
Abstract
Transcriptionally silenced heterochromatin bearing methylation of histone H3 on lysine 9 (H3K9me) is critical for maintaining organismal viability and tissue integrity. Here we show that in addition to ensuring H3K9me, MET-2, the Caenorhabditis elegans homolog of the SETDB1 histone methyltransferase, has a noncatalytic function that contributes to gene repression. Subnuclear foci of MET-2 coincide with H3K9me deposition, yet these foci also form when MET-2 is catalytically deficient and H3K9me is compromised. Whereas met-2 deletion triggers a loss of silencing and increased histone acetylation, foci of catalytically deficient MET-2 maintain silencing of a subset of genes, blocking acetylation on H3K9 and H3K27. In normal development, this noncatalytic MET-2 activity helps to maintain fertility. Under heat stress MET-2 foci disperse, coinciding with increased acetylation and transcriptional derepression. Our study suggests that the noncatalytic, focus-forming function of this SETDB1-like protein and its intrinsically disordered cofactor LIN-65 is physiologically relevant.
Similar content being viewed by others


Main
The proper segregation of active and inactive regions of the genome during differentiation is critical for the establishment and maintenance of tissue and genome integrity in eukaryotes. Heterochromatic regions of the genome are condensed and sequestered at the nuclear periphery or around nucleoli and are typically not transcribed1,2. Constitutive heterochromatin is associated with H3K9me, which is necessary for the repression of satellite repeats and transposable elements3. Tissue-specific genes can also be repressed by H3K9me (refs. 4,5,6). Finally, H3K9me mediates chromatin anchoring at the nuclear periphery7 and helps prevent interaction between euchromatic and heterochromatic compartments8,9. In mammals, the loss of H3K9me is associated with aging, cancer and the impaired maintenance of tissue integrity10,11,12,13.
The enzymes that establish and maintain heterochromatin form subnuclear clusters or foci in many species14,15,16,17,18,19. In budding yeast, a function has been attributed to these stable perinuclear foci containing the Silent Information Regulator complex: the clustering promotes histone deacetylation and contributes to the stability and spread of repressed chromatin19. In mammals, the H3K9 histone methyltransferases (HMTs) SUV39H and SETDB1 remain bound to clusters of heterochromatin throughout the cell cycle20,21. In the case of SUV39H, this localized accumulation of the HMT requires an N-terminal chromodomain, which recognizes H3K9me2/me3 and thereby sequesters the enzyme at its chromatin targets21. To date, however, no function was assigned to the localized retention of H3K9 HMTs.
Recently, heterochromatic focus formation has been proposed to depend on disordered protein domains and multivalent interactions that promote local phase separation, as exemplified by the H3K9me reader HP1 protein22,23. In Caenorhabditis elegans, however, foci of MET-2, the SETDB1 homolog responsible for H3K9me deposition, and its disordered domain cofactor LIN-65, remain stable despite downregulation of the worm HP1 homologs14. Moreover, MET-2, unlike SUV39H, has no chromodomain. We therefore set out to examine what drives the focal clustering of heterochromatin proteins such as MET-2, and the relationship of such foci to histone H3K9me.
Although several C. elegans proteins have been implicated in methylation of H3K9 (refs. 24,25,26,27), only the double deletion of genes encoding two SET domain-containing proteins, MET-2 and SET-25, eliminates all detectable H3K9me, both in embryos6,7,28 and at later stages of somatic development6,7,29. The loss of H3K9me compromises the transcriptional repression of tissue-specific genes and repetitive elements6,30, although no other common histone methylation marks were altered upon met-2 and set-25 ablation7. The SET domain of MET-2 has 50% identity to that of SETDB1 (refs. 18,28,31,32), which has been shown to methylate histone H3K9 both in vivo33 and in vitro34,35. Moreover, MET-2 harbors a domain organization very similar to SETDB1, including a methyl CpG binding domain, Pre-SET domain and bifurcated SET domain at the C terminus (Fig. 1a). Nonetheless, the demonstration of HMT activity for recombinant MET-2 has been hindered because the purification of this large, partially unstructured protein with its cofactor, LIN-65, has proven difficult. We will therefore refer to MET-2 as a putative H3K9 HMT.

a, Domain layout of LIN-65, MET-2 and ARLE-14 proteins adapted from SMART(EMBL). IDR, intrinsically disordered region; CC, coiled-coil; CysR, cysteine-rich. b, Representative volcano plot from WT embryos and iBAQ (intensity-based absolute quantification) values for each indicated protein from immunoprecipitation–MS/MS using MET-2::FLAG::mCherry as bait. Bars indicate the mean and dots represent individual measurements. c, Representative live images of MET-2::mCherry in WT, arle-14Δ(tm6845) or lin-65Δ embryos and quantification of MET-2 foci. Scale bar, 5 µm. N = 3, n = 30. Nuclei: WT = 840, arle-14Δ = 961. d, Correlation between met-2Δ and arle-14Δ FC in gene expression (log2FC) compared with WT. Colors marking significantly changed genes (FDR < 0.001; log2FC > 1): met-2, dark red; arle-14, turquoise; common genes, black. Pearson correlation coefficient (r) is displayed. e, Venn diagram showing overlap of lin-65Δ-, arle-14Δ- and met-2Δ-derepressed genes. lin-65Δ data from ref. 14. f, Representative western blot and quantification of H3K9me2 normalized to histone H2B in WT, lin-65;arle-14 embryos and sibling single mutants. N = 4. Image (*) marks residual H3K9me2 signal. P(met-2Δ, lin-65Δ) = 2.4 × 10−3, P(met-2Δ, arle-14Δ) = 9.0 × 10−3, P(lin-65Δ, lin-65Δ;arle-14Δ) = 2.6 × 10−3, P(arle-14Δ, lin-65Δ;arle-14Δ) = 9.0 × 10−3. g, Progeny number per worm at 25 °C in WT, met-2Δ, lin-65Δ, arle-14Δ and arle-14Δ set-25Δ(n5021). N = 3, n = 60. Independent experiments comparing progeny number between WT and set-25Δ are included as a reference. N = 2, n = 40. P(WT, met-2Δ) = 6.7 × 10−13, P(WT, arle-14Δ) = 7.3 × 10−13, P(met-2Δ, lin-65Δ) = 0.998, P(met-2Δ, arle-14Δ) = 6.7 × 10−13, P(lin-65Δ, arle-14Δ) = 6.7 × 10−13, P(arle-14Δ, arle-14Δ set-25Δ) = 0.0002 by one-way analysis of variance (ANOVA), followed by Tukey post hoc test. ***P < 0.0001, ****P < 0.00001; a.u., arbitrary units; IP, immunoprecipitation.
In cells, MET-2 and its two cofactors, the intrinsically disordered protein LIN-65 (refs. 14,18), and the poorly characterized but conserved ARLE-14, form foci that coincide with H3K9 methylation14,18. Not only the MET-2 partners, but also their regulatory interactions, are conserved in flies (Windei and CG14464 bind dSETDB1)36,37 and mammals (ATF7IP and ARLE14EP bind SETDB1)38,39,40. Ablation of lin-65 (or atf7ip) leads to a loss of MET-2 (or SETDB1) in nuclei, respectively14,18,41, and in worms, lin-65 deletion compromises the formation of MET-2 foci and derepresses a large subset of MET-2-sensitive targets14,18. Consistent with the notion that the unstructured domains present in LIN-65 contribute to MET-2 focus formation, these foci are sensitive to interventions that disrupt phase-separated condensates, such as treatment with 1,6-hexanediol and heat14.
Here we examined the mechanisms of MET-2-mediated heterochromatin silencing and its impact on development and fertility. Unexpectedly, we discovered a function for MET-2 protein that operates alongside its role in H3K9 methylation and correlates with focus formation. Restoring foci with a catalytic-deficient MET-2 maintained low acetylation levels on heterochromatin and rescued somatic developmental rates, mitigating inappropriate transcription and infertility. This was achieved in an ARLE-14-dependent manner. We argue that MET-2 promotes gene repression through both catalytic and noncatalytic functions, the latter coinciding with heterochromatic MET-2 focus formation.
Results
To understand how MET-2 is regulated, we examined in greater detail its interaction with ARLE-14 and LIN-65 (refs. 14,18). LIN-65, similar to its homolog ATF7IP, is composed of two very large regions of disorder surrounding a coiled-coil domain (Fig. 1a). Reports in flies and mammals have shown this coiled-coil domain binds the N terminus of SETDB1 (refs. 16,42). ARL14EP, the mammalian homolog of ARLE-14, requires its conserved cysteine-rich C-terminal domain to bind SETDB1 (ref. 43). We examined the interdependence of MET-2 complex formation using mutants and mass spectrometry. In wild-type (WT, N2) embryos expressing MET-2::FLAG::mCherry, only ARLE-14 and LIN-65 were highly enriched in the MET-2 pulldown (Fig. 1b)14,18. Neither the ablation of lin-65 (lin-65Δ(gw1465)) nor of arle-14 (arle-14Δ(tm6845)) affected binding of the other cofactor to MET-2. We conclude that ARLE-14 and LIN-65 interact independently with MET-2.
Loss of lin-65 impairs both MET-2 nuclear accumulation and concentration into subnuclear foci (Fig. 1c)41, and thus largely phenocopies met-2 deletion14,18. To distinguish roles of nuclear accumulation from focus formation, we introduced a nuclear localization signal (NLS) to the endogenously tagged met-2::mCherry locus by CRISPR (NLS::met-2). The NLS::MET-2::mCherry nucleoplasmic signal was stronger than the WT MET-2::mCherry that traffics between the cytosol and nucleus14,18, but in both cases foci were clearly distinguishable (Extended Data Fig. 1a). Whereas the ablation of lin-65 eliminated the nuclear enrichment of WT MET-2::mCherry, the NLS::MET-2 fusion protein remained nuclear but failed to form foci in the absence of LIN-65. Western blotting confirmed that H3K9me2 levels were not altered by the NLS tag on MET-2 as long as LIN-65 was present, and dropped similarly upon deletion of lin-65 (Extended Data Fig. 1b). We conclude that nuclear accumulation of MET-2 is not sufficient for H3K9 HMT activity and focus formation, and that LIN-65 promotes both.
In contrast to LIN-65, ARLE-14 is not required for general silencing of repetitive elements14,18, and its loss compromised neither the nuclear import of MET-2 nor the number of foci formed (Fig. 1c). Nevertheless, embryos carrying either arle-14Δ or two other independent arle-14 mutant alleles lost a majority of H3K9me2, as shown by quantitative fluorescence and western blots (Extended Data Fig. 1c,d). The impacts of lin-65Δ and of arle-14Δ on H3K9me2 levels in embryos were similar. Whereas we previously found that MET-2 stability is reduced in embryos lacking lin-65 (ref. 14), the loss of arle-14 did not reduce MET-2::mCherry levels (Extended Data Fig. 1e). These data argue that both ARLE-14 and LIN-65 promote MET-2 methylation activity, while only LIN-65 is essential for MET-2 focus formation.
Because the loss of H3K9me is associated with a failure to silence heterochromatin5,6,33,44, we examined gene expression in arle-14Δ using RNA sequencing (RNA-seq). Remarkably, only 14% of the genes derepressed by loss of met-2 (met-2Δ genes) were upregulated in arle-14Δ (Fig. 1d,e), while 59% of the met-2Δ-derepressed genes were upregulated in lin-65Δ. These data suggested that residual low levels of H3K9me2 in combination with the preserved accumulation of MET-2 into foci (promoted by LIN-65 in the arle-14 mutant) restricts gene derepression.
Because arle-14 and lin-65 mutants had different effects on transcription and focus formation, we performed epistasis analysis for the two genes, monitoring H3K9 methylation. We found that the lin-65Δ;arle-14Δ double mutant displayed a greater loss of H3K9me2 than either single mutant alone, achieving the same drop in methylation as in met-2Δ embryos (Fig. 1f). Consistent with the fact that ARLE-14 and LIN-65 interact with MET-2 independently of each other, we conclude that they function in parallel to promote MET-2’s role in H3K9 methylation.
C. elegans animals lacking all H3K9me reproduced efficiently when cultivated at 20 °C or below, yet the loss of met-2 strongly accentuated the reduced fertility of worms at 25 °C, coincident with elevated levels of p53-dependent germline apoptosis (refs. 6,30). This loss of germline arises largely from the formation of R-loops at repeat elements, which are promiscuously transcribed upon loss of met-2 (refs. 6,30). To test whether ARLE-14 and LIN-65 differentially affect worm fertility, we quantified the number of progeny in arle-14Δ and lin-65Δ mutants. We observed an enhanced loss of viable progeny in both met-2Δ and lin-65Δ animals at 25 °C, while the arle-14Δ population was only modestly decreased in comparison with WT worms, despite having a near equal drop in H3K9me2 (Fig. 1g and Extended Data Fig. 1f). The striking difference in the impact of lin-65Δ and arle-14Δ on fertility was substantiated independently at 20 °C (ref. 14, and see Fig. 4a below). The residual fertility in arle-14Δ animals was not dependent on SET-25, which is required for all H3K9me3 in embryos7,28,30, as the arle-14Δ set-25Δ double mutant was no less fertile than arle-14Δ alone (Fig. 1g).
We note that in both lin-65Δ and arle-14Δ embryos substantially more H3K9me2 is retained than in met-2Δ animals (Fig. 1f and Extended Data Fig. 1d), yet lin-65Δ phenotypes parallel those of met-2Δ more closely than those of arle-14Δ. This suggested that H3K9me2 and met-2Δ might not correlate strictly for all phenotypic read-outs, raising the possibility that MET-2 has functions beyond H3K9me deposition. These functions nonetheless require LIN-65 and its incumbent focus formation.
Catalytically deficient MET-2 attenuates defects of met-2Δ
To test directly whether MET-2 has a function beyond ensuring H3K9me deposition, we used CRISPR-mediated mutagenesis to compromise the active site of MET-2’s SET domain. The C1237A mutation met-2(gw1660) (hereafter met-2-CD) alters a highly conserved motif that is essential for the catalytic activity of SET domain proteins generally45,46,47 (Fig. 2a), and specifically of SETDB1 (ref. 48). Consistently, we find that H3K9me2 and H3K9me3 levels dropped to the same level in met-2-CD as in met-2Δ (Fig. 2b–d and Extended Data Fig. 2a,b). Moreover, H3K9me2 levels were as low in met-2Δ set-25Δ as in met-2-CD set-25Δ double mutants (Fig. 2c). Mass spectrometry suggested that H3K9me1 might not be completely abolished in the met-2-CD strain (Extended Data Fig. 2b), although this potential difference in H3K9me1 could not be detected by western blot (Extended Data Fig. 2a), nor was met-2-CD sufficient to preserve H3K9me1/2/3-dependent perinuclear anchoring of a well-characterized heterochromatic reporter (Extended Data Fig. 2c)7,49. Taken together, we conclude that met-2-CD is catalytically deficient for H3K9 methylation yet does not compromise MET-2 protein levels (Extended Data Fig. 2d) or MET-2’s ability to bind ARLE-14 or LIN-65 (Extended Data Fig. 2e). This mutant made it possible to monitor H3K9me2-independent functions of MET-2.

a, Allele map of met-2 locus showing deletion allele met-2Δ(n4256) as black bar and point mutant met-2-CD(gw1660). Amino acid change is shown in parentheses. Putative stop codon for met-2Δ is shown with an ‘X’. The SET domain is indicated with light red. b,c, Representative western blot and quantification of H3K9me2 in WT, met-2Δ and met-2-CD (b) or WT, met-2Δ, met-2Δ set-25Δ and met-2-CD set-25Δ (c). N = 3. NS, nonsignificant by two-sided t-test. d, Immunofluorescence and quantification of H3K9me2 normalized to H4 in WT, met-2Δ and met-2-CD. Scale bar, 5 µm. N = 3, n nuclei(embryos): WT = 1,542(49), met-2Δ = 985(51), met-2-CD = 819(39). Median and quartiles shown. NS, not significant by one-way ANOVA, followed by Tukey post hoc test. e, Live imaging of embryos expressing either WT MET-2 or MET-2-CD::mCherry and quantification of MET-2 foci per nucleus. Scale bar, 5 µm. N = 3, nuclei(embryos): met-2::mCherry = 569(30), met-2-CD::mCherry = 613(30). NS, not significant by two-sided Wilcoxon signed-rank test. f, Brood sizes of the indicated strains at 25 °C. N = 2, n = 40. P(WT, met-2Δ) = 4.7 × 10−14, P(WT, met-2-CD) = 0.0002, P(met-2-CD, met-2Δ)=1.04 × 10−13, P(met-2Δ set-25Δ, met-2-CD set-25Δ) = 1.05 × 10−13, by one-way ANOVA, followed by Tukey post hoc test. g, Percentage of synchronized L1s that develop into L4 larvae 42 h after re-feeding at 20 °C. N = 4. P(WT, met-2Δ) = 4.7 × 10−5, P(WT, lin-65Δ;met-2-CD) = 7.2 × 10−4, P(met-2Δ, met-2-CD) = 5.5 × 10−4, P(met-2-CD, lin-65Δ;met-2-CD) = 0.015; ***P < 0.0001, ****P < 0.00001.
We introduced the met-2-CD mutation (C1237A) into the met-2::mCherry locus to track catalytically deficient MET-2 in live embryos. We found that MET-2-CD forms foci, and the number of foci did not differ from WT MET-2 (Fig. 2e). Both WT and MET-2-CD foci were dependent on LIN-65, and thus we concluded that MET-2 foci can form independently of HMT catalytic activity. This allowed us to distinguish the physiological effects arising from a loss of H3K9me2 from those caused by the dispersion of MET-2 foci.
Because fertility is sensitive to the loss of H3K9me, we next scored progeny in met-2-CD animals. Despite being H3K9me-deficient, the met-2-CD population retained higher levels of fertility than met-2Δ animals, and this was independent of set-25 (Fig. 2f). Previous work has shown that met-2Δ animals also display a stochastic delay in development30, which we quantified by scoring the number of L1 larvae that develop to the L4 larval stage by 42 h in a synchronized culture at 20 °C. In contrast to met-2Δ, met-2-CD animals had no notable developmental delay (Fig. 2g). Moreover, the rescue of development timing was reduced upon lin-65 ablation (Fig. 2g). In conclusion, a LIN-65-dependent function of MET-2-CD is important for normal development in both somatic and germline tissues, suggesting a role for MET-2 foci that is independent of MET-2’s HMT activity.
MET-2-CD can repress heterochromatic genes
It was unclear whether MET-2-CD would be sufficient to preserve gene repression, despite the drop in H3K9me2 found in this mutant. We performed transcriptome analysis in met-2-CD, met-2Δ and WT early embryos. Importantly, most of the genes upregulated in met-2Δ were also upregulated in met-2-CD, highlighting the importance of H3K9me in maintaining repression (Fig. 3a, ‘common’ genes, N = 1225). However, there were 271 met-2Δ targets that were upregulated in met-2Δ but not in met-2-CD (‘met-2Δ-specific’ genes) and 179 genes that exhibited a mitigated response (derepression was more than 2-fold lower in met-2-CD than in met-2Δ, ‘met-2Δ ≫ met-2-CD’, Fig. 3b). In other words, 28% of met-2 target genes retained complete or partial repression in met-2-CD embryos.

a, Correlation of RNA-seq log2 FCs between met-2Δ and met-2-CD relative to WT embryos. Genes significantly upregulated to WT in both met-2Δ and met-2-CD are colored in black (FDR < 0.01; FC > 2). Genes that are upregulated to WT and >2-fold higher in the met-2Δ compared with met-2-CD are colored in red (FDR < 0.01). Dotted line demarcates met-2Δ-specific genes (FDR < 0.01 and FC > 2 in met-2Δ/WT, and FC < 0.1 in met-2-CD/WT). Pearson correlation coefficient (r) is displayed. N = 3. b, log2FC of met-2Δ-specific genes, met-2Δ»met-2-CD and common genes in met-2Δ and met-2-CD. c, Representative western blot and quantification comparing H3K9ac levels in WT, met-2Δ and met-2-CD embryos. N = 4. P(met-2Δ, met-2-CD) = 0.038, P(WT, met-2Δ) = 0.0078. d, Immunofluorescence and quantification of mean H3K27ac signal per nucleus normalized to H4 in WT, met-2Δ and met-2-CD. Scale bar, 5 µm. N = 3, n nuclei(embryos): WT = 780(44), met-2Δ = 778(47), met-2-CD = 765(38). Median and quartiles shown. P(WT, met-2Δ) < 2.0 × 10−16, P(WT, met-2-CD) = 1.1 × 10−5, P(met-2Δ, met-2-CD) < 2.0 × 10−16 by two-sided Wilcoxon signed-rank test. e, H3K27ac ChIP-seq in met-2Δ or met-2-CD early embryos compared with WT. Scatterplot correlates the mean number of reads per peak (normalized to input, log2) in met-2Δ or met-2-CD with WT. Differentially enriched peaks for each genotype versus WT are highlighted in color (FDR < 0.01). N = 3. f, H3K27ac ChIP-seq exemplar region of Chr II is shown (N = 3). Pink box highlights a peak with gain of acetylation in met-2Δ embryos but not in met-2-CD. g, Venn diagram of H3K27ac differentially enriched peaks in ChIP-seq strains shown in e compared with WT (FDR < 0.01). h, Expression heatmap and pan-H3ac ChIP-qPCR at representative met-2Δ-specific genes, common genes or the met-2 independent gene unc-119. Bars indicate the mean and dots represent individual measurements. N = 3 (met-2Δ N = 2). ChIP-qPCR, Chromatin Immunoprecipitation followed by quantitative real-time PCR; *P < 0.01, **P < 0.001, ****P < 0.00001.
Heterochromatin is not only marked by H3K9me but is also characterized by a general hypoacetylation of histone tails. In contrast, gene activation is associated with increased histone tail acetylation. Intriguingly, mass spectrometry showed a global increase in H3K9 acetylation in met-2Δ embryos that was absent in met-2-CD embryos (Extended Data Fig. 2b). We confirmed this result by western blot and immunofluorescence, showing that both histone H3 and H4 acetylation (Extended Data Fig. 3a), including H3K9ac and H3K27ac (Fig. 3c,d), were increased in met-2Δ embryos, but not in met-2-CD embryos. These results suggested that the aberrant gain of acetylation in met-2Δ animals is not an inevitable consequence of H3K9me loss.
To see which loci gain histone acetylation in met-2 mutants, we performed Chromatin immunoprecipitation followed by genome-wide sequencing (ChIP-seq) in early embryos. We concentrated on H3K27ac due to its well-described role in transcriptional activation and enhancer function50. Relative to WT embryos, we defined 565 differentially enriched H3K27ac peaks in met-2Δ, while only 38 changes were detected in met-2-CD (Fig. 3e,f). All but one of the rare met-2-CD peaks were also observed in met-2Δ embryos (Fig. 3g). In addition, we examined the enrichment for pan-histone H3 acetylation (H3ac) at a subset of met-2Δ-specific genes by Chromatin Immunoprecipitation followed by quantitative real-time PCR (ChIP-qPCR). Compared with WT, these loci were enriched for H3ac in met-2Δ but not in met-2-CD embryos (Fig. 3h), whereas for common targets or a nonrepressed control, H3ac enrichment was very similar in met-2Δ and met-2-CD strains (Fig. 3h). The lack of acetylation in the met-2-CD embryos was not linked to residual H3K9me1, as the two mutants (met-2-CD and met-2Δ) had equally low levels of H3K9me1 at these loci (Extended Data Fig. 3b). Instead, the data suggested that MET-2 contributes to transcriptional repression and blocks the acetylation of specific loci, independent of its HMT activity.
ARLE-14 promotes MET-2 association with chromatin
After determining that catalytic-dependent and -independent roles for MET-2 exist in gene repression, we sought to see if ARLE-14 plays a role in the noncatalytic MET-2 function. If ARLE-14 simply promotes MET-2 HMT activity, met-2-CD would be epistatic with arle-14. We examined fertility at both 20 °C and 25 °C in arle-14Δ met-2-CD double mutant animals. Remarkably, at 20 °C neither arle-14Δ nor met-2-CD single mutants had significantly fewer progeny than WT animals (Fig. 4a). However, the arle-14Δ met-2-CD population showed a synthetic loss of viable offspring at both temperatures, phenocopying met-2Δ mutants (Fig. 4a and Extended Data Fig. 4a). This was surprising, because arle-14Δ did not affect foci of WT MET-2 (Figs. 1c and 2e). Nonetheless, we found that arle-14 was required both for MET-2-CD::mCherry foci number and the overall nuclear abundance of the HMT (Fig. 4b–d), an effect that was enhanced at 25 °C (Extended Data Fig. 4b,c). These data argued that ARLE-14 acts redundantly with H3K9me to stabilize MET-2 in foci.

a, Brood sizes of the indicated strains at 20 °C. N = 2, n = 40. P(WT, met-2Δ) < 2.0 × 10−16, P(WT, met-2-CD) = not significant, P(WT, arle-14Δ) = not significant, P(WT, arle-14Δ met-2-CD) = 1.94 × 10−12, P(met-2Δ, met-2-CD) = 8.2 × 10−14, by one-way ANOVA, followed by Tukey post hoc test. b–d, Live imaging of expressing either WT MET-2::mCherry or MET-2-CD::mCherry ± arle-14 RNAi (b), and quantification of MET-2 nuclear signal P(WT(vector), WT(arle-14)) = 1.0 × 10−12, P(WT(vector), met-2-CD(vector)) < 2.0 × 10−16, P(WT(vector), met-2-CD(arle-14)) = 5.7 × 10−7, P(met-2-CD(vector), met-2-CD(arle-14)) < 2.0 × 10−16 (c) and MET-2 foci per nucleus (d). P(met-2-CD(vector), met-2-CD(arle-14 RNAi)) = 0.041. Scale bar, 5 µm. N = 3, n = 45 embryos. Nuclei: met-2::mCherry: vector = 816, arle-14 RNAi = 1,199, met-2-CD::mCherry: vector = 1,056, arle-14 RNAi = 1,787. P values calculated by two-sided Wilcoxon signed-rank test. e, mCherry ChIP-qPCR at representative met-2Δ-specific genes, common genes or the met-2-independent gene unc-119. Bars indicate the mean and dots represent individual measurements. N = 3. Statistical comparison of arle-14Δ met-2-CD with met-2-CD: C55C3.3 P = 0.05, col-166 P = 0.74, bcl-11 P = 0.004, F10D11.6 P = 0.009, grl-16 P = 0.34, glf-1 P = 0.0024, nep-17 P = 0.05, unc-119 P = 0.15, Student’s t-test. f, heatmap of log2 FC of met-2Δ-specific genes, met-2Δ»met-2-CD genes and common genes in met-2Δ, met-2-CD, arle-14Δ and arle-14Δ met-2-CD relative to WT. g, distribution of log2 FC of the same comparisons as in (f) displayed as violin plots. met-2Δ-specific: P(met-2Δ, arle-14Δ met-2-CD) < 2.0 × 10−16, P(met-2-CD, arle-14Δ met-2-CD) < 2.0 × 10−16, P(arle-14Δ, arle-14Δ met-2-CD) < 2.0 × 10−16; met-2Δ»met-2-CD: P(met-2Δ, arle-14Δ met-2-CD) < 2.0 × 10−16, P(met-2-CD, arle-14Δ met-2-CD) = 0.00012, P(arle-14Δ, arle-14Δ met-2-CD) < 2.0 × 10−16; common: P(met-2Δ, arle-14Δ met-2-CD) < 2.0 × 10−16, P(met-2-CD, arle-14Δ met-2-CD) < 2.0 × 10−16, P(arle-14Δ, arle-14Δ met-2-CD) < 2.0 × 10−16 by two-sided Wilcox rank sum test. N = 3. Boxplots show median, boxes 50% and whiskers 90% of each group. NS, not significant; *P < 0.01, **** and #### P < 0.00001.
Although the function of worm ARLE-14 is unclear, recent evidence in human cells suggested that the human homolog, ARL14EP, may facilitate SETDB1 interaction with chromatin43. To see if ARLE-14 promotes MET-2 binding to chromatin, we performed ChIP-qPCR using antibodies against MET-2::mCherry in both WT and met-2-CD backgrounds, both in the presence and absence of ARLE-14. MET-2-CD showed similar enrichment as WT MET-2 at five of seven tested targets (Fig. 4e). Loss of arle-14 showed little effect on WT MET-2::mCherry enrichment at most MET-2 gene targets (with the exception of col-166), but arle-14 did lead to the loss of MET-2-CD at these same sites (Fig. 4e). Consistently, the loss of ARLE-14 in met-2-CD embryos led to the upregulation of met-2Δ-specific targets based on RNA-seq data, which was not observed in arle-14Δ alone (Fig. 4f,g). Taken together, these data suggested that ARLE-14 acts redundantly with H3K9me to stabilize both MET-2 foci and its association with MET-2-controlled genes. Thus, arle-14 is needed for the noncatalytic function of MET-2 on both physiological and molecular levels.
Acute heat stress dissociates MET-2 foci
Having shown by genetics that MET-2 has a noncatalytic function that correlates with its concentration into foci, we wondered whether foci dynamics might be physiologically relevant in animals containing normal levels of H3K9me. We previously showed that MET-2 foci disperse upon transient exposure to 37 °C heat shock (HS) (Fig. 5a and Extended Data Fig. 5a)14, coincident with the canonical heat shock response (HSR)51. Intriguingly, although the HSR is clearly associated with well-described signaling pathways that dampen transcription while specifically upregulating protein chaperone genes52,53, earlier studies in other species also linked acute heat stress to the transcriptional derepression of silent chromatin54,55,56,57,58. Thus, HS conditions provided a physiological system in which we could monitor a transcriptional response that coincides with the induced dispersion of MET-2 foci, without genetic alteration.

a, Representative images of MET-2::mCherry foci in live WT embryos cultured normally (20 °C) or heat-stressed (HS; 37 °C, 60 min). Scale bar, 5 µm. N = 3. b, RNA-seq comparing the gene expression in RPKM (reads per kilobase million) of HS treatment or met-2Δ relative to WT(20 °C) early embryos. Genes marked in light purple and dark red are upregulated in 37 °C-treated embryos and met-2Δ, respectively, relative to WT(20 °C) (FDR < 0.01; log2FC > 2). N = 3. c,d, Venn diagram of derepressed genes (c) and correlation between HS-treated and met-2Δ log2FC in gene expression compared with WT(20 °C) (d). WT 37 °C versus met-2Δ: P < 2.2 × 10−16 by two-sided Fisher’s exact test. Pearson correlation coefficient (r) is displayed. e, Immunofluorescence and quantification of H3K9me2 per nucleus normalized to H4 in WT(20 °C) and 30-min HS-treated embryos. Scale bar, 5 µm. N = 3, n nuclei(embryos): 20 °C = 1,522(51), 37 °C = 1,139(46). Median and quartiles shown. P < 2.0 × 10−16 by two-sided Wilcoxon signed-rank test. f,g, Representative western blots and quantification of H3K9me2 (f) or H3ac (g) normalized to MRG-1 in WT(20 °C) and HS-treated WT embryos. HS: 37 °C, 30 min (f) or 60 min (g). H3K9me2 N = 5, P = 1.1 × 10−4; pan-H3ac N = 4, P = 0.0012; H3K9ac N = 5, P = 0.025; H3K27ac N = 4, P = 0.018 by two-sided t-test. h, H3K27ac ChIP-seq HS-treated early embryos compared with WT(20 °C). Differentially enriched peaks are highlighted in color (FDR < 0.01). N = 3. i, Venn diagram of H3K27ac differentially enriched peaks in HS treatment versus met-2 mutants shown in Fig. 5h and Fig. 3e relative to WT (20 °C) (FDR < 0.01). WT 37 °C versus met-2Δ: P < 2.2 × 10−16 by two-sided Fisher’s exact test. *P < 0.01, **P < 0.001, ***P < 0.0001, ****P < 0.00001.
We performed RNA-seq on early embryos exposed to either normal temperature (20 °C) or HS (1 h at 37 °C). We observed a broad upregulation of genes at 37 °C relative to embryos kept at 20 °C (2026 genes up, 314 genes down, fold change (FC) > 4, false discovery rate (FDR) < 0.01, Fig. 5b). Consistent with the dispersion of MET-2 foci, we found that most HS-upregulated genes, similar to met-2Δ-upregulated genes, had very low expression levels in embryos cultured at 20 °C (80% reads per kilobase million < 4 at 20 °C Fig. 5b). Strikingly, we observed a positive correlation (r = 0.65) between the transcriptional changes induced by 37 °C versus those induced by the loss of met-2 at 20 °C versus WT (20 °C) embryos (Fig. 5b–d). In fact, 76% of the met-2Δ target genes were upregulated under HS conditions, constituting 56% of all HS-derepressed genes (Fig. 5c). The majority of the shared targets were genes that are normally expressed in postembryonic, differentiated tissues (Extended Data Fig. 5b). These data suggest that embryos exposed to HS not only lose MET-2 foci but also derepress many genes that are normally repressed by MET-2 (Fig. 5c,d). On the other hand, met-2Δ embryos respond to HS similarly to WT animals with respect to global gene expression, HSP gene induction and HSF-1 foci formation (Extended Data Fig. 5c–f).
Because met-2Δ mutants present enhanced phenotypes at 25 °C (refs. 6,30), we wondered whether the shift from 20 °C to 25 °C also induces a similar stress response as exposure to 37 °C. However, no HSF-1 foci formed in either WT or met-2Δ embryos at 25 °C, nor did the transcriptome of animals grown at 25 °C correlate with the pattern of genes induced by HS (Extended Data Fig. 5f,g). Thus, the loss of fertility at 25 °C is not a heat shock response.
Given that the loss of MET-2 foci correlates with a loss of H3K9me (Extended Data Fig. 1b), we examined how H3K9me levels change during 30 min at 37 °C. Indeed, embryos exposed to HS displayed a drop in H3K9me2 to 45% or 50% of untreated levels, as measured by western blot or immunofluorescence, respectively (Fig. 5e,f). Whereas the met-2 messenger RNA level upon HS was unchanged (log2FC = −0.094), we scored a ~24% reduction in MET-2 protein level (Extended Data Fig. 6c), which may influence chromatin states. We examined regions that bear H3K9me2 in embryos at 20 °C by ChIP-seq, to see if H3K9me2 is also reduced in a localized manner after exposure to 37 °C for 1 h. Indeed, we observed a general reduction in read counts for H3K9me2 to 63% of level scored at 20 °C (Extended Data Fig. 6a).
Whereas the transcriptional induction following HS was stronger than what we observed in met-2Δ, the loss of H3K9me2 was less. Given our earlier data that link the loss of MET-2 foci with increased acetylation levels, we checked to see if histone acetylation changes during HS. Western blots showed that HS-treated embryos have a substantial increase in pan-H3ac, including increases in H3K9ac and, more modestly, H3K27ac (Fig. 5g). Taken together, our results argue that HS-induced dispersion of MET-2 foci coincides with a partial destabilization of H3K9me2, a strong gain of H3ac and widespread transcriptional activation of loci silenced by MET-2 under normal conditions.
To compare the changes during HS to those incurred by met-2Δ, we examined changes in H3K27ac in WT embryos (HS versus 20 °C) by ChIP-seq, and compared these peaks with data from our met-2Δ and met-2-CD animals. HS (30 min at 37 °C) led to an enrichment of H3K27ac on 1646 peaks (Fig. 5h). Importantly, 49.7% of the H3K27ac peaks in met-2Δ were shared with those in HS embryos (Fig. 5i). Nonetheless, there were three times as many peaks after HS as in unstressed met-2Δ embryos (1646 in HS versus 565 in met-2), consistent with the idea that HS mobilizes a broad transcriptional response, as well as destabilizing MET-2 foci. Other regions of the genome of HS-treated embryos actually lost H3K27ac, reflecting the fact that a certain degree of transcriptional downregulation occurs during the canonical HSR59. The sites that gained acetylation in both HS and met-2Δ embryos were mainly promoters and repeats, while met-2-CD embryos showed almost no gain in acetylation over WT (Extended Data Fig. 6b). Intriguingly, the met-2 target genes that remain fully or partially repressed in the met-2-CD background (presumably because they remain in foci) showed particularly pronounced derepression during HS-induced loss of focus formation (Extended Data Fig. 6d,e). In other words, the dissolution of MET-2 foci upon HS is coupled with increased acetylation and gene derepression, beyond the program induced by HSF-1, reinforcing the correlation we found between MET-2-CD foci and transcriptional repression.
Discussion
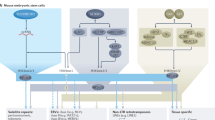
The sequestration and self-association of heterochromatin into foci has been observed since the early days of microscopy1. While these foci coincided with HP1 and with H3K9me-specific HMTs, it was unclear whether the localized concentration of these proteins simply enhanced the propagation of H3K9 methylation, or served another role. Using a mutant of a key residue in the SET domain of the SETDB1-like enzyme MET-2 (met-2-CD), we have been able to distinguish a repressive role for MET-2 focus formation distinct from the catalytic function of MET-2’s SET domain. H3K9me is clearly the key regulatory mark for the stable silencing of lineage-specific genes and repetitive elements across animal species, and the majority of loci repressed by heterochromatin in worms depend on the activity of MET-2’s SET domain. Yet the careful analysis of the met-2-CD allele has revealed a second, noncatalytic role for MET-2 that requires its binding partners LIN-65 (H.s. ATF7IP) and ARLE-14 (H.s. ARL14EP), and which correlates with the accumulation of MET-2 and its ligands in subnuclear foci (Fig. 6).

The primary function of the SETDB1-like enzyme, MET-2, is to mediate H3K9 methylation (1). Its loss leads to extensive gene and repeat derepression. The concentration of MET-2, LIN-65 and ARLE-14 into nuclear foci requires LIN-65 and represses transcription by H3K9me-independent inhibition of histone acetylation in parallel to ensuring efficient H3K9 methylation (2). ARLE-14 stabilizes MET-2 foci at chromatin redundantly with H3K9me. HATs, histone acetyl transferases; KDMs, histone demethylases; TFs, transcription factors.
LIN-65 is necessary for focus formation, while ARLE-14 may stabilize MET-2 interaction with chromatin within foci, acting redundantly with H3K9me with respect to MET-2 recruitment and focus formation (ref. 14) (Fig. 6). Foci of MET-2-CD appear to be sufficient to block local acetylation and mitigate inappropriate transcription of selected developmentally regulated genes, in an ARLE-14-dependent manner. We infer that MET-2 foci not only correlate with efficient H3K9me, but they create a semirepressed state that is refractory to histone acetylation. Consistent with the repressive function, MET-2-CD was able to largely preserve fertility and somatic development in the absence of MET-2’s catalytic activity under normal growth conditions (20–25 °C). For this function, however, it requires ARLE-14.
The comparison of the met-2Δ and met-2-CD transcriptomes showed that even among the loci that were derepressed in both met-2Δ and met-2-CD embryos, the transcriptional levels of many genes were higher in met-2Δ. Genes that retained full or partial repression constitute 28% of met-2 target genes. This argues that H3K9me and MET-2’s noncatalytic function silence genes in a partially additive manner, reinforcing each other to maintain heterochromatic repression.
The mechanism of focus-mediated repression appears to be largely structural: we found that MET-2-CD foci were able to block the increase in histone tail acetylation found in met-2Δ mutants. We note that similar to H3K9me, histone hypoacetylation is a hallmark of heterochromatin across eukaryotic species60,61. In Schizosaccharomyces pombe the histone deacetylases (HDACs) Clr3 and Sir2 are essential to restrict RNA Pol II access to heterochromatin62,63, and the treatment of murine cells with HDAC inhibitors resulted in a widespread loss of heterochromatic repression64 and impaired the establishment of lineage-specific expression patterns in differentiating mouse embryos65. Finally, histone acetylation itself is known to promote chromatin decompaction, weaken the interaction between histones and DNA, and antagonize phase separation of chromatin arrays in vitro66,67,68,69. Therefore, MET-2-CD’s ability to prevent hyperacetylation could explain how it maintains the semirepressed state that we observe by RNA-seq. MET-2-CD may block acetyltransferase access directly or recruit the factors, such as HDACs, that ensure that heterochromatin remains silent.
We cannot yet determine whether MET-2/LIN-65/ARLE-14 preserve hypoacetylation directly or indirectly. Although no HDACs were enriched in our MET-2 pulldown14 (Fig. 1b), it was recently suggested that MET-2 can physically associate with HDA-1 (HDAC1) in a SUMOylation-dependent manner70. In addition to its physical interaction with LIN-65 and ARLE-14, and potentially HDAC1, MET-2 interacts genetically with the H3K4 demethylase SPR-5 (ref. 71), the DREAM complex72 and SMARC-1 chromatin remodelers73. While we do not exclude it, the hypothesis that these factors serve as effectors of MET-2’s noncatalytic function is made less likely by the fact that each of the others was shown to act either redundantly with MET-2 or through H3K9me. Alternatively, recent investigations into the function of subnuclear condensates suggest that these foci may physically impair the access of transcription activating complexes, such as RNA Pol II, Mediator, basal transcription factors or histone acetyltransferases74. Because foci of MET-2 and its cofactors share properties of condensates, they may also block recruitment of the transcription machinery and thereby inhibit the activation and acetylation of silenced genes, despite reduced levels of H3K9me.
We note that we were unsuccessful in expressing recombinant MET-2 or LIN-65, potentially due to their large size and highly disordered domains75,76. This precluded in vitro tests with recombinant protein for direct binding and/or HMT activity, which would complement the extensive genetic and biochemical evidence implicating MET-2 as the primary H3K9me1/me2 HMT in C. elegans embryos7,15,28,30,32,71,77,78,79,80,81,82. Nevertheless, we were able to demonstrate H3K9me activity of MET-2 recovered in a pull-down assay. We note that MET-2-CD has the same protein stability, localization, dynamics and protein interactions as WT MET-2, and yet H3K9me2 levels drop strikingly. This supports a direct role of MET-2 catalytic activity in H3K9me deposition, and a direct role for MET-2-CD in a secondary repressive function associated with LIN-65 dependent focus formation.
We find that ARLE-14 becomes essential to stabilize the MET-2-CD interaction with chromatin and its accumulation in foci, when catalytic function is compromised (Fig. 4). Consistently, the ARLE-14 homolog ARL14EP has been shown in vitro to bind H3 peptides in a SETDB1-independent manner43. We therefore propose that ARLE-14 helps recruit MET-2 to chromatin, acting independently of the H3K9me mark. It is possible that ARLE-14 has other roles in gene activation that are independent of MET-2, or else that ARLE-14 also prevents MET-2 mistargeting, given that some genes actually acquire, rather than lose, repression upon loss of arle-14 (Fig. 4f).
Our study is not the first to suggest noncatalytic functions for a diverse group of HMTs. In mammalian cells, the H3K9 HMT G9a is able to promote repression by recruiting DNA methylation enzymes, independently of its catalytic activity44,83. Similarly, mammalian MLL3 and MLL4 do not require the enzymes’ H3K4 methylation activity to maintain enhancer activation in mice84. We speculate that in the case of MET-2, the noncatalytic function may allow for multiple layers of repressive control that respond to different stimuli and react with different kinetics, an example of which is the cellular response to heat stress (HS, 30 min to 1 h at 37 °C). HS resulted in hyperacetylation and activation of silenced genes, coincident with MET-2 focus dispersion, even though the loss of genome-wide H3K9me was less severe than that triggered by arle-14Δ or met-2Δ strains. The dissolution of MET-2 foci by HS illustrates a rapid transcriptional response that promotes chromatin opening while retaining H3K9me, potentially allowing a quick post-HS recovery of the prestress transcriptome.
In summary, we have uncoupled the effect of MET-2 concentration into subnuclear foci from its lysine methyltransferase activity. While H3K9me deposition is the primary driver of gene silencing, both H3K9me and MET-2 focus formation contribute to the repression of transcription and may mutually reinforce the heterochromatic state. Whereas MET-2 foci do not require the HP1-like factors HPL-1 and HPL-2 (ref. 14), they do depend on LIN-65, a protein rich in low-complexity sequence and disordered domains14,18. They are also stabilized by ARLE-14 in a manner redundant with H3K9 methylation, and they attenuate histone tail acetylation. This indicates that the SETDB1-like complex containing MET-2, ARLE-14 and LIN-65 contributes to gene repression beyond the targeted methylation of histone H3K9, with physiological consequences for organismal growth and fertility.
Methods
Strains and maintenance
Strains used in this study are listed in Supplementary Table 1. Unless otherwise indicated, experiments were performed using early embryos isolated from animals cultured at 20 °C. Embryos were isolated by standard bleaching procedures; embryos isolated from heat-stressed adults were isolated in buffers prewarmed to 37 °C. The conditions of HS are indicated for each experiment. RNA interference (RNAi) experiments were performed as previously described14. Briefly, synchronized L1s were fed HT115 bacteria expressing double stranded RNA (dsRNA) on nematode growth media supplemented with 1 mM IPTG and 100 µg ml−1 carbenicillin. Embryos isolated from these animals were analyzed as indicated. Synchronized animals fed either vector or arle-14 RNAi were grown to L4 stage at 20 °C before being shifted to 25 °C overnight, and their progeny were imaged (Extended Data Fig. 4).
We used CRISPR as previously described18,85,86 to generate alleles NLS::met-2(gw1786), arle-14(gw1584) and met-2(gw1660). CRISPR RNA (crRNA) and homology directed repair (HDR) repair templates are listed in Supplementary Table 2. arle-14(gw1584) was later confirmed not to be a null allele by mass spectrometry.
Live microscopy and immunofluorescence
Imaging was performed as previously described14. Briefly, all images were captured using Visiview software on a confocal spinning-disk microscope, the AxioImager M1 (Zeiss) with a Yokogawa CSU-X1 scanhead (Yokogawa), a Rolera Thunder camera (Photometrics) and an α plan-NEOFLUAR ×100/1.45 oil objective (Zeiss). Live embryos were mounted on 2% agarose pads in M9 buffer. For immunofluorescence, embryos were fixed for 5 min in 2% formaldehyde, then transferred to poly-l-lysine-coated slides and snap-frozen. Embryos were freeze-cracked and immediately treated with 100% ethanol precooled to −20 °C for 2 min then allowed to dry. After three 5-min washes using PBS with 0.25% Triton X-100 (PBS-X), slides were blocked in PBS-X + 2% milk for 1 h at room temperature and subsequently exposed to antibodies overnight at 4 °C in a humid chamber. After washing as above, slides were treated at room temperature with secondary antibodies diluted in PBS-X + 2% milk. Antibody dilutions are as follows: 1:500 mouse anti-H3K9me2 (MBL), 1:5,000 mouse anti-H3K27ac (gift from H. Kimura), 1:500 rabbit anti-H4 (Abcam), 1:1,000 goat anti-mouse Alexa Fluor 488 (A11001; Invitrogen) and 1:1,000 donkey anti-rabbit Alexa Fluor 555 (A31572; Invitrogen). After DAPI (1:2,000) staining in PBS-X for 10 min, and three washes, slides were mounted with ProLong Gold Antifade (Thermo Fisher Scientific). Images were deconvolved using the Huygens remote manager (http://www.huygens-rm.org/wp/). Nuclear mean intensity and number of foci were quantified using KNIME87. Nuclei were first identified using MET-2::mCherry fluorescence, then foci were detected with a Laplacian-of-Gaussian detector from TrackMate (fmi-ij2-plugins-0.2.5, https://doi.org/10.5281/zenodo.1173536)88.
Western immunoblotting
All lysates were prepared at 4 °C and first processed using a Fast Prep-24 5G Benchtop Homogenizer (MP Biomedicals) using a 4:1 ratio of lysate to 0.5-mm zirconia/silicon beads (BioSpec). For histone methylation analysis, embryos were lysed in RIPA buffer. Lysates were treated with 5 µl of benzonase (Sigma) for 1 h at 4 °C with rotation. For acetylation analysis, embryos were lysed in TAP buffer (150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 0.5% NP-40, 1 mM EDTA, 10% glycerol, 2× cOmplete-EDTA-free protease inhibitors (Roche), 1× Deacetylase Inhibitor (Active Motif) and 1 mM DTT) and sonicated using a Bioruptor Plus (Diagenode) for 14 cycles, 15 s on, 30 s off. TAP lysates were cleared at 21,000g for 10 min. Then, 10 µg of total protein was separated on Bis-Tris gels (Bio-Rad or Invitrogen) using MES buffer and transferred to 0.2-µm PVDF (Bio-Rad). Membranes were blocked with Protein-Free Blocking Buffer (Pierce) or PBS plus 0.5% Tween-20 (PBS-T) with 5% powdered milk (Sigma). Antibody dilutions: 1:2,000 mouse anti-H3K9me2 (MBL), 1:1,000 rabbit anti-RFP antibody, preadsorbed (Rockland), 1:100,000 mouse anti-H3K9me3 (MBL), 1:60,000 rabbit anti-H3K9me1 (ab8896; Abcam), 1:40,000 mouse anti-H3K9acetyl (a gift from H. Kimura), 1:40,000 rabbit anti-acetyl-Histone H3 (Millipore), 1:2,000 rabbit anti-acetyl-Histone H4 (Millipore), 1:20,000 rabbit anti-H2B (Abcam) and 1:40,000 rabbit anti-MRG-1 (49130002; Novus Biologicals). After overnight rotation with antibodies diluted in blocking buffer at 4 °C, blots were washed three times in PBS-T, re-blocked and exposed to HRP-conjugated secondary antibodies for 1 h (goat anti-mouse IgG HRP (1:10,000; Jackson ImmunoResearch 115-035-146), goat rabbit IgG HRP (1:20,000; Jackson ImmunoResearch 111-035-144)). After three further washes in PBS-T, ECL (Millipore) signal was detected with an Imager 600 (GE).
Mass spectrometry
For immunoprecipitation experiments, embryos isolated by bleaching gravid adults were lysed at 4 °C in TAP buffer. Proteins from homogenized, sonicated and cleared TAP lysates (above) were immunoprecipitated with anti-FLAG-M2 beads (Sigma). Proteins were digested, bound to beads and subjected to LC–MS/MS exactly as described previously14. Intensity-based absolute quantification values were used as a measure of protein abundance in immunoprecipitation89. For H3K9 post-translational modification profiling, we employed the Histone Purification Kit Mini (Active Motif). Embryos isolated by bleaching were homogenized as above in histone extraction buffer. After overnight extraction at 4 °C with rotation, we proceeded with histone purification according to the manufacturer’s instructions. Purified histones were propionylated twice before and once after overnight trypsin digestion to prevent cleavage at lysine residues and facilitate separation of peptides. Samples were dried in a speedvac and resuspended in 0.1% trifluoroacetic acid (TFA), 2% acetonitrile. LC–MS was performed as follows: samples were loaded onto a PepMap 100 C18 2-cm trap (Thermo Fisher) using an EASY nLC-1000 system (Thermo Fisher). On-line peptide separation was then performed on a 15-cm EASY-Spray C18 column (ES801, Thermo Fisher) by applying a linear gradient of increasing acetonitrile concentration at a flow rate of 150 nl min−1. An Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) was operated by alternating between MS1 survey scan mode, recording spectra at 120,000 resolution in the Orbitrap, and data-independent parallel reaction monitoring (PRM) mode, by targeting previously identified doubly charged histone H3 peptide isoforms of the sequence ‘KSTGGKAPR’ (9–17) containing lysines K9 and K14 (H3K9_K14me0 at m/z = 507.2873, H3K9me2K14ac at m/z = 514.2951, H3K9acK14ac at m/z = 521.2847, H3K9acK14me0 at m/z = 28.2926, H3K9me3K14me0 at m/z = 528.3107, H3K9me1K14ac at m/z = 535.3004, H3K9me1K14me0 at m/z = 542.3082, H3K4me0 at m/z = 522.2982, with ‘me0’ = in vitro propionylated, me1 = methyl + in vitro propionylated), and two endogenously unmodified reference peptide precursors (YRPGTVALR (41–49) at m/z = 544.8109 and YQKSTELLIR_K56me0 (54–63, with K56me0 = in vitro propionylated lysine) at m/z = 681.8817) for higher-energy collisional dissociation (HCD) fragmentation at a normalized collision energy of 33%, recorded at 15,000 resolution in the Orbitrap analyzer. Acquired data from strains indicated in Supplementary Table 1 were loaded into Skyline software (v.20.2.0.343)90, diagnostic fragment (z = 1) ions y6, y7 and y8 for each histone H3K9 peptide isoform (mark) of interest were selected and retention time windows were manually chosen to avoid quantifying background noise from coeluting isoforms or other unrelated analytes. Integrated fragment ion abundances were aggregated by peptide and histone mark for all samples and replicates and then divided by the total sum of all histone H3 peptide abundances per sample.
Developmental phenotypes
For brood size, L4 animals grown from egg at the indicated temperature were singled out onto individual plates. They were transferred to fresh plates every 24 h until egg laying ceased, after which their total progeny was counted. For developmental rate, synchronized L1s were plated and the numbers of L4 and pre-L4 (delayed) animals were counted 42 h post feeding.
RNA-seq
RNA was extracted from early embryos using Trizol as previously described6. Embryos were freeze-cracked five times, then RNA was extracted with choloroform followed by isopropanol precipitation. Further purification was performed with the RNA Clean and Concentrator kit (Zymo). Libraries were produced using the Smart-Seq2 mRNA sequencing kit (Illumina). Ribosomal RNA was depleted using Ribo-Zero Gold kit (Epicentre), and libraries were produced using the Total RNA Sequencing Scriptseq kit (Illumina). Equimolar amounts of indexed libraries were pooled and sequenced on a HiSeq 2500 (Illumina).
Reads were analyzed as described previously6. Adapters were trimmed using Trimmomatic v.0.39. Reads were aligned to the C. elegans genome (ce10) with the R package QuasR v.1.22.0 (www.bioconductor.org/packages/2.12/bioc/html/QuasR.html; ref. 91). The command ‘proj < -qAlign(‘samples.txt’,‘BSgenome. Celegans.UCSC.ce10’, splicedAlignment = TRUE)’ instructs hisat2 (ref. 92) to align using default parameters, considering unique reads for genes and genome-wide distribution. Count tables of reads mapping within annotated exons in WormBase (WS220) were constructed using the qCount function of the QuasR package to quantify the number of reads in each window (qCount(proj,GRange_object,orientation = ‘same’)) and normalized by division by the total number of reads in each library and multiplied by the average library size. Transformation into log2 space was performed after the addition of a pseudocount of 8 to minimize large changes in abundance FC caused by low count numbers. The EdgeR package v.3.24 was applied to select genes with differential transcript abundances between indicated genotypes (contrasts) based on FDRs for genes. Replica correlations are shown in Extended Data Fig. 6f,g. Annotation of tissue and cell type expression is based on annotation tables provided by the tissue atlas93.
ChIP
ChIP experiments were performed as previously described6. In brief: early embryos were harvested from synchronized animals in three replicates. Then, 40 μg of chromatin was incubated overnight with 2 μg of anti-H3K9me2 (MBL), anti-H3K27ac (Abcam, ab177178), anti-H3K9me1 (Abcam, ab176880) or anti-acetyl-Histone H3 (Millipore) antibody coupled to Dynabeads Sheep Anti-Rabbit IgG (Invitrogen), in FA buffer (50 mM HEPES/KOH pH 7.5, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 150 mM NaCl)) containing 1% SDS. For MET-2::mCherry ChIP, 100 μg of chromatin was incubated with RFP-Trap magnetic agarose (Chromotek). Antibody-bound chromatin was washed 3 × 5 min with FA buffer; 5 min with FA buffer with 1 M NaCl; 10 min with FA buffer with 500 mM NaCl; 5 min with TEL buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0) and 2 × 5 min with Tris-EDTA-buffer. Complexes were eluted in 1% SDS in Tris-EDTA buffer with 250 mM NaCl at 65 °C for 15 min. Samples and inputs were treated with 20 μg of RNAse A for 30 min at 37 °C and 20 μg of proteinase K for 1 h at 55 °C. Crosslinks were reversed by overnight incubation at 65 °C. DNA was subsequently purified using Zymo DNA purification columns (Zymo Research). Primers used in quantitative PCR are listed in Supplementary Table 2.
Libraries were prepared as previously described6 using the NEBNext Ultra DNA Library Prep kit for Illumina (NEB no. 7370) and the NEBNext Multiplex Oligos for Illumina (NEB no. E7335), according to the manufacturer’s recommendations, without size selection. Libraries were indexed and amplified using 12 PCR cycles, following the manufacturer’s recommendations. Libraries were further purified with Agencourt AmPure XP beads (Beckman no. A63881). Library size range and concentration were determined using a BioAnalyzer 2100 (Agilent Technologies) and Qubit (Invitrogen) instrument, respectively. Equimolar amounts of indexed libraries were pooled and sequenced on a HiSeq 2500 (Illumina) in rapid mode (Paired-End 50). Reads were aligned to the C. elegans genome (ce10) with the R package QuasR v.1.22.0 (www.bioconductor.org/packages/2.12/bioc/html/QuasR.html). The command ‘proj < -qAlign(‘samples.txt’,‘BSgenome. Celegans.UCSC.ce10’)’ instructs bowtie to align using the parameters ‘-m 1–best–strata–phred33-quals’, considering unique reads for genes and genome-wide distribution.
Read density was calculated by tiling the genome into 500-base-pair (bp) nonoverlapping windows and using the qCount function of the QuasR package to quantify the number of reads in each window (qCount(proj,GRange_object,orientation = ‘any’)). H3K9me-positive domains were determined as regions with a consecutive enrichment of H3K9me2 over input. For gene quantification, gene annotation from WormBase was used (WS220). Quantitation for each gene was performed by counting the reads overlapping the exons. Genome annotation was based on BSgenome.Celegans.UCSC.ce10 package (https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Celegans.UCSC.ce10.html). Differences in read depths between samples were normalized by dividing each sample by total reads and multiplying by average library size. The various count tables used throughout this study were normalized according to the total genome count. log2 expression levels were determined after addition of a pseudocount of 2 (y = log2(x + 2)) to minimize large changes in FC caused by low count numbers. Results are displayed as the mean enrichment of immunoprecipitation − input (log2). H3K27ac peaks were called using MACS2 (ref. 94) with the following parameters: -g 93260000 --broad. Differential peaks among the genotypes were called using the Diffbind package v.3.0.11 (http://bioconductor.org/packages/release/bioc/html/DiffBind.html)95, normalizing on the total read counts and using the implemented DESeq2 analysis with standard parameters. Read density was calculated by tiling the genome into 500-bp nonoverlapping windows, or specifically at promoters (defined as 1 kilobase upstream and 100 bp downstream of transcription start site), and using the qCount function of the QuasR package to quantify the number of reads in each window (qCount(proj,GRange_object,orientation = ‘any’)). Differences in read depths between samples were normalized by dividing each sample by total reads and multiplying by average library size. The various count tables used throughout this study were normalized according to the total genome count.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All genome-wide datasets from this study have been uploaded to the Gene Expression Omnibus (GEO) under accession number GSE168925, GSE122341, and to the Sequence Read Archive (SRA) under SRP080806. Other numerical measurements are provided as Source data with this paper.
Change history
15 April 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41594-022-00776-w
References
Akhtar, A. & Gasser, S. M. The nuclear envelope and transcriptional control. Nat. Rev. Genet. 8, 507–517 (2007).
Padeken, J. & Heun, P. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr. Opin. Cell Biol. 28, 54–60 (2014).
Padeken, J., Zeller, P. & Gasser, S. M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 31, 12–19 (2015).
Nicetto, D. et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 363, 294–297 (2019).
Peters, A. H. et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 (2001).
Zeller, P. et al. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 48, 1385–1395 (2016).
Towbin, B. D. et al. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150, 934–947 (2012).
Bian, Q., Anderson, E. C., Yang, Q. & Meyer, B. J. Histone H3K9 methylation promotes formation of genome compartments in Caenorhabditis elegans via chromosome compaction and perinuclear anchoring. Proc. Natl Acad. Sci. USA 117, 11459–11470 (2020).
Mizuguchi, T. et al. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516, 432–435 (2014).
Zhang, W. et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015).
Liu, J. et al. HP1α mediates defective heterochromatin repair and accelerates senescence in Zmpste24-deficient cells. Cell Cycle 13, 1237–1247 (2014).
Chen, M. W. et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840 (2010).
Rowbotham, S. P. et al. H3K9 methyltransferases and demethylases control lung tumor-propagating cells and lung cancer progression. Nat. Commun. 9, 4559 (2018).
Delaney, C. E. et al. Heterochromatic foci and transcriptional repression by an unstructured MET-2/SETDB1 co-factor LIN-65. J. Cell Biol. 218, 820–838 (2019).
Padeken, J. et al. Argonaute NRDE-3 and MBT domain protein LIN-61 redundantly recruit an H3K9me3 HMT to prevent embryonic lethality and transposon expression. Genes Dev. 35, 82–101 (2021).
Tsusaka, T., Shimura, C. & Shinkai, Y. ATF7IP regulates SETDB1 nuclear localization and increases its ubiquitination. EMBO Rep. 20, e48297 (2019).
Melcher, M. et al. Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol. Cell. Biol. 20, 3728–3741 (2000).
Mutlu, B. et al. Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation in C. elegans embryos. Sci. Adv. 4, eaat6224 (2018).
Gasser, S. M., Hediger, F., Taddei, A., Neumann, F. R. & Gartenberg, M. R. The function of telomere clustering in yeast: the circe effect. Cold Spring Harb. Symp. Quant. Biol. 69, 327–337 (2004).
Ginno, P. A., Burger, L., Seebacher, J., Iesmantavicius, V. & Schübeler, D. Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat. Commun. 9, 4048 (2018).
Müller-Ott, K. et al. Specificity, propagation, and memory of pericentric heterochromatin. Mol. Syst. Biol. 10, 746 (2014).
Larson, A. G. et al. Liquid droplet formation by HP1ɑ suggests a role for phase separation in heterochromatin. Nature 547, 236–240 (2017).
Strom, A. R. et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017).
Greer, E. L. et al. A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep. 7, 113–126 (2014).
Kalinava, N., Ni, J. Z., Peterman, K., Chen, E. & Gu, S. G. Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans. Epigenetics Chromatin 10, 6 (2017).
Schwartz-Orbach, L. et al. Caenorhabditis elegans nuclear RNAi factor SET-32 deposits the transgenerational histone modification, H3K23me3. eLife 9, e54309 (2020).
Wang, W. et al. SET-9 and SET-26 are H3K4me3 readers and play critical roles in germline development and longevity. eLife 7, e34970 (2018).
Garrigues, J. M., Sidoli, S., Garcia, B. A. & Strome, S. Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res. 25, 76–88 (2015).
Methot, S. P. et al. H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nat. Cell Biol. 23, 1163–1175 (2021).
Padeken, J. et al. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 33, 436–451 (2019).
Andersen, E. C. & Horvitz, H. R. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development 134, 2991–2999 (2007).
Bessler, J. B., Andersen, E. C. & Villeneuve, A. M. Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLoS Genet. 6, e1000830 (2010).
Dodge, J. E., Kang, Y. K., Beppu, H., Lei, H. & Li, E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 24, 2478–2486 (2004).
Wang, H. et al. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 12, 475–487 (2003).
Funyu, T., Kanemaru, Y., Onoda, H. & Arita, K. Preparation of the ubiquitination-triggered active form of SETDB1 in Escherichia coli for biochemical and structural analyses. J. Biochem. 170, 655–662 (2021).
Koch, C. M., Honemann-Capito, M., Egger-Adam, D. & Wodarz, A. Windei, the Drosophila homolog of mAM/MCAF1, is an essential cofactor of the H3K9 methyl transferase dSETDB1/Eggless in germ line development. PLoS Genet. 5, e1000644 (2009).
Guruharsha, K. G. et al. A protein complex network of Drosophila melanogaster. Cell 147, 690–703 (2011).
Ichimura, T. et al. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J. Biol. Chem. 280, 13928–13935 (2005).
Rolland, T. et al. A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014).
Hein, M. Y. et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 (2015).
Timms, R. T., Tchasovnikarova, I. A., Antrobus, R., Dougan, G. & Lehner, P. J. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep. 17, 653–659 (2016).
Osumi, K., Sato, K., Murano, K., Siomi, H. & Siomi, M. C. Essential roles of Windei and nuclear monoubiquitination of Eggless/SETDB1 in transposon silencing. EMBO Rep. 20, e48296 (2019).
Peter, C. J. et al. In vivo epigenetic editing of Sema6a promoter reverses transcallosal dysconnectivity caused by C11orf46/Arl14ep risk gene. Nat. Commun. 10, 4112 (2019).
Tachibana, M., Matsumura, Y., Fukuda, M., Kimura, H. & Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 27, 2681–2690 (2008).
Zhang, X. et al. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell 12, 177–185 (2003).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Landry, J. et al. Set2-catalyzed methylation of histone H3 represses basal expression of GAL4 in Saccharomyces cerevisiae. Mol. Cell. Biol. 23, 5972–5978 (2003).
Schultz, D. C., Ayyanathan, K., Negorev, D., Maul, G. G. & Rauscher, F. J. 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932 (2002).
Meister, P., Towbin, B. D., Pike, B. L., Ponti, A. & Gasser, S. M. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev. 24, 766–782 (2010).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Pirkkala, L., Nykänen, P. & Sistonen, L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 15, 1118–1131 (2001).
Good, S. & van Oosten-Hawle, P. in Protein Homeostasis Diseases (ed. Pey, A. L.) Ch. 3 (Academic Press, 2020).
Morimoto, R. I. & Nollen, E. A. A. in Handbook of Cell Signaling (eds. Bradshaw, R. A. & Dennis, E. A.) Ch. 310 (Academic Press, 2003).
Jolly, C. et al. Stress-induced transcription of satellite III repeats. J. Cell Biol. 164, 25–33 (2004).
Rizzi, N. et al. Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol. Biol. Cell 15, 543–551 (2004).
Valgardsdottir, R. et al. Structural and functional characterization of noncoding repetitive RNAs transcribed in stressed human cells. Mol. Biol. Cell 16, 2597–2604 (2005).
Horard, B. et al. Global analysis of DNA methylation and transcription of human repetitive sequences. Epigenetics 4, 339–350 (2009).
Kurhanewicz, N. A., Dinwiddie, D., Bush, Z. D. & Libuda, D. E. Elevated temperatures cause transposon-associated DNA damage in C. elegans spermatocytes. Curr. Biol. 30, 5007–5017.e4 (2020).
Neueder, A. et al. HSF1-dependent and -independent regulation of the mammalian in vivo heat shock response and its impairment in Huntington’s disease mouse models. Sci. Rep. 7, 12556 (2017).
He, H. et al. Coordinated regulation of heterochromatin inheritance by Dpb3-Dpb4 complex. Proc. Natl Acad. Sci. USA 114, 12524–12529 (2017).
Dillon, N. Heterochromatin structure and function. Biol. Cell 96, 631–637 (2004).
Alper, B. J. et al. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. EMBO J. 32, 2321–2335 (2013).
Yamada, T., Fischle, W., Sugiyama, T., Allis, C. D. & Grewal, S. I. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol. Cell 20, 173–185 (2005).
Taddei, A., Maison, C., Roche, D. & Almouzni, G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol. 3, 114–120 (2001).
Terranova, R., Sauer, S., Merkenschlager, M. & Fisher, A. G. The reorganisation of constitutive heterochromatin in differentiating muscle requires HDAC activity. Exp. Cell. Res. 310, 344–356 (2005).
Hizume, K. et al. Nano-scale analyses of the chromatin decompaction induced by histone acetylation. Arch. Histol. Cytol. 73, 149–163 (2010).
Kadonaga, J. T. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 92, 307–313 (1998).
Tse, C., Sera, T., Wolffe, A. P. & Hansen, J. C. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18, 4629–4638 (1998).
Gibson, B. A. et al. Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e21 (2019).
Kim, H. et al. HDAC1 SUMOylation promotes Argonaute-directed transcriptional silencing in C. elegans. eLife 10, e63299 (2021).
Kerr, S. C., Ruppersburg, C. C., Francis, J. W. & Katz, D. J. SPR-5 and MET-2 function cooperatively to reestablish an epigenetic ground state during passage through the germ line. Proc. Natl Acad. Sci. USA 111, 9509–9514 (2014).
Rechtsteiner, A. et al. Repression of germline genes in Caenorhabditis elegans somatic tissues by H3K9 dimethylation of their promoters. Genetics 212, 125–140 (2019).
Yang, B. et al. A DNA repair protein and histone methyltransferase interact to promote genome stability in the Caenorhabditis elegans germ line. PLoS Genet. 15, e1007992 (2019).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Graether, S. P. Troubleshooting guide to expressing intrinsically disordered proteins for use in NMR experiments. Front. Mol. Biosci. 5, 118 (2018).
Singh, K. K. & Graether, S. P. Expression and purification of an intrinsically disordered protein. Methods Mol. Biol. 2141, 181–194 (2020).
Andersen, E. C. & Horvitz, H. R. Two C. elegans histone methyltransferases repress lin-3EGF transcription to inhibit vulval development. Development 134, 2991–2999 (2007).
Fields, B. D. & Kennedy, S. Chromatin compaction by small RNAs and the nuclear RNAi machinery in C. elegans. Sci. Rep. 9, 9030 (2019).
Houri-Zeevi, L., Korem Kohanim, Y., Antonova, O. & Rechavi, O. Three rules explain transgenerational small RNA inheritance in C. elegans. Cell 182, 1186–1197.e12 (2020).
Lev, I. et al. MET-2-dependent H3K9 methylation suppresses transgenerational small RNA inheritance. Curr. Biol. 27, 1138–1147 (2017).
McMurchy, A. N. et al. A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife 6, e21666 (2017).
Tian, Y. et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208 (2016).
Jiang, Q. et al. G9a plays distinct roles in maintaining DNA methylation, retrotransposon silencing, and chromatin looping. Cell Rep. 33, 108315 (2020).
Dorighi, K. M. et al. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol. Cell 66, 568–576.e4 (2017).
Paix, A., Folkmann, A. & Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods 121-122, 86–93 (2017).
Dokshin, G. A., Ghanta, K. S., Piscopo, K. M. & Mello, C. C. Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210, 781–787 (2018).
Preisach, C., Burkhardt, H., Schmidt-Thieme, L. & Decker, R. (eds) Data Analysis, Machine Learning and Applications (Springer, 2008).
Tinevez, J. Y. et al. TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Pino, L. K. et al. The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244 (2020).
Gaidatzis, D., Lerch, A., Hahne, F. & Stadler, M. B. QuasR: quantification and annotation of short reads in R. Bioinformatics 31, 1130–1132 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008).
Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012).
Acknowledgements
We thank H. Kimura for monoclonal antibodies; I. Katic and L. Xu of the FMI worm facility; J. Eglinger and L. Gelman of the FMI imaging and microscopy facility; S. Smallwood, S. Aluri and S. Thiry from the FMI Functional Genomics facility; and the FMI Protein Analysis facility for their support. We thank the Caenorhabditis Genetics Center of the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440) for strains. We thank D. Cabianca for helpful comments. J.P. and S.P.M. were supported by long-term EMBO fellowships. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Epiherigans—grant agreement no. 743312 to S.M.G.). S.M.G. also thanks the Swiss National Science Foundation (grant no. 31003A_176286) and the Novartis Research Foundation for support.
Author information
Authors and Affiliations
Contributions
C.E.D., S.P.M. and J.P. planned and performed experiments, carried out analysis where necessary and helped make figures and write the text. V.K., J.S. and D.H. planned and performed experiments and carried out analysis. S.M.G. evaluated data, provided advice, secured funding and helped write the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural and Molecular Biology thanks David Katz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Beth Moorefield was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ARLE-14 acts in parallel with LIN-65 to promote H3K9me.
(a) Representative images of live embryos expressing MET-2::mCherry from the endogenous met-2 locus, either as wild-type (WT) or tagged with a nuclear localization sequence (NLS), + /- lin-65Δ(gw1465) deletion and quantification of MET-2 foci using automated image analysis. Foci number was not determinable in lin-65;met-2::mCherry due to the lack of MET-2::mCherry nuclear enrichment. Scale bar = 5 µm. N = 3, n = 30. Nuclei: met-2::mCherry = 700, NLS::met-2::mCherry = 594, lin-65Δ;met-2::mCherry = n.d., lin-65Δ;met-2::mcherry = 557. ****p-adj = 1.8*10-11 by one-way ANOVA, followed by Tukey post-hoc test. (b) Representative image of Western blot and quantification of H3K9me2 mean signal intensity normalized to MRG-1 in total cell extracts from strains shown in (A). N = 3. **p(met-2::mCherry, lin-65Δ;met-2::mcherry)=2.4*10-5, p(NLS::met-2::mCherry, lin-65Δ;met-2::mCherry)=8.7*10-3, ns = not significant by two-sided t-test. (c) Immunofluorescence and quantification of H3K9me2 intensity per nucleus normalized to H4 in embryos. Scale bar = 5 µm. N = 3, nuclei(embryos): WT = 885(37), met-2Δ = 985(51), and arle-14(tm6845) = 826(44). Median and quartiles shown. ***p-adj < 2.0*10-16 by one-way ANOVA, followed by Tukey post-hoc test. (d) Representative western blot and quantification of H3K9me2 in three independent arle-14 mutants, met-2Δ, and lin-65Δ. N = 3. Statistics measure each mutant compared to WT. **p vs WT: met-2Δ = 4.5*10-6, arle-14(tm6748)=3.4*10-5, arle-14(gw1584)=3.4*10-5, arle-14(tm6845)=1.8*10-4, lin-65Δ = 1.1*10-3, ns = not significant by two-sided t-test. (e) Representative Western blot and quantification of MET-2::mcherry in WT and arle-14(gw1584) embryos. (-) indicates no mCherry tag present. N = 3. ns = not signficant by two-sided t-test. (f) Number of progeny per worm at 25 °C comparing arle-14(tm6748) to WT and met-2Δ. N = 2, n = 40. p(WT, met-2Δ)=1.34*10-14, p(met-2Δ, arle-14(tm6748))=2.8*10-14 by one-way ANOVA, followed by Tukey post-hoc test.
Extended Data Fig. 2 MET-2-CD deficient in HMT activity is stable in expression and interactions.
(a) Representative Western blot and quantification of H3K9me1 and H3K9me3 in WT, met-2Δ, and met-2-CD. H3K9me1 N = 3, H3K9me3 N = 4. ns=non-significant by two-sided t-test. (b) Percentage of H3K9me1/me2/me3 and H3K9ac level in WT, met-2Δ, met-2-CD and met-2 set-25 relative to WT detected by mass spectrometry normalized to the total H3K9 peptides detected. Bars indicate the mean and dots represent individual measurements. N = 3. (c) Scheme of the gwIs4 heterochromatic reporter and zoning assay with quantification of array position in the indicated genotypes. We selected a single plane from a 3D stack in which the GFP signal was brightest for scoring distance from the periphery95. Bars indicate the average and dots represent mean % from individual experiments. N = 3. foci(embryos): WT = 699(50), met-2Δ = 869(57), met-2-CD = 850(59), met-2Δ set-25Δ = 819(51), met-2-CD set-25Δ = 820(56), (d) Representative Western blots and quantification measuring mCherry signal in WT (untagged), MET-2::mCherry, and MET-2-CD::mCherry). N = 3. ns = not significant by two-sided t-test. (e) iBAQ values showing enrichment of MET-2, ARLE-14, and LIN-65 from immunoprecipitation-MS/MS using LIN-65::GFP as bait in WT or met-2-CD embryos. Bars indicate the mean and dots represent individual measurements.
Extended Data Fig. 3 MET-2-CD inhibits acetylation independent of H3K9me.
(a) Representative Western blots and quantification comparing WT, met-2Δ, and met-2-CD embryo acetylation levels. panH3ac N = 3, p(WT, met-2Δ) = 0.011, p(WT, met-2-CD) = 6.4*10-3, p(met-2Δ, met-2-CD) = 3.2*10-3; panH4ac N = 4, p(WT, met-2Δ) = 7.4*10-3, p(met-2Δ, met-2-CD) = 2.3*10-3 by two-sided t-test. (b) H3K9me1 ChIP-qPCR at representative met-2Δ-specific genes, common genes, or the met-2 independent gene unc-119. Bars indicate the mean and dots represent individual measurements. N = 3.
Extended Data Fig. 4 ARLE-14 is required for fertility and nuclear enrichment of MET-2 in met-2-CD animals.
(a) Brood sizes of the indicated strains at 25 °C. N = 3, n = 60. ns = not significant, p(WT,met-2Δ)< 2.0*10-16, p(WT,met-2-CD) = 4.2*10-6, p(met-2Δ,met-2-CD) < 2.0*10-16, p(met-2Δ, arle-14Δ met-2-CD) = 0.77,by one-way ANOVA, followed by Tukey post-hoc test. (b-c) Live imaging of embryos expressing either WT MET-2::mCherry or MET-2-CD::mCherry + /- arle-14 RNAi grown at 25 °C and (c) quantification of MET-2 nuclear signal. p(WT(vector), WT(arle-14))=not significant, p(WT(vector), met-2-CD(vector))=1.1*10-9, p(WT(vector),met-2-CD(arle-14)) < 2.0*10-16, p(met-2-CD(vector),met-2-CD(arle-14))<2.0*10-16 Low nuclear mCherry signal precluded foci quantification. Scale bar = 5 µm. N = 3 N = 3, n = 45 embryos. nuclei: met-2::mCherry: vector = 884, arle-14 RNAi = 1823, met-2-CD::mCherry: vector = 1076, arle-14 RNAi = 878. ns = not significant, ****p-adj < 10-4 compared to WT vector, ####p-adj < 10-4 compared to MET-2-CD vector by two-sided Wilcoxon signed-rank test.
Extended Data Fig. 5 Loss of met-2 or heat stress activates transcription in an HSF-1 independent manner.
(a) Quantification of foci loss shown in Fig. 5a, from ref. 14. ****p-adj=1.5*10-9 by two-sided Wilcoxon signed-rank test. (b) Genes de-repressed in both met-2Δ mutant and WT(37 °C) early embryos (Fig. 5d, black circles) are normally expressed in differentiated tissues92. Heatmap shows the expression (RPKM) in WT L3 larvae of each derepressed gene (columns) in individual tissues (rows). (c) Correlation of gene expression in WT and met-2Δ embryos both treated with HS (37 °C, 1 hr) relative to WT(20 °C). Pearson correlation coefficient (r) is displayed. (d) Expression changes of all genes designated Heat Shock Protein (hsp) in WT(37 °C) and met-2Δ relative to WT(20 °C). Pink box = log2 FC > 2 in both WT-HS and met-2Δ−20 °C. (e) Representative images of HSF-1::GFP protein in live WT or met-2Δ embryos + /- HS. HS: 37 °C, 30 min. Scale bar = 5 µm. (f) Fraction of embryos exhibiting HSF-1::GFP foci after 30 min at the indicated temperature. N = 2, n embryos = 20-60 per temperature. (g) Correlation of expression between embryos treated with HS (37 °C, 1 h) and embryos from parents grown at 25 °C6, both compared to WT(20 °C). N = 3.
Extended Data Fig. 6 Heat stress leads to permissive transcriptional state.
(a) Comparison of H3K9me2 genome wide enrichment (500 bp tiles) over input (log2) between HS (37 °C, 60 min) and WT(20 °C) embryos via ChIP-seq (N = 2). Boxplots show median, boxes 50% and whiskers 90% of each group. Correlation between replicates: HS = 0.97, WT(20 °C) = 0.98. (b) Percentage of defined genomic elements that overlap with peaks differentially enriched for H3K27ac in HS treatment (37 °C, 60 min), met-2Δ, and met-2-CD embryos relative to WT(20 °C). (c) Representative Western blot and quantification of MET-2::mcherry in embryos + /- HS: 37 °C, 60 min. N = 3. *p = 0.050 by two-sided t-test. (d-e) Log2 FC of met-2Δ-specific genes, met-2Δ»met-2-CD, and common genes in HS (37 °C, 60 min), met-2Δ and met-2-CD and (d) mean fold change observed in HS in each gene set. N = 3. Boxplots show median, boxes 50% and whiskers 90% of each group. (f-g) Correlations between RNAseq replicas for (e) WT and met-2Δ + /- HS (37 °C, 60 min), and (f) WT, met-2Δ, met-2-CD, arle-14Δ, and arle-14Δ met-2-CD.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Delaney, C.E., Methot, S.P., Kalck, V. et al. SETDB1-like MET-2 promotes transcriptional silencing and development independently of its H3K9me-associated catalytic activity. Nat Struct Mol Biol 29, 85–96 (2022). https://doi.org/10.1038/s41594-021-00712-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-021-00712-4
This article is cited by
-
Fasting shapes chromatin architecture through an mTOR/RNA Pol I axis
Nature Cell Biology (2024)
-
SetDB1 and Su(var)3-9 are essential for late stages of larval development of Drosophila melanogaster
Chromosome Research (2023)
-
Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance
Nature Reviews Molecular Cell Biology (2022)
