
Abstract
The Middle East respiratory syndrome coronavirus (MERS-CoV) causes severe and often lethal respiratory illness in humans, and no vaccines or specific treatments are available. Infections are initiated via binding of the MERS-CoV spike (S) glycoprotein to sialosides and dipeptidyl-peptidase 4 (the attachment and entry receptors, respectively). To understand MERS-CoV engagement of sialylated receptors, we determined the cryo-EM structures of S in complex with 5-N-acetyl neuraminic acid, 5-N-glycolyl neuraminic acid, sialyl-LewisX, α2,3-sialyl-N-acetyl-lactosamine and α2,6-sialyl-N-acetyl-lactosamine at 2.7–3.0 Å resolution. We show that recognition occurs via a conserved groove that is essential for MERS-CoV S-mediated attachment to sialosides and entry into human airway epithelial cells. Our data illuminate MERS-CoV S sialoside specificity and suggest that selectivity for α2,3-linked over α2,6-linked receptors results from enhanced interactions with the former class of oligosaccharides. This study provides a structural framework explaining MERS-CoV attachment to sialoside receptors and identifies a site of potential vulnerability to inhibitors of viral entry.
Similar content being viewed by others

Main
The Middle East respiratory syndrome coronavirus (MERS-CoV) was discovered in 2012 on the Arabian Peninsula as the causative agent of acute respiratory distress (and renal failure), with an unusually high fatality rate of ~35% (ref. 1). The vast majority of the 2,482 confirmed cases were reported in the Middle East, although the virus was introduced into 27 countries. A total of 203 new MERS-CoV cases were reported in 2019 and there is currently no vaccine or cure for MERS or any coronavirus-induced diseases in humans. Zoonotic MERS-CoV transmission from dromedary camels (the host reservoir for this virus) along with human-to-human transmission are the two known modes of infection2,3. Future emergence of (zoonotic) coronaviruses is likely to occur, given the large reservoir in bats and birds, underscoring the importance of studying these pathogens4,5,6,7.
MERS-CoV is an enveloped Nidovirus decorated with homotrimers of the spike (S) glycoprotein that mediates entry into host cells. S is the major antigen present at the viral surface and is the target of neutralizing antibodies during infection as well as the focus of vaccine design. Recent cryo-EM structures of MERS-CoV and related coronavirus S ectodomain trimers provided snapshots of this key protein in prefusion8,9,10,11,12,13,14,15 and postfusion16 conformations, receptor-bound states17,18,19 and in complex with neutralizing antibodies20,21. MERS-CoV S is composed of an N-terminal S1 subunit, which is folded as four domains (A–D) and mediates attachment to dipeptidyl-peptidase 4 (DPP4, the host receptor)22, and a C-terminal S2 subunit that merges the viral and cellular membranes to initiate infection. MERS-CoV S is cleaved at the junction between the S1 and S2 subunits either during viral biogenesis or upon encounter of target cells23,24,25,26. A second cleavage site, designated S2′, is found upstream of the fusion peptide in the S2 subunit and is processed at the onset of membrane fusion. Both cleavage sites participate in enhancing viral entry and modulating host range and cell tropism23,24,27.
Sialic acids (derivatives of neuraminic acid) are ubiquitous carbohydrates found as terminal residues on glycoproteins and glycolipids decorating the surface of eukaryotic cells. Neuraminic acid modifications, along with the formation of specific glycosidic linkages, result in a wide chemical variety of sialoglycoconjugates across cell types, tissues and animal species28. As a result, differential sialoside recognition can profoundly impact the zoonotic transmission, tropism and virulence of many viruses. For example, a few amino acid substitutions in influenza virus hemagglutinins account for the preference switch from avian enteric tract to human respiratory tract sialylated receptors29,30,31.
MERS-CoV primarily infects human lung epithelial cells upon interacting with DPP4 (ref. 22). Crystal structures of the MERS-CoV S domain B in complex with the DPP4 ectodomain32,33, along with cryo-EM structures of the MERS-CoV S ectodomain trimer15,20,21, have furthered our understanding of the mechanism of DPP4 engagement. In addition to attachment to DPP4, we recently showed that MERS-CoV infection of human airway epithelial cells involves low-affinity interactions with sialosides, using the S glycoprotein domain A, as depletion of sialic acid from the cell surface dampened viral entry34. A binding preference was found for α2,3-linked over α2,6-linked sialosides, and these interactions were hindered by 9-O-acetylation or 5-N-glycolylation of the terminal neuraminic acid34. Furthermore, we found that MERS-CoV S sialylated receptors are abundant in the camel nasal respiratory epithelium and the human lung alveoli, which coincides with DPP4 expression and the sites of MERS-CoV replication in these mammals34,35. As sialoside modifications, linkages and distribution vary among and within host species, the selectivity of MERS-CoV S for certain sialoside glycotopes may provide a determinant of host and tissue tropism of this zoonotic pathogen. These findings provided a plausible rationale for explaining that equine DPP4 could support MERS-CoV infection of cultured cells, although horses were found to be resistant to experimental MERS-CoV infection (despite the presence of DPP4 in their respiratory tract)36,37,38. Although sialoside attachment appears to be a key step modulating MERS-CoV infection, the location of the sialoside-binding site, the interactions involved in ligand recognition and the molecular basis for receptor specificity remain unknown. This information is crucial for understanding viral tropism and infectivity, assessing the zoonotic potential of MERS-CoV and related coronaviruses, and providing a blueprint for the design of coronavirus inhibitors.
To understand the structural basis of MERS-CoV attachment to and specificity for host sialosides, we determined cryo-EM structures of the S glycoprotein ectodomain trimer in complex with 5-N-acetyl neuraminic acid (Neu5Ac), 5-N-glycolyl neuraminic acid (Neu5Gc), sialyl-LewisX (sLeX), α2,3-sialyl-N-acetyl-lactosamine (2,3-SLN) and α2,6-sialyl-N-acetyl-lactosamine (2,6-SLN) at 2.7, 3.0, 2.7, 2.7 and 2.9 Å resolution, respectively. We demonstrate that the receptor binds in a groove located at the surface of domain A that is distinct from the 9-O-acetyl sialoside-engagement site identified for HCoV-OC43 and related β-1 coronavirus S glycoproteins19,39. We further show that the residues involved in Neu5Ac recognition are conserved across MERS-CoV isolates and essential for MERS-CoV S-mediated hemagglutination of human erythrocytes and entry into human airway epithelial cells. Our data rationalize MERS-CoV S attachment to neuraminic acids that are not 9-O-acetylated or 5-N-glycolylated and suggest an explanation for the selectivity for α2,3-linked over α2,6-linked sialosides. This study provides a structural framework for understanding MERS-CoV S sialoglycan receptor engagement and specificity, illuminates host range and cell tropism, and identifies a site of vulnerability accessible to neutralizing antibodies and small-molecule inhibitors.
Results
A sialoside-binding site in the MERS-CoV S glycoprotein
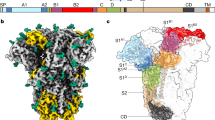
To identify the MERS-CoV S sialoside-recognition site, we determined a cryo-EM structure of the MERS-CoV S 2P stabilized ectodomain trimer21 incubated with 100 mM Neu5Ac at 2.7 Å resolution (Fig. 1a, Extended Data Figs. 1a and 2a–g and Table 1). The presence of the sialoside stabilized a large fraction of MERS-CoV S trimers in the closed state, with all three DPP4-binding B domains adopting a down conformation, obeying three-fold symmetry. We did not detect major conformational changes compared to closed protomers of the LCA60 Fab-bound MERS-CoV S structure20 (Cα r.m.s. deviation (r.m.s.d.) of 1.35 Å for 1,145 aligned residues). The resolution estimate of our map is supported by the detection of ordered water molecules40 interacting with the S glycoprotein (Extended Data Fig. 1f,g) and by the observation of folic acid bound to S, as previously identified using X-ray crystallography and mass spectrometry of isolated domain A21. The density is equally well resolved for the ligand and the surrounding S amino acid residues, allowing unambiguous identification of the binding site and docking of Neu5Ac in the map (Extended Data Fig. 1d). The ligand interacts with a groove located at the periphery of domain A that is distinct from the HCoV-OC43 S 9-O-acetyl sialoside-binding site19 (Figs. 1a–e and 2a–c). The absence of N-linked glycans in the immediate vicinity of the binding groove probably facilitates unobstructed engagement of sialoglycoconjugates at the surface of target cells (Fig. 2a). The ligand-binding site is located ~50 and ~75 Å away from the DPP4-binding site of a neighboring B domain in the closed or open state, respectively (Fig. 1b).

a, Molecular surface representation of the MERS-CoV S ectodomain trimer, with each protomer in a different color. The Neu5Ac ligands are rendered as spheres. b, Spatial relationships between the attachment (sialoside) and entry (DDP4) receptor-binding sites are illustrated using a composite model obtained from the cryo-EM structure shown in a, the cryo-EM structure of MERS-CoV S bound to the LCA60 neutralizing antibody Fab fragment (PDB 6NB3)20 and a crystal structure of the MERS-CoV S domain B bound to DPP4 (PDB 4KR0)32. The sialoside-binding site is located ~50 and ~75 Å away from the DPP4-binding site of a neighboring B domain in the closed or open state, respectively. c, Surface representation of the ligand-binding site colored by electrostatic potential from −12 (red) to +10 (blue) kBT/ec. Neu5Ac is shown in stick representation. d,e, Two orthogonal views of the sialoside-binding site rendered as a ribbon diagram with the side chains of key surrounding residues shown as sticks. Neu5Ac is rendered as sticks and the corresponding region of cryo-EM density is shown as a blue mesh contoured at 5.5σ. Dashed lines show selected electrostatic interactions formed between MERS-CoV S amino acid residues and the ligand. In all panels, nitrogen and oxygen atoms are colored blue and red, respectively, while carbon atoms are colored gray (Neu5Ac) or pink (MERS-CoV S). Glycans in MERS-CoV S have been omitted for clarity.

a,b, Molecular surface representation of the MERS-CoV S (a) and HCoV-OC43 S (b) A domains bound to Neu5Ac and 5-N-acetyl,9-O-acetyl neuraminic acid α-methyl glycoside (9-O-Ac-Neu5Ac, PDB 6NZK19), respectively, and oriented identically. The sialosides are rendered as sticks with carbon, nitrogen and oxygen atoms colored gray, blue and red, respectively. N-linked glycans observed in the cryo-EM reconstructions are rendered as dark blue spheres. c, Ribbon diagrams of superimposed MERS-CoV S (pink) and HCoV-OC43 S (gold) A domains with bound sialosides. The sialosides are rendered as sticks with carbon, nitrogen and oxygen atoms colored pink (MERS-CoV S) or gold (HCoV-OC43 S), blue and red, respectively.
The ligand C1 carboxylate electrostatically interacts with the Ser133 side chain hydroxyl, and the 5-nitrogen atom is hydrogen-bonded to the Ile132 backbone carbonyl (Fig. 1c–e). The Neu5Ac hydroxyl group at position 8 is hydrogen-bonded to the Arg307 side chain guanidium whereas the hydroxyl group at position 9 interacts with the Arg307 guanidium as well as the Ala92 backbone amide and carbonyl oxygen via hydrogen bonding (Fig. 1c–e). The ligand N-acetyl methyl inserts into a hydrophobic depression defined by residues Phe39, Phe101, Ile131 and Ile132 whereas the N-acetyl carbonyl oxygen is hydrogen-bonded to the Gln36 side chain amide (Fig. 1c–e). The sialoside buries 280 Å2 of it surface upon MERS-CoV S engagement, corresponding to 61% of the ligand total accessible surface area, with most interactions occurring on the same side of the ligand. Bioinformatics analysis of MERS-CoV isolates shows that the residues contacting the sialoside or participating in the formation of the binding groove are conserved. Specifically, Gln36, Phe39, Phe101, Ile132 and R307 are strictly conserved in the 284 MERS-CoV isolates sequenced so far. The H91Y substitution is observed in four MERS-CoV isolates (human/Korea/Seoul/168–1–2015 GB, ALB08311; human/Korea/Seoul/168-2-2015 GB, ALB08322; human/KOR/Seoul/014-2015 GB, ANC28634.1; human/Riyadh_1337/KSA/2014 GB, AMW90853.1), the S133N or S133R substitutions are present in two isolates (human/England/4/2013 GB, AJD81440.1 and Hu/Quaseem-KSA-18012872/2018 GB, QBF80608.1, respectively) and the Q304K substitution is observed in 1 isolate (camel/UAE/D1164.11/2014 GB, AJG44080.1).
The architecture of the binding groove suggests 9-O-acetylation of the neuraminic acid could not be sterically accommodated (Extended Data Fig. 3a), in agreement with the previously reported distinct specificity of MERS-CoV S compared to HCoV-OC43 S34. To assess if 5-N-glycolylation of the neuraminic acid could be tolerated, we determined a cryo-EM structure of MERS-CoV S 2P incubated with 100 mM Neu5Gc at 3.0 Å resolution (Extended Data Figs. 1b, 3b–f and 4a–d and Table 1). Comparison of this map with the MERS-CoV S/Neu5Ac map shows that the Neu5Gc density is weaker than that of the Neu5Ac and the glycolyl hydroxyl group could not be resolved, even after extensive focused 3D classification of the data (Extended Data Fig. 3b–f). This observation is in line with the reported MERS-CoV S binding preference to Neu5Ac compared to Neu5Gc based on the lack of hemagglutination of (Neu5Gc-rich) horse erythrocytes and absence of binding to Neu5Gc-containing sialosides on a glycan array34. The structure suggests the MERS-CoV S hydrophobic pocket defined by Phe39, Phe101, Ile131 and Ile132 could sterically and/or chemically disfavor accommodation of the 5-N-glycolyl hydroxyl group in the conformation observed in virtually all Neu5Gc-containing protein complex structures (Extended Data Fig. 3b–f). The high concentrations of sialosides used for the cryo-EM experiments, however, allowed us to visualize Neu5Gc bound to MERS-CoV S, much in the same way that crystal structures of influenza virus hemagglutinins in complex with mismatched receptor analogs or of rhesus rotavirus VP8* bound to Neu5Gc were obtained in spite of very weak or lack of detectable interactions in solution29,30,41. Because the equine respiratory tract is abundantly decorated with Neu5Gc-containing oligosaccharides42, these data collectively suggest a plausible explanation for the resistance of horses to experimental MERS-CoV infection, despite the fact MERS-CoV S can use equine DPP4 for promoting entry into target cells in vitro34,36,38.
The sialoside binding site is required for S-mediated viral attachment and entry
To evaluate the importance of the interactions identified by cryo-EM for sialoside recognition, we substituted with alanine individual MERS-CoV S amino acid residues interacting with Neu5Ac and assessed the effect on domain A-mediated hemagglutination of human erythrocytes. As previously described, the wild-type MERS-CoV S domain A fused to human immunoglobulin Fc only promoted hemagglutination of human erythrocytes when multimerized on lumazine synthase nanoparticles34, indicative of enhancement of low-affinity interactions via avidity (Fig. 3a and Extended Data Fig. 5a). Complete inhibition of hemagglutination was observed, however, for each binding site mutant tested, that is, F39A, H91A, I132A, S133A and R307A (Fig. 3a and Extended Data Fig. 5a). These results validated the structural observations by demonstrating that recognition of Neu5Ac via these residues is necessary for MERS-CoV S-mediated attachment to sialosides.

a, Hemagglutination assay using MERS-CoV S wild type (WT) or mutants. Wells positive for hemagglutination (HA) are encircled. F39A, H91A, I132A, S133A or R307A substitutions completely abrogated domain A-Fc-mediated hemagglutination of human erythrocytes. Mock-treated erythrocytes, absence of the protein A-fused lumazine synthase (pA-LS) or replacement of domain A with domain B (Dom. B) were used as negative controls. The assays were performed four times and a representative experiment is shown. b, MERS-CoV S F39A, H91A, S133A or R307A substitutions inhibited entry of pseudotyped murine leukemia viral particles into human airway Calu-3 cells. Data are normalized relative to wild type and shown as mean and s.d. of n = 3 pseudovirus experiments (technical replicates).
To further assess the functional relevance of the identified interactions, we interrogated the impact of individual domain A substitutions on MERS-CoV S-mediated entry into target cells. We used the murine leukemia virus platform with a luciferase reporter43 for quantifying entry of particles pseudotyped with either wild-type or mutant MERS-CoV S into human airway Calu-3 cells. The S133A and R307A substitutions abrogated entry of pseudotyped particles due to disruption of the aforementioned electrostatic interactions involving the Neu5Ac C1 carboxylate and glycerol side chain, respectively (Fig. 3b and Extended Data Fig. 5b). The F39A and H91A mutants showed reduced infectivity of the pseudotyped particles by more than 90%, probably due to disruption of the ligand-binding groove and loss of favorable van der Waals or electrostatic interactions (Fig. 3b and Extended Data Fig. 5b). These findings demonstrate the key role of the identified residues for interacting with Neu5Ac in the context of a full-length, membrane-embedded MERS-CoV S glycoprotein and show that attachment to sialoside receptors using the binding site identified by cryo-EM is essential for promoting MERS-CoV S-mediated entry into human airway Calu-3 cells. Furthermore, as sialidase treatment of Calu-3 cells34 or mutations of the S sialoside-recognition site abrogated MERS-CoV S-mediated entry, it is likely that sialoside attachment either precedes DPP4 engagement or occurs simultaneously.
MERS-CoV S specificity for α2,3-linked sialoside receptors
We previously showed that the MERS-CoV S domain A preferentially interacts with α2,3-linked, compared to α2,6-linked, sialosides, with sulfated sLeX being the preferred binder34. To understand the architectural principles underlying this specificity, we determined three cryo-EM structures of MERS-CoV S 2P in the presence of 50 mM sLeX (Extended Data Figs. 1c and 6 and Table 1), 70 mM 2,3-SLN (Extended Data Figs. 1d and 7, and Table 1) or 70 mM 2,6-SLN (Extended Data Figs. 1e and 8 and Table 1) at 2.7, 2.7 and 2.9 Å resolution, respectively.
The cryo-EM reconstruction obtained for MERS-CoV S in complex with sLeX resolves the terminal Neu5Ac linked to galactose, via a 2,3-glycosidic bond adopting a cis configuration, as well as the N-acetyl glucosamine (GlcNAc) and fucose rings at lower contour level due to conformational heterogeneity of the ligand (Fig. 4a,b). Engagement of the Neu5Ac moiety of the sLeX involves identical interactions to those observed in our structure of MERS-CoV S in complex with Neu5Ac (Fig. 1c–e). This finding, and the detection of a galactose moiety linked to the oxygen at position 2 of Neu5Ac, validate our docking of the ligand in the cryo-EM density. The sLeX trisaccharide core (Neu5Ac(α2–3)galactose(β1–4)GlcNAc) adopts a roughly linear conformation when bound to MERS-CoV S (Fig. 4a,b), as is the case for influenza virus hemagglutinins in complex with α2,3-sialosides29,31,44. The MERS-CoV S structure in complex with 2,3-SLN shows a very similar pose of the ligand to that observed for sLeX, although the GlcNAc is even more conformationally heterogeneous in the trisaccharide compared to sLeX and was therefore not modeled (Fig. 4c,d).

a–f, Enlarged views of the MERS-CoV S structures in complex with sLeX (a,b), 2,3-SLN (c,d) and 2,6-SLN (e,f). The cryo-EM densities corresponding to the ligands are shown as a blue mesh contoured at 3.7σ (a), 3.2σ (b), 5.8σ (c,d) and 4.5σ (e,f). g,h, The ligand-binding site with the sLeX and 2,6-SLN-bound MERS-CoV S structures superimposed, showing the distinct orientation of the two ligands relative to domain A. The asterisks indicate the proximity of the sLeX GlcNAc to the binding site, whereas the 2,6-SLN GlcNAc points away towards the solvent. In a–d, the A domain is rendered as a ribbon diagram with selected side chains of residues surrounding the sialoside shown as sticks. The ligands are rendered as sticks with atoms colored by elements (carbon, cyan (sLeX), green (2,3-SLN) or orange (2,6-SLN); nitrogen, blue; oxygen, red). Neu5Ac, 5-N-acetyl neuraminic acid; GlcNAc, N-acetyl glucosamine; Fuc, fucose; Gal, galactose.
The cryo-EM map obtained for MERS-CoV S bound to 2,6-SLN resolves the Neu5Ac and galactose moieties, linked via a 2,6-glycosidic bond adopting a cis configuration, and the GlcNAc ring at lower contour level due to conformational variability of the ligand (Fig. 4e,f). The 2,6-SLN has a U-shaped conformation, comparable to reports on influenza virus hemagglutinins29,30, that positions the GlcNAc moiety away from the binding groove and the rest of the S glycoprotein (Fig. 4e,f).
The cryo-EM structures suggest an explanation for the finding that sulfated sLeX is the preferred MERS-CoV S sialoside ligand34. Although the sLeX conformational dynamic limited its resolvability, the galactose and the GlcNAc moieties appear to be positioned within contact distance of the MERS-CoV S domain A due to the ligand conformation (Fig. 4a–d,g,h). Furthermore, sulfation of the GlcNAc hydroxyl group at position 6 could putatively allow formation of electrostatic interactions with the Gln304 side chain or neighboring residues (Fig. 4g,h), as described for influenza virus A/Vietnam/1194/2004 hemagglutinin44. The bent architecture of 2,6-SLN, however, orients the GlcNAc away from the binding site, hindering contacts with S (which appear limited to the Neu5Ac and galactose moieties). We therefore hypothesize that the trend of preferential attachment to α2,3- over α2,6-sialosides results from enhanced interactions with the former type of ligand, leading MERS-CoV to target cell types presenting such sialylated receptors at their surface.
Discussion
We show here that sialosides bind to MERS-CoV S using a site distinct from the one observed for HCoV-OC43 S in complex with 9-O-acetyl sialylated receptors19,39, despite the fact that the bound ligands are only separated by a few ångstroms from each other when superimposing the two structures. Although the β-sandwich architecture of domain A is conserved among all coronaviruses, distinct variable loops are grafted onto it, and neither the MERS-CoV S nor the HCoV-OC43 S sialoside-binding site is conserved in the infectious bronchitis virus S glycoprotein10, which is known to engage α2,3-sialosides at the surface of avian host cells45. These findings suggest coronavirus S glycoproteins have independently evolved the ability to recognize sialosides using domain A, similar to their ability to engage different proteinaceous receptors via modifications of domain B. Our results therefore set coronavirus S glycoproteins apart from influenza virus A hemagglutinins, which recognize the Neu5Ac moiety through a largely conserved binding site29,30. We hypothesize that coronaviruses that utilize both an entry receptor and an attachment receptor for infection, such as MERS-CoV or transmissible gastroenteritis virus, have more evolutionary freedom with respect to their ability to attach to sialoglycans than viruses only using an entry receptor. This could have led to the independent loss and/or acquisition of a sialoside-recognition function as an adaption to new hosts upon cross-species transmission of distinct viruses. The ability to interact with sialoglycoconjugates might be a recent MERS-CoV S acquisition, as neither HKU4 S1 nor HKU5 S1 could hemagglutinate human erythrocytes34. This modular evolutionary plasticity underlies coronavirus cross-species transmission, switching of host cell types and viral fitness.
We previously suggested that MERS-CoV S interacts with sialoside receptors through lower-affinity interactions than influenza A/California/04/2009 hemagglutinin (containing the T200A and E227A substitutions), as multimerization with a lumazine synthase nanoparticle was required to detect binding of a dimeric MERS-CoV S domain A-Fc construct but not of a dimeric hemagglutinin ectodomain-Fc construct34. Because influenza A virus utilizes neuraminidase to promote the release of progeny virions from host cells, off-target cells or decoys, the enhanced affinity for sialosides is balanced with a receptor-destroying enzyme. Similarly, HCoV-OC43 possesses a hemagglutinin esterase with receptor-destroying activity46 to achieve the same function. Because MERS-CoV does not encode an equivalent enzyme, the binding dynamic of viral particles to sialoside receptors is entirely based on the energetics of these reversible interactions34. The strong dependence on interactions with sialosides for MERS-CoV entry into human airway Calu-3 cells is therefore probably a result of the high concentration of sialylated receptors34, favoring virus adsorption through avidity and potentially increasing the likelihood of binding to DPP4, leading to viral entry. Similar roles have been proposed for carcinoembryonic antigen-related cell adhesion molecule 5 (ref. 47) and binding immunoglobulin protein48.
Although MERS-CoV S is densely decorated with N-linked glycans, the sialoside-recognition site is unobstructed, a possible requirement for receptor engagement. This finding is in marked contrast with the conformational masking and/or glycan shielding of the domain B entry receptor-binding motifs observed for several coronavirus S glycoproteins, including MERS-CoV S13,15,18,20,21. We recently identified an antibody-blocking domain A-mediated hemagglutination of erythrocytes with in vitro neutralizing activity of MERS-CoV S pseudovirions and partial in vivo prophylactic protection of mice challenged with a lethal dose of MERS-CoV49. These findings, along with our mutagenesis data, indicate that the sialoside-binding groove identified here represents a key site of vulnerability to inhibitors of MERS-CoV infection that could be targeted for the future development of therapeutics.
Methods
Cell lines
HEK293F is a cell line adapted to grow in suspension (Life Technologies). HEK293T/17 cells (ATCC, catalog no. CRL-11268) and human hepatic Huh-7 cells (National Institutes of Biomedical Innovation, Health and Nutrition, catalog no. JCRB0403) are adherent cells. Cell lines were not authenticated or tested for mycoplasma contamination.
Protein expression and purification
The MERS-CoV S 2P ectodomain was produced in 500 ml HEK293F cells grown in suspension using FreeStyle 293 expression medium (Life Technologies) at 37 °C in a humidified 8% (vol/vol) CO2 incubator rotating at 130 r.p.m. The cultures were transfected using 293fectin (ThermoFisher Scientific) with cells grown to a density of one million cells per ml and cultivated for three days. The supernatants were harvested and cells resuspended for another three days, yielding two harvests. Clarified supernatants were purified using a 5 ml Cobalt affinity column (Takara). Purified protein was concentrated and flash-frozen in Tris-saline (50 mM Tris, pH 8.0, 150 mM NaCl) before cryo-EM analysis.
Protein A-fused lumazine synthase nanoparticles (a self-assembling 60-meric lumazine synthase nanoparticle, N-terminally extended with the immunoglobulin Fc-binding domain of the Staphylococcus aureus protein A) and Fc-tagged domain A (residues 19–357) or Fc-tagged domain B (residues 358–588) of the MERS-CoV S protein (GB, YP_009047204.1) were expressed and purified as previously described34. Purified proteins were analyzed on a 12% (wt/vol) SDS–PAGE gel under reducing conditions and stained with GelCodeBlue stain reagent (Thermo Scientific).
Cryo-electron microscopy sample preparation and data collection
Incubation of 1 μM MERS-CoV S 2P stabilized ectodomain trimer with 100 mM Neu5Ac (Millipore 110138), 100 mM Neu5Gc (Miilipore 362000), 50 mM sLeX (Carbosynth OS04058), 70 mM 2,3-SLN (Carbosynth OS06484) or 70 mM 2,6-SLN (Carbosynth OS09313) was performed overnight at 4 °C. We used 100 mM Neu5Ac based on our previous work on HCoV-OC43 S19 and work on influenza virus30, whereas the lower concentrations used for larger ligands were determined empirically based on the contrast of cryo-EM micrographs. A 3 μl volume of 0.16 mg ml−1 MERS S-sialoglycan complex was loaded onto a freshly glow discharged (30 s at 20 mA) lacey carbon grid with a thin layer of evaporated continuous carbon before plunge freezing using a vitrobot MarkIV (ThermoFisher Scientific) using a blot force of −1 and 2.5 s blot time at 100% humidity and 25 °C. Data were acquired using an FEI Titan Krios transmission electron microscope operated at 300 kV and equipped with a Gatan K2 Summit direct detector and Gatan Quantum GIF energy filter, operated in zero-loss mode with a slit width of 20 eV. Automated data collection was carried out using Leginon50 at a nominal magnification of ×130,000 and a pixel size of 0.525 Å. The dose rate was adjusted to 8 counts per pixel per second, and each video was acquired in super-resolution mode fractionated in 50 frames of 200 ms.
Cryo-electron microscopy data processing
Video frame alignment, estimation of the microscope contrast-transfer function parameters, particle picking and extraction were carried out using Warp51. Particle images were extracted with a box size of 800 binned to 400 yielding a pixel size of 1.05 Å. For each dataset, two rounds of reference-free two-dimensional (2D) classification were performed using Relion52 to select well-defined particle images. Subsequently, two rounds of 3D classification with 50 iterations each (angular sampling of 7.5° for 25 iterations and 1.8° with local search for 25 iterations) were carried out using Relion without imposing symmetry to select closed trimers with the three B domains in the down conformation. 3D refinements imposing C3 symmetry were carried out using non-uniform refinement in cryoSPARC53 using an initial model generated by converting a modified version of PDB 6NB3 (ref. 20) with all three B domains closed to a map low-pass filtered to 30 Å resolution. Particle images were subjected to the Bayesian polishing procedure implemented in Relion3.0 (ref. 54) before performing another round of 3D refinement in Relion followed by per-particle defocus refinement and, again, 3D refinement in Relion. The particles were then subjected to 3D classification without refining angles and shifts using a soft mask delineating the ligand-binding site and with a tau value of 60. Final 3D refinements were carried out using non-uniform refinement in cryoSPARC. Local resolution estimation, filtering and sharpening were carried out using cryoSPARC. Reported resolutions are based on the gold-standard Fourier shell correlation (FSC) of 0.143 criterion and Fourier shell correlation curves were corrected for the effects of soft masking by high-resolution noise substitution55.
Model building
We used UCSF Chimera56 to fit the MERS-CoV S atomic model (PDB 6NB3) into the cryo-EM map followed by subsequent manually rebuilding using Coot57,58. N-linked glycans were hand-built into the density where visible, and then refined and relaxed using Rosetta59,60,61,62,63,64. Glycan refinement relied on a dedicated Rosetta protocol, which uses physically realistic geometries based on prior knowledge of saccharide chemical properties62, and was aided by using both sharpened and unsharpened maps. The final models were analyzed using MolProbity65, EMRinger66, Privateer67 and PISA68. Figures were generated using UCSF ChimeraX69.
Pseudovirus entry assay
Murine leukemia virus pseudotyped with MERS-CoV S was prepared as previously described43. HEK293T/17 cells were co-transfected with a full-length MERS-CoV S or MERS-CoV S mutant encoding plasmid, a murine leukemia virus Gag-Pol packaging construct and the murine leukemia virus transfer vector encoding a luciferase reporter using the Lipofectamine 2000 or 3000 transfection reagent (Life Technologies) according to the manufacturer’s instructions. Cells were incubated for 5–12 h at 37 °C and 8% CO2 with transfection medium. Cells were then washed with DMEM twice and DMEM containing 10% (vol/vol) FBS was added for 72 h. The supernatants were then harvested and filtered through 0.45 μm membranes before concentration with a 30 kDa centrifugal concentrator membrane (Amicon).
Calu-3 cells grown in DMEM containing 10% (vol/vol) FBS and 1% (vol/vol) PenStrep were plated into 96-well plates following trypsinization at a density of ~3.2 × 104 cells per ml and allowed to grow for 48 h at 37 °C with 8% (vol/vol) CO2. The cells were washed three times with DMEM before addition of 40 μl of MERS-CoV S or MERS-CoV S mutant pseudovirus for 2 h at 37 °C with 8% (vol/vol) CO2. Following the 2 h incubation, 40 μl of 20% (vol/vol) FBS and 2% (vol/vol) PenStrep was added to the wells and the plate was incubated for 72 h. ONE-Glo-EX was added in equivolume (~75 μl to account for evaporation) and the luciferase counts were read on a Varioskan plate reader.
Western blotting
SDS–PAGE (4×) loading buffer was added to all concentrated pseudovirus samples. The samples were run on a 4–15% (wt/vol) gradient Tris-glycine gel (BioRad) and transferred to PVDF membranes. An anti-S2 MERS-CoV S purified 1.6c7 primary antibody49 (1:500 dilution) and an Alexa Fluor 680-conjugated goat anti-human secondary antibody (1:40,000 dilution, Jackson Laboratory) were used for western blotting. A LI-COR processor was used to develop images.
Hemagglutination assay
High-sensitivity nanoparticle-based hemagglutination assays were performed as previously described34. Briefly, wild-type or mutant MERS-CoV S domain A-Fc (starting at 6.77 μg per well) was complexed with protein A-fused lumazine synthase nanoparticles at a 0.6:1 molar ratio for 30 min on ice. Mixtures were subsequently serially diluted (twofold dilutions) and mixed 1:1 with human erythrocytes (0.5% (wt/vol) in phosphate buffered saline) in V-bottomed, 96-well plates. Hemagglutination was scored after 2 h of incubation at 4 °C.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The cryo-EM maps (sharpened and unsharpened) and atomic models have been deposited to the EMDB and wwPDB under accession numbers EMD-20542 and PDB 6Q04 (MERS-CoV S + Neu5Ac), EMD-20829 (MERS-CoV S + Neu5Gc), EMD-20543 and PDB 6Q05 (MERS-CoV S + sLeX), EMD-20544 and PDB 6Q06 (MERS-CoV S + 2,3-SLN) and EMD-20545 and PDB 6Q07 (MERS-CoV S + 2,6-SLN). Source Data are available online for Extended Data Fig. 5.
References
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012).
Haagmans, B. L. et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 14, 140–145 (2014).
Sabir, J. S. et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351, 81–84 (2016).
Woo, P. C. et al. Molecular diversity of coronaviruses in bats. Virology 351, 180–187 (2006).
Menachery, V. D. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513 (2015).
Menachery, V. D. et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl Acad. Sci. USA 113, 3048–3053 (2016).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Gui, M. et al. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 27, 119–129 (2017).
Kirchdoerfer, R. N. et al. Pre-fusion structure of a human coronavirus spike protein. Nature 531, 118–121 (2016).
Shang, J. et al. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 14, e1007009 (2018).
Shang, J. et al. Cryo-EM structure of porcine delta coronavirus spike protein in the pre-fusion state. J. Virol. 92, e01556–17 (2018).
Walls, A. C. et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114–117 (2016).
Walls, A. C. et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 23, 899–905 (2016).
Xiong, X. et al. Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J. Virol. 92, e01628–17 (2017).
Yuan, Y. et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 8, 15092 (2017).
Walls, A. C. et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl Acad. Sci. USA 114, 11157–11162 (2017).
Kirchdoerfer, R. N. et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 8, 15701 (2018).
Song, W., Gui, M., Wang, X. & Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 14, e1007236 (2018).
Tortorici, M. A. et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 26, 481–489 (2019).
Walls, A. C. et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 176, 1026–1039 (2019).
Pallesen, J. et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl Acad. Sci. USA 114, E7348–E7357 (2017).
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013).
Park, J. E. et al. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl Acad. Sci. USA 113, 12262–12267 (2016).
Millet, J. K. & Whittaker, G. R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl Acad. Sci. USA 111, 15214–15219 (2014).
Earnest, J. T. et al. The tetraspanin CD9 facilitates MERS-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 13, e1006546 (2017).
Earnest, J. T., Hantak, M. P., Park, J. E. & Gallagher, T. Coronavirus and influenza virus proteolytic priming takes place in tetraspanin-enriched membrane microdomains. J. Virol. 89, 6093–6104 (2015).
Yang, Y. et al. Two mutations were critical for bat-to-human transmission of Middle East respiratory syndrome coronavirus. J. Virol. 89, 9119–9123 (2015).
Stencel-Baerenwald, J. E., Reiss, K., Reiter, D. M., Stehle, T. & Dermody, T. S. The sweet spot: defining virus–sialic acid interactions. Nat. Rev. Microbiol. 12, 739–749 (2014).
Xiong, X. et al. Receptor binding by a ferret-transmissible H5 avian influenza virus. Nature 497, 392–396 (2013).
Xiong, X. et al. Receptor binding by an H7N9 influenza virus from humans. Nature 499, 496–499 (2013).
Liu, J. et al. Structures of receptor complexes formed by hemagglutinins from the Asian influenza pandemic of 1957. Proc. Natl Acad. Sci. USA 106, 17175–17180 (2009).
Lu, G. et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500, 227–231 (2013).
Wang, N. et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 23, 986–993 (2013).
Li, W. et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl Acad. Sci. USA 114, E8508–E8517 (2017).
Widagdo, W. et al. Differential expression of the Middle East respiratory syndrome coronavirus receptor in the upper respiratory tracts of humans and dromedary camels. J. Virol. 90, 4838–4842 (2016).
Vergara-Alert, J. et al. Livestock susceptibility to infection with Middle East respiratory syndrome coronavirus. Emerg. Infect. Dis. 23, 232–240 (2017).
Barlan, A. et al. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J. Virol. 88, 4953–4961 (2014).
Meyer, B. et al. Serologic assessment of possibility for MERS-CoV infection in equids. Emerg. Infect. Dis. 21, 181–182 (2015).
Hulswit, R. J. G. et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl Acad. Sci. USA 116, 2681–2690 (2019).
Campbell, M. G., Veesler, D., Cheng, A., Potter, C. S. & Carragher, B. 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife 4, e06380 (2015).
Yu, X. et al. Structural basis of rotavirus strain preference toward N-acetyl- or N-glycolylneuraminic acid-containing receptors. J. Virol. 86, 13456–13466 (2012).
Suzuki, Y. et al. Sialic acid species as a determinant of the host range of influenza A viruses. J. Virol. 74, 11825–11831 (2000).
Millet, J. K. & Whittaker, G. R. Murine leukemia virus (MLV)-based coronavirus spike-pseudotyped particle production and infection. Bio Protoc. 6, e2035 (2016).
Xiong, X. et al. Recognition of sulphated and fucosylated receptor sialosides by A/Vietnam/1194/2004 (H5N1) influenza virus. Virus Res. 178, 12–14 (2013).
Wickramasinghe, I. N., de Vries, R. P., Grone, A., de Haan, C. A. & Verheije, M. H. Binding of avian coronavirus spike proteins to host factors reflects virus tropism and pathogenicity. J. Virol. 85, 8903–8912 (2011).
Bakkers, M. J. et al. Betacoronavirus adaptation to humans involved progressive loss of hemagglutinin-esterase lectin activity. Cell Host Microbe 21, 356–366 (2017).
Chan, C. M. et al. Carcinoembryonic antigen-related cell adhesion molecule 5 is an important surface attachment factor that facilitates entry of Middle East respiratory syndrome coronavirus. J. Virol. 90, 9114–9127 (2016).
Chu, H. et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 293, 11709–11726 (2018).
Widjaja, I. et al. Towards a solution to MERS: protective human monoclonal antibodies targeting different domains and functions of the MERS-coronavirus spike glycoprotein. Emerg. Microbes Infect. 8, 516–530 (2019).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Goddard, T. D., Huang, C. C. & Ferrin, T. E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007).
Brown, A. et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D 71, 136–153 (2015).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of COOT. Acta Crystallogr. D 66, 486–501 (2010).
DiMaio, F., Leaver-Fay, A., Bradley, P., Baker, D. & Andre, I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One 6, e20450 (2011).
DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
Frenz, B., Walls, A. C., Egelman, E. H., Veesler, D. & DiMaio, F. RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800 (2017).
Frenz, B. D. R. et al. Automatically fixing errors in glycoprotein structures with Rosetta. Structure 27, 134–139 (2019).
Walls, A. et al. Crucial steps in the structure determination of a coronavirus spike glycoprotein using cryo-electron microscopy. Protein Sci. 26, 113–121 (2017).
Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Agirre, J. et al. Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 (2015).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences (R01GM120553, D.V.), the National Institute of Allergy and Infectious Diseases (HHSN272201700059C, D.V.), a Pew Biomedical Scholars Award (D.V.), an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (D.V.) and a Swiss National Science Foundation PostDoc Mobility fellowship (M.M.S.). M.A.T. acknowledges support from the Institut Pasteur. This work was also supported by the Arnold and Mabel Beckman cryo-EM center at the University of Washington.
Author information
Authors and Affiliations
Contributions
B.-J.B., F.D. and D.V. supervised the research. Y.-J.P., A.C.W., Z.W., M.M.S., W.L., M.A.T., B.-J.B. and D.V. designed the experiments. A.C.W. and M.M.S. expressed and purified the MERS-CoV S ectodomain. Y.-J.P. and Z.W. performed cryo-EM sample preparation and data collection. Y.-J.P., Z.W. and D.V. processed the cryo-EM data. Y.-J.P., Z.W., F.D. and D.V. built and refined the atomic models. A.C.W. and Z.W. performed S mutagenesis. A.C.W. carried out the pseudovirus assays. W.L. carried out the hemagglutination assays. Y.-J.P., A.C.W., W.L., B.-J.B. and D.V. analyzed the data. Y.-J.P., A.C.W. and D.V. prepared the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Inês Chen was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Chemical structures of the sialosides used for structural studies.
a, Neu5Ac. b, Neu5Gc. c, sLeX. d, 2,3-SLN. e, 2,6-SLN.
Extended Data Fig. 2 CryoEM analysis of the MERS-CoV S glycoprotein in complex with Neu5Ac at 2.7 Å resolution.
a,b, Representative electron micrograph (a) and class averages (b) for the Neu5Ac-bound MERS-CoV S structure. c, Gold-standard (blue) and map/model (red) Fourier shell correlation curves. The 0.143 and 0.5 cutoffs are indicated by horizontal dashed lines. d, Local resolution map calculated using cryoSPARC. The Neu5Ac ligand is estimated to be resolved at 3Å resolution. e–g, Representative cryoEM densities shown as blue mesh with the corresponding atomic model rendered as sticks colored grey, blue and red for carbon, nitrogen and oxygen atoms, respectively. Dashed bonds indicate hydrogen bonds with ordered water molecules.
Extended Data Fig. 3 Structural basis for the MERS-CoV S selectivity for neuraminic acids that are not 9-O-acetylated or 5-N-glycolylated.
a, Superimposition of the 5-N-acetyl,9-O-acetyl neuraminic acid α-methyl glycoside (9-O-Ac-Neu5Ac, green) from the HCoV-OC43 S holo structure (PDB 6NZK19) to the bound Neu5Ac from the MERS-CoV S/Neu5Ac complex structure suggests the MERS-CoV S sialoside-binding site could not sterically accommodate the 9-O-acetyl group in this conformation. The distances between the 9-O-acetyl group and His91 or Ala92 are indicated. b, The MERS-CoV S sialoside-binding site rendered as a ribbon diagram with the side chains of key surrounding residues shown. Neu5Ac is rendered with the corresponding region of cryoEM density from the MERS-CoV S/Neu5Ac complex structure (low-pass filtered to 3Å and scaled to the MERS-CoV S/Neu5Gc map) shown as a blue mesh contoured at 6σ. c–f, The cryoEM structure of MERS-CoV S in complex with Neu5Gc shows weaker density for the sialoside (blue mesh contoured at 6σ), indicating poor steric and/or chemical accommodation of the 5-N-glycolyl hydroxyl group in the hydrophobic pocket defined by Phe39, Phe101, Ile131 and Ile132. In panels (c-e) Neu5Gc was modeled based on the porcine rotavirus CRW-8 VP8* holo structure (PDB 3TAY41) which corresponds to the conformation observed in 1 out of 8 structures available in the protein data bank with a Neu5Gc analogue. In panel f, Neu5Gc was modeled based on the rhesus rotavirus VP8* holo structure (PDB 3TB041) which is the only structure in the protein data bank featuring Neu5Gc with the 5-N-glycolyl group in an alternate orientation. The distance between the 5-N-glycolyl hydroxyl group and Gln36 is indicated.
Extended Data Fig. 4 CryoEM analysis of the MERS-CoV S glycoprotein in complex with Neu5Gc at 3.0 Å resolution.
a,b, Representative electron micrograph (a) and class averages (b) for the Neu5Gc-bound MERS-CoV S structure. c, Gold-standard Fourier shell correlation curve. The 0.143 cut-off is indicated by horizontal dashed lines. d, Representative density shown as blue mesh with the corresponding atomic model rendered as sticks colored grey, blue and red for carbon, nitrogen and oxygen atoms, respectively.
Extended Data Fig. 5 SDS-PAGE and Western blot analyses.
a, SDS-PAGE analysis of purified wild type or mutants MERS-CoV S domain A fused to human immunoglobulin Fc. Two micrograms of each protein was loaded. b, Western-blot analysis of murine leukemia viral particles pseudotyped with wild type or mutants MERS-CoV S using an anti-MERS-CoV S1 polyclonal antibody. Uncropped blot image is available as source data.
Extended Data Fig. 6 CryoEM analysis of the MERS-CoV S glycoprotein in complex with sLeX at 2.7 Å resolution.
a,b, Representative electron micrograph (a) and class averages (b) for the sLeX-bound MERS-CoV S structure. c, Gold-standard (blue) and map/model (red) Fourier shell correlation curves. The 0.143 and 0.5 cutoffs are indicated by horizontal dashed lines. d, Representative density shown as blue mesh with the corresponding atomic model rendered as sticks colored grey, blue and red for carbon, nitrogen and oxygen atoms, respectively.
Extended Data Fig. 7 CryoEM analysis of the MERS-CoV S glycoprotein in complex with 2,3-SLN at 2.7 Å resolution.
a,b, Representative electron micrograph (a) and class averages (b) for the 2,3-SLN-bound MERS-CoV S structure. c, Gold-standard (blue) and map/model (red) Fourier shell correlation curves. The 0.143 and 0.5 cutoffs are indicated by horizontal dashed lines. d, Representative density shown as blue mesh with the corresponding atomic model rendered as sticks colored grey, blue and red for carbon, nitrogen and oxygen atoms, respectively.
Extended Data Fig. 8 CryoEM analysis of the MERS-CoV S glycoprotein in complex with 2,6-SLN at 2.9 Å resolution.
a,b, Representative electron micrograph (a) and class averages (b) for the 2,6-SLN-bound MERS-CoV S structure. c, Gold-standard (blue) and map/model (red) Fourier shell correlation curves. The 0.143 and 0.5 cutoffs are indicated by horizontal dashed lines. d, Representative density shown as blue mesh with the corresponding atomic model rendered as sticks colored grey, blue and red for carbon, nitrogen and oxygen atoms, respectively.
Supplementary information
Source data
Rights and permissions
About this article

Cite this article
Park, YJ., Walls, A.C., Wang, Z. et al. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat Struct Mol Biol 26, 1151–1157 (2019). https://doi.org/10.1038/s41594-019-0334-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-019-0334-7
This article is cited by
-
Cationic crosslinked carbon dots-adjuvanted intranasal vaccine induces protective immunity against Omicron-included SARS-CoV-2 variants
Nature Communications (2023)
-
Automatic and accurate ligand structure determination guided by cryo-electron microscopy maps
Nature Communications (2023)
-
Identification of a Sialosides binding pocket in early-lineage SARS-CoV-2 via an optimized STD NMR method: a novel explanation for coronavirus virulence and zoonosis
Molecular Biomedicine (2022)
-
Antibodies to combat viral infections: development strategies and progress
Nature Reviews Drug Discovery (2022)
-
Structural definition of a pan-sarbecovirus neutralizing epitope on the spike S2 subunit
Communications Biology (2022)
