PHENIX48, essentially as described6 (CCmask 0.74; CCbox 0.79; CCpeaks 0.39; CCvol 0.72; mean CCcarbohydrates 0.62). Finally, the model was completed by fusing it with EGF I–III/β-hairpin coordinates extracted from the top AlphaFold2 model of the whole UMOD branch, flexibly fit into a cryo-EM map of the same protein region (overall nominal resolution 6.1 Å)6 using Namdinator49 (CCmask 0.59; CCbox 0.76; CCpeaks 0.43; CCvol 0.56; mean CCcarbohydrates 0.60). Following further rebuilding and real-space refinement against a composite map of full-length UMOD generated by multibody refinement6 (Extended Data Fig. 6), performed using the starting model as a reference for generating torsion restraints, protein geometry and carbohydrate structure were validated using PHENIX50/MolProbity (Supplementary Table 3) and Privateer; model-to-map validation was carried out with PHENIX (CCmask 0.75; CCbox 0.81; CCpeaks 0.48; CCvol 0.73; mean CCcarbohydrates 0.77). The final model consists of 1,127 protein residues, corresponding to a complete chain (chain A, D25–F587) and two half chains (chain B, S444–F587; chain C, D25–S444) that together recapitulate all the protein-protein interactions in the UMOD filament, as well as 84 N-glycan residues.
The model of the UMOD branch + EGF IV/FimHL complex was generated by manually docking the crystallographic structure of FimHL bound to trimannose (chains A and F of PDB ID 6GTW)51 into the difference density between the cryo-EM maps of the FimH-bound and free UMOD branch + EGF IV (calculated using TEMPy:DiffMap52 and masked around the decoy module region), so that the lectin made an equivalent interaction with the α1,3 branch of the high-mannose glycan attached to UMOD N275. After optimizing the position of FimHL against the difference map by rigid-body refinement, introducing A27V, S62A substitutions to match the sequence of FimH from UPEC UTI89 variant A27V and rebuilding the other residues of the N275 glycan, the whole complex was finally subjected to real-space refinement with restraints generated using the starting coordinates as a reference (Supplementary Table 3).
Sequence-structure analysis
Structure-based sequence alignments, generated using MAFFT53 as implemented in ConSurf54, were rendered with ESPript55. For calculating consensus information at different thresholds, a ConSurf alignment that sampled homologs of the GP2 branch domain with 35–95% identities was first pruned of incomplete sequences (yielding a final set of 129 aligned sequences) and then processed with MView56.
GDT_TS scores were calculated using the AS2TS server57 and possible structural similarities were assessed using Dali58. Secondary structure was assigned using STRIDE59; structural figures were generated with PyMOL (Schrödinger, LLC) and UCSF Chimera/ChimeraX60.
Site specific N-glycosylation analysis by liquid chromatography–tandem mass spectrometry
The His-tagged GP2 branch region purified from the conditioned medium of HEK293T cells was denatured, reduced and alkylated before digestion with either sequencing-grade AspN or with pepsin/chymotrypsin. The digests were analyzed on an Ultimate 3000 nanoLC system online coupled to a QExactive mass spectrometer (ThermoFisher Scientific). Raw data was analyzed by ByonicTM (Protein Metrics Inc.) set to identify glycopeptides from the fragmented parent ion. The acceptance criterion was a false discovery rate on the protein level below 1%. Peptide and glycan sequences were analyzed by ByonicTM from the higher-energy C-trap dissociation (HCD) spectra and verified manually.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The UniProt (https://www.uniprot.org/) IDs for hGP2 and hUMOD are P55259 and P07911, respectively; the IDs of other sequences reported in the alignment of Extended Data Fig. 1b are Q9D733 (mGP2), Q91X17 (mUMOD), Q8WWZ8 (hLZP), Q8R4V5 (mLZP), Q8N2E2 (hVWDE) and Q6DFV8 (mVWDE). The Electron Microscopy Data Bank (EMDB; https://www.ebi.ac.uk/emdb/) ID of the UMOD filament map used for assembling the composite map shown in this work is EMD-10553; the UMOD filament core and FimHL/trimannose coordinates used as starting models can be retrieved from the Protein Data Bank (PDB; https://www.rcsb.org/) with IDs 6TQK and 6GTW, respectively. Structure factors and atomic models for the P1, P212121 and C2 crystal forms of the GP2 decoy domain have been deposited in the PDB with accession codes 7P6R, 7P6S and 7P6T, respectively. Cryo-EM density maps of full-length UMOD and the UMOD branch + EGF IV/FimHL complex have been deposited in the EMDB with accession codes EMD-13378 and EMD-13794, respectively; the corresponding coordinates have been deposited in the PDB with accession codes 7PFP and 7Q3N. Source data are provided with this paper.
Code availability
The Python code for filament picking is available at: https://doi.org/10.5281/zenodo.5807535.
References
Kobayashi, K., Yanagihara, K., Ishiguro, K. & Fukuoka, S. GP2/THP gene family of self-binding, GPI-anchored proteins forms a cluster at chromosome 7F1 region in mouse genome. Biochem. Biophys. Res. Commun. 322, 659–664 (2004).
Pak, J., Pu, Y., Zhang, Z. T., Hasty, D. L. & Wu, X. R. Tamm-Horsfall protein binds to type 1 fimbriated Escherichia coli and prevents E. coli from binding to uroplakin Ia and Ib receptors. J. Biol. Chem. 276, 9924–9930 (2001).
Hase, K. et al. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune response. Nature 462, 226–230 (2009).
Devuyst, O., Olinger, E. & Rampoldi, L. Uromodulin: from physiology to rare and complex kidney disorders. Nat. Rev. Nephrol. 13, 525–544 (2017).
Kurashima, Y. et al. Pancreatic glycoprotein 2 is a first line of defense for mucosal protection in intestinal inflammation. Nat. Commun. 12, 1067 (2021).
Stsiapanava, A. et al. Cryo-EM structure of native human uromodulin, a zona pellucida module polymer. EMBO J. 39, e106807 (2020).
Stanisich, J. J. et al. The cryo-EM structure of the human uromodulin filament core reveals a unique assembly mechanism. eLife 9, e60265 (2020).
Weiss, G. L. et al. Architecture and function of human uromodulin filaments in urinary tract infections. Science 369, 1005–1010 (2020).
Yang, H., Wu, C., Zhao, S. & Guo, J. Identification and characterization of D8C, a novel domain present in liver-specific LZP, uromodulin and glycoprotein 2, mutated in familial juvenile hyperuricaemic nephropathy. FEBS Lett. 578, 236–238 (2004).
Yu, S. & Lowe, A. W. The pancreatic zymogen granule membrane protein, GP2, binds Escherichia coli Type 1 fimbriae. BMC Gastroenterol. 9, 58 (2009).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
van Rooijen, J. J., Voskamp, A. F., Kamerling, J. P. & Vliegenthart, J. F. Glycosylation sites and site-specific glycosylation in human Tamm-Horsfall glycoprotein. Glycobiology 9, 21–30 (1999).
Danzer, C. et al. Comprehensive description of the N-glycoproteome of mouse pancreatic β-cells and human islets. J. Proteome Res. 11, 1598–1608 (2012).
Huang, Z. Q. & Sanders, P. W. Localization of a single binding site for immunoglobulin light chains on human Tamm-Horsfall glycoprotein. J. Clin. Invest. 99, 732–736 (1997).
Armenteros, J. J. A. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Raj, I. et al. Structural basis of egg coat–sperm recognition at fertilization. Cell 169, 1315–1326.e17 (2017).
Briggs, D. C. & Hohenester, E. Structural basis for the initiation of glycosaminoglycan biosynthesis by human xylosyltransferase 1. Structure 26, 801–809.e3 (2018).
Pulido, D., Hussain, S.-A. & Hohenester, E. Crystal structure of the heterotrimeric integrin-binding region of laminin-111. Structure 25, 530–535 (2017).
Bokhove, M. et al. Easy mammalian expression and crystallography of maltose-binding protein-fused human proteins. J. Struct. Biol. 194, 1–7 (2016).
DuBridge, R. B. et al. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7, 379–387 (1987).
Aricescu, A. R., Lu, W. & Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 (2006).
Serafini-Cessi, F., Bellabarba, G., Malagolini, N. & Dall’Olio, F. Rapid isolation of Tamm-Horsfall glycoprotein (uromodulin) from human urine. J. Immunol. Methods 120, 185–189 (1989).
Kalas, V. et al. Evolutionary fine-tuning of conformational ensembles in FimH during host–pathogen interactions. Sci. Adv. 3, e1601944 (2017).
Gorrec, F. The MORPHEUS II protein crystallization screen. Acta Crystallogr. F Struct. Biol. Commun. 71, 831–837 (2015).
Nurizzo, D. et al. The ID23-1 structural biology beamline at the ESRF. J. Synchrotron Radiat. 13, 227–238 (2006).
McCarthy, A. A. et al. ID30B – a versatile beamline for macromolecular crystallography experiments at the ESRF. J. Synchrotron Radiat. 25, 1249–1260 (2018).
Oscarsson, M. et al. MXCuBE2: the dawn of MXCuBE collaboration. J. Synchrotron Radiat. 26, 393–405 (2019).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Karplus, P. A. & Diederichs, K. Assessing and maximizing data quality in macromolecular crystallography. Curr. Opin. Struct. Biol. 34, 60–68 (2015).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Terwilliger, T. C. et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 (2008).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Agirre, J. et al. Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 (2015).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Sharov, G., Morado, D. R., Carroni, M. & de la Rosa-Trevín, J. M. Using RELION software within the Scipion framework. Acta Crystallogr. D Struct. Biol. 77, 403–410 (2021).
Xu, C. Cryo-EM-filament-picker. Zenodo https://doi.org/10.5281/zenodo.5807535 (2021).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 86, 2.9.1–2.9.37 (2016).
Shen, M.-Y. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006).
Krieger, E. et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77, 114–122 (2009).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Kidmose, R. T. et al. Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019).
Afonine, P. V. et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 74, 814–840 (2018).
Sauer, M. M. et al. Binding of the bacterial adhesin FimH to its natural, multivalent high-mannose type glycan targets. J. Am. Chem. Soc. 141, 936–944 (2019).
Joseph, A. P. et al. Comparing cryo-EM reconstructions and validating atomic model fit using difference maps. J. Chem. Inf. Model. 60, 2552–2560 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Brown, N. P., Leroy, C. & Sander, C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14, 380–381 (1998).
Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 31, 3370–3374 (2003).
Holm, L. Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42 (2020).
Frishman, D. & Argos, P. Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 (1995).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Fukuoka, S. Molecular cloning and sequences of cDNAs encoding α (large) and β (small) isoforms of human pancreatic zymogen granule membrane-associated protein GP2. Biochim. Biophys. Acta 1491, 376–380 (2000).
Jovine, L., Qi, H., Williams, Z., Litscher, E. & Wassarman, P. M. The ZP domain is a conserved module for polymerization of extracellular proteins. Nat. Cell Biol. 4, 457–461 (2002).
Shen, H.-L. et al. Liver-specific ZP domain-containing protein (LZP) as a new partner of Tamm-Horsfall protein harbors on renal tubules. Mol. Cell. Biochem. 321, 73–83 (2009).
Leigh, N. D. et al. von Willebrand factor D and EGF domains is an evolutionarily conserved and required feature of blastemas capable of multitissue appendage regeneration. Evol. Dev. 22, 297–311 (2020).
Fritz, B. A., Poppel, C. S., Fei, M. W. & Lowe, A. W. Processing of the major pancreatic zymogen granule membrane protein, GP2. Pancreas 24, 336–343 (2002).
Acknowledgements
We thank D. Briggs (The Francis Crick Institute, London) for advice on transient transfection of Expi293F cells; A. Vegvari (Karolinska Institutet Proteomics Biomedicum core facility) for the MS analysis of the FimHL bands; the Plateforme d’Analyses des Glycoconjugués (PAGés) and the Plateforme d’Analyse Protéomique et de Protéines Modifiés (P3M) for GP2 N65 glycan LC-MS/MS; the staff of the European Synchrotron Radiation Facility (ESRF; Grenoble) and the Swedish National Cryo-EM Facility (Stockholm) for help with X-ray and cryo-electron microscopy data collection and preprocessing; A. Zemla (Lawrence Livermore National Laboratory, Livermore) for help with GDT_TS calculations; and T. Terwilliger (New Mexico Consortium, Los Alamos) for discussion. This work was supported by the Swedish Research Council (project grants 2016-03999 and 2020-04936 to L.J.), the Karolinska Institutet Research Foundation (grant 2016fobi50035 to L.J.), the Knut and Alice Wallenberg Foundation (project grant 2018.0042 to L.J.) and the Ministry of Health, Singapore, NMRC grant (MOH-000382-00 to B.W.).
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
A.S., S.N. and L.H. expressed and purified proteins. A.S. and S.N. carried out protein-protein interaction experiments. A.S., L.J. and D.d.S. performed crystallographic research. K.T. and J.J. generated AlphaFold2 models. C.X., B.W., L.J., M.C. and A.S. performed cryo-EM research. N.Y. analyzed protein glycosylation by mass spectrometry. L.J. coordinated the study and wrote the manuscript with A.S., based on input from all other coauthors.
Corresponding author
Ethics declarations
Competing interests
J.J. has filed provisional patent applications relating to machine learning for predicting protein structures. The other authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Beth Moorefield was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Structure of the GP2 N-terminal branch and its relation with the corresponding regions of UMOD and additional mammalian proteins.
a, Domain architecture of mature human GP2 and UMOD. Domains are indicated by their acronyms, except for UMOD epidermal growth factor (EGF) domains that are labeled according to their roman number, the single EGF domain of GP2 (corresponding to UMOD EGF IV) that is labeled as ‘E’ and the β-hairpin of the decoy module (‘β’). The UMOD D10C epitope recognized by Bence-Jones proteins (BJP)14 is shown as a green stripe. Black and magenta inverted tripods indicate the N-glycosylation sites of GP2 and UMOD, respectively, with the high-mannose chains attached to GP2 N65 (this study) and UMOD N2758,12 colored cyan. The position corresponding to the alternative 3’ splice site generating the β isoform of GP2 (T178 | D179)61 and the elastase cleavage site of UMOD (S291 | S292)62 are indicated by vertical blue and orange arrows, respectively. b, Alignment of D10C domain sequences from human (h) and murine (m) homologues of GP2 and UMOD, as well as liver-specific zona pellucida protein (LZP/OIT3, a molecule that can also interact with UMOD in the kidney and urine63) and von Willebrand factor D and EGF domain-containing protein (VWDE; a protein involved in appendage regeneration in a variety of vertebrate species64). Identical residues are highlighted in white and shaded in red; conserved residues are red and marked by blue frames when clustered. Consensuses at different sequence identity thresholds, based on a comprehensive alignment of homologous sequences, are also reported (bold uppercase characters: amino acids with the same one-letter code; regular lowercase characters: l, [I,V,L]; h, [F,Y,W,H,I,V,L]; + , [H,K,R]; -, [D,E]; p, [Q,N,S,T,C,H,K,R,D,E]; u, [G,A,S]; s, [G,A,S,V,T,D,N,P,C]; t, [G,A,S,Q,N,S,T,C,H,K,R, D,E]; (.), any amino acid). GP2 secondary structure elements, rainbow-colored from blue (N-terminus) to red (C-terminus), and disulfide bond connectivity are shown above and below the alignment, respectively. Other elements are labeled as in (a), with a green box indicating the BJP epitope14. Black bold numbers above the alignment indicate hGP2 residues; light grey numbers between parentheses refer to the corresponding hUMOD residues. c, Cartoon representation of the GP2 decoy module, rainbow-colored following the same scheme used for the secondary structure elements of (b). Disulfide bonds are represented as grey sticks. d, Topology and disulfide connectivity diagram of the decoy module.
Extended Data Fig. 2 The isolated GP2 branch and the corresponding decoy module of UMOD bind FimHL.
a, For assessing whether the lectin domain of FimH is able to bind in vitro to the branch of GP2 or the equivalent region of UMOD (corresponding to the respective decoy modules, see main text), untagged FimHL was expressed in E. coli and a crude periplasmic extract was prepared. n = 2. b, SEC analysis of the material eluted after incubating purified His-tagged GP2 or UMOD decoy modules bound to IMAC beads with the FimHL-containing E. coli periplasmic extract (magenta curves). In both cases, reducing SDS-PAGE of peak fractions and tandem mass spectrometry (MS/MS) of the corresponding ~15 kDa bands show the presence of complexes between the decoy modules and the bacterial adhesin, indicating that the former are able to selectively recognize the latter among the pool of periplasmic proteins. SEC elution profiles of the GP2 and UMOD decoy domains by themselves are also shown (light blue curves), and a low-molecular weight contaminant peak is indicated by *. GP2 decoy module, UMOD decoy module: n = 3; GP2 decoy module/FimHL, UMOD decoy module/FimHL, n = 2. c, Control SEC profile of unbound His-tagged FimHL with SDS-PAGE analysis of the peak. § indicates minor high-molecular weight contaminants eluting with or close to the void volume. n = 3.
Extended Data Fig. 3 AlphaFold2 model phasing of the GP2 branch P1 X-ray data.
a, Superposition of five AlphaFold2 models of the GP2 N-terminal branch indicates the presence of three distinct units, with a central hairpin-like region (residues D45-F60; orange box) separating an N-terminal low-confidence region (residues V29-L44; red box) from a C-terminal globular domain (residues D61-S181; green box). An ensemble corresponding to the latter was used as search model for MR. b-c, Electron density for an Endo H cleavage-derived N-acetylglucosamine residue attached to N122 (b) and the hairpin region (c), two GP2 elements not included in the MR search ensemble. Fourier maps at different stages of the structure determination process are shown, contoured at the indicated levels.
Extended Data Fig. 4 Comparison of the predicted and experimental structures of the human GP2 branch region.
The crystallographic models, shown as semi-transparent cartoons colored in black (P1), grey (P212121) and white (C2), are superimposed on the top AlphaFold2 model, colored from blue to red according to a 100-(per-residue confidence (pLDDT11)) scale that ranges from 0 (blue; maximum confidence) to 100 (red; minimum confidence). Note how the low-confidence prediction for the N-terminal region of the GP2 branch matches the observations that the corresponding residues are largely structurally disordered in the different crystal forms of the protein (whose first resolved residues, S41/Y42 (P1 chains A/B), Y42 (P212121) or L44 (C2) are indicated) and apparently proteolytically removed from mature native GP265. Similarly, two protein regions that display relative structural flexibility in the GP2 crystals, the β-hairpin and part of the long loop connecting 310 helix B to conserved Cys 2 (white box), contain residues predicted with lower confidence by AlphaFold2.
Extended Data Fig. 5 Pathogenic substitutions in the D10C domain affect clusters of highly conserved residues.
a-b, GP2 D10C residues corresponding to UMOD amino acids mutated in kidney disease patients (panel a, red) are largely clustered into two highly conserved protein regions (panel b). Sequence conservation is represented using a color spectrum ranging from green (lowest conservation) to violet (highest conservation). c-g, Alternative representation of the structural details shown in Fig. 1c–g, with residues colored by sequence conservation.
Extended Data Fig. 6 Assembly of the composite map of full-length UMOD.
Multi-body refinement of the UMOD filament core plus D10C domain (left path) and the whole UMOD branch (right path) were performed separately. Helical symmetry was applied to the filament core plus D10C, after the best homogenous filamentous segments were selected based on 2D classes. Meanwhile, the particles with the better contrast, more extended branch features were independently selected, locally 3D classified and refined, without helical symmetry. The final composite map was assembled by merging copies of the branch with the filament core plus D10C.
Extended Data Fig. 7 Inactivation of the N65 glycosylation site of GP2 impairs the interaction between the protein’s decoy module and FimHL.
a, The FimH-binding high-mannose glycan attached to UMOD N275 is located in the groove between the β-hairpin and D10C domain moieties of the protein’s decoy module (left panel). Although this sequon is not conserved in the decoy module of GP2, the groove of the latter contains a different, but closely spaced, N-glycosylation site at position 65 (right panel). b, SEC analysis of the material eluted after incubating an E. coli periplasmic extract containing untagged FimHL with wild-type or N65A mutant GP2 decoy modules immobilized on IMAC beads (left panels). Reducing SDS-PAGE analysis of the corresponding peak fractions (right panels) shows that FimHL binds to the wild-type GP2 decoy module but not to the N65A mutant. n = 2.
Extended Data Fig. 8 Mass spectrometric analysis of GP2 glycopeptides detects the oligomannose-5 structure attached to N65.
Supporting MS2 spectrum of precursor m/z 1170.46, 61DPCQNYTLL69, carrying oligomannose-5 (HexNAc2Hex5). Prepared by Asp-N digestion of the GP2 branch purified from HEK293T cells. N-glycan structures are depicted following the Consortium for Functional Glycomics (CFG) notation: HexNAc, N-acetylglucosamine (blue square); Hex, mannose (green circle). The cysteine residue is carbamidomethylated. Detected peptide-backbone fragment ions are presented in the peptide sequence. Interestingly, complex-type carbohydrate structures were also found to be attached to N65. This is consistent with the observation that, although UMOD N275 and GP2 N65 are both located in the groove between the β-hairpin and the D10C domain of the respective decoy modules, N65 is relatively more exposed than N275 in the structure (Extended Data Fig. 7a), making the N65 glycan chains more susceptible to modification.
Extended Data Fig. 9 3D reconstruction of the UMOD branch/FimHL complex.
Identification, isolation and local refinement of a single UMOD branch unit bound to one copy of FimHL. After incubation with an excess concentration of FimHL, UMOD filaments were subjected to cryo-EM analysis. Following filament autopicking by an in-house script, highly heterogenous filament segments were sorted by performing cryoSPARC 2D class runs, after binning. Segment coordinates from good 2D classes were then extracted and re-imported into RELION. After iterative 3D classification with and without applying helical symmetry, the segments with higher FimHL occupancy were selected and grouped into different sub-classes. Segments representing a single branch unit of the best UMOD/FimHL sub-class were extracted and used for 3D reconstruction of the density of UMOD bound to FimHL. In the bottom left panel, the extra density of FimHL in the UMOD branch/FimHL complex could be identified in the 2D class images. Red arrows point to the location of FimHL.
Supplementary information
Source data
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Unprocessed gel.
Source Data Extended Data Fig. 2
Unprocessed gels.
Source Data Extended Data Fig. 7
Unprocessed gels.
Source Data Extended Data Fig. 9
Unprocessed gel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stsiapanava, A., Xu, C., Nishio, S. et al. Structure of the decoy module of human glycoprotein 2 and uromodulin and its interaction with bacterial adhesin FimH. Nat Struct Mol Biol 29, 190–193 (2022). https://doi.org/10.1038/s41594-022-00729-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-022-00729-3
This article is cited by
-
Advances in uromodulin biology and potential clinical applications
Nature Reviews Nephrology (2024)
-
Machine learning in preclinical drug discovery
Nature Chemical Biology (2024)
-
Glycoprotein 2 as a gut gate keeper for mucosal equilibrium between inflammation and immunity
Seminars in Immunopathology (2024)
-
AlphaFold2 and its applications in the fields of biology and medicine
Signal Transduction and Targeted Therapy (2023)
