
Abstract
Competitive coevolution between microbes and viruses has led to the diversification of CRISPR–Cas defense systems against infectious agents. By analyzing metagenomic terabase datasets, we identified two compact families (775 to 803 amino acids (aa)) of CRISPR–Cas ribonucleases from hypersaline samples, named Cas13X and Cas13Y. We engineered Cas13X.1 (775 aa) for RNA interference experiments in mammalian cell lines. We found Cas13X.1 could tolerate single-nucleotide mismatches in RNA recognition, facilitating prophylactic RNA virus inhibition. Moreover, a minimal RNA base editor, composed of engineered deaminase (385 aa) and truncated Cas13X.1 (445 aa), exhibited robust editing efficiency and high specificity to induce RNA base conversions. Our results suggest that there exist untapped bacterial defense systems in natural microbes that can function efficiently in mammalian cells, and thus potentially are useful for RNA-editing-based research.
Similar content being viewed by others


Main
CRISPR–Cas systems have been classified into six subtypes and numerous orthologs in the wide spectrum of the microbial community1. Recent identification of compact CRISPR systems in uncultured microbes for type II and V families further broadens our knowledge on widespread coevolution between diverse CRISPR machinery and infectious agents2,3,4. Moreover, compact CRISPR effectors are highly preferred to generate CRISPR-based therapeutic modalities due to the in vivo delivery constraints of adeno-associated virus (AAV), commonly used for the treatment of durable diseases5. In contrast with the DNA-targeting activity of Cas9 and Cas12, Cas13 is a single effector recently identified in type VI CRISPR systems for RNA-guided RNA-interfering activity6,7. CRISPR–Cas13 empowers versatile applications for RNA research in both mammalian cells and plants, such as live imaging, RNA degradation, base editing and nucleic acid detection8. Numerous Cas13 effectors divided into four families have been identified previously; however, the uncharacterized space of CRISPR–Cas13 systems in natural microbes remained elusive.
Here, we identified two compact families of CRISPR–Cas13 in metagenomic datasets and engineered them for RNA degradation and RNA base conversion in mammalian cells.
Results
Identification of type VI-X and VI-Y Cas ribonuclease families
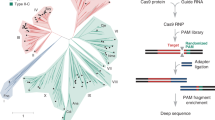
We developed a computational pipeline (Extended Data Fig. 1a) to search for previously uncharacterized CRISPR–Cas13 systems from metagenomic datasets. Using the CRISPR array as a search anchor, we first obtained metagenomic assemblies from the JGI database9 and adapted existing algorithms for de novo CRISPR array detection6. This led to the identification of 340,425 putative CRISPR repeat arrays. Up to 10 kilobases (kb) of genomic DNA sequence flanking each CRISPR array was extracted to further identify predicted protein-coding genes in the immediate vicinity. To identify compact Cas13 effectors, we searched among 250,901 candidate proteins with 400–900-aa residues and within 10 protein-coding genes associated with the CRISPR array, and found 24,959 proteins containing two RxxxxH motifs of the HEPN ribonuclease domain separately located at the N and C termini of the protein. Among RxxxxH motif-containing proteins, 64 contained two RxxxxH motifs of the three following types: RNxxxH, RHxxxH and RQxxxH. These three types were also found in the majority of previously known Cas13 (Extended Data Fig. 1b). Based on the fact that reported CRISPR–Cas13 systems have a single CRISPR RNA (crRNA) with conserved stem-loop structure10, we identified 31 Cas13 candidates. After excluding proteins with known functions in the National Center for Biotechnology Information (NCBI) non-redundant protein (NR) database, we obtained six candidate Cas13 proteins (Supplementary Tables 1 and 2). Further alignment of the six proteins back to the original pool of 24,959 proteins yielded one more candidate protein with RNxxxH and RxxxxH motifs. Furthermore, BLAST searches detected no sequence similarity of the identified Cas13 proteins with any of the previously identified type VI effector proteins in the NCBI NR database based on an E value cutoff of 1 × 10−10 (ref. 11) (Supplementary Table 3). By analyzing protein sequence similarity among the seven Cas13 variants, two distinct groups were found, corroborated by HEPN domain alignment results (Extended Data Fig. 1d,e and Supplementary Table 4). We therefore classify the seven proteins (size ranging from 775 to 803 aa) into two Cas13 families, including two members (‘Cas13X.1’, ‘Cas13X.2’) in VI-X, and five members (‘Cas13Y.1’ to ‘Cas13Y.5’) in VI-Y (Fig. 1a,b).

a, Maximum-likelihood tree of Cas13X, Cas13Y and previously reported Cas13a (refs. 17,32,33), Cas13b (refs. 16,34), Cas13c and Cas13d (ref. 15). Commonly used family members and protein sizes are shown in parentheses. The evolutionary distance scale of 0.5 is shown. b, Maximum-likelihood phylogenetic tree of Cas13X and Cas13Y proteins identified in this study, with the full Cas13X and Cas13Y CRISPR loci drawn along with conserved HEPN RNase domains. Blue and green rectangles indicate Cas13 proteins and CRISPR DRs, respectively. Gray diamonds denote spacer sequences. c, Predicted secondary RNA structures of DR sequences for Cas13X and Cas13Y proteins. d, Heatmap of mCherry protein knockdown activity of Cas13X and Cas13Y orthologs in HEK293T cells using pre-crRNA or crRNA. e, Effect of pre-crRNA versus crRNA and NLS versus no NLS on knockdown activity of Cas13X.1. Normalized MFI, mean fluorescence intensity relative to the nontargeting condition (n = 3). All values shown are mean ± s.e.m. P values are by two-sided unpaired t-test.
To investigate potential targets of natural crRNA in the CRISPR loci (Supplementary Table 5), we conducted a target search in metagenomic datasets of Cas13X and Cas13Y as well as NCBI viral databases. Fifteen potential target sequences including three perfect matches were identified from metagenomic datasets for crRNA arrays associated with Cas13X.1, Cas13X.2, Cas13Y.2 and Cas13Y.4 (Supplementary Table 6). Moreover, one perfect match associated with the ‘Ga0307438_1084463’ contig from the Cas13f.5 sample has very high similarity (E value < 1 × 10−100) with the DNA adenine methyltransferase gene previously reported to be carried by prophages in some bacteria12,13,14. From the GenBank-phage, RefSeq-plasmid and IMG/VR databases, four crRNAs in the array associated with Cas13X.2, Cas13Y.2 and Cas13Y.4 matched potential target sequences that have 3–5 mismatches with spacer sequences. These potential target sequences are from plasmids found in natural microorganisms such as Borrelia miyamotoi, Anabaena variabilis, Nostoc sp. and Acinetobacter pittii (Supplementary Table 6). It is likely that Cas13X and Cas13Y are deployed by their host to prevent invasion of these mobile genetic elements containing the target sequences.
Engineering Cas13X.1 for RNA interference in mammalian cells
To identify highly active Cas13 orthologs, we use a eukaryotic cell-based mCherry reporter system (Extended Data Fig. 2a) to examine the RNA-targeting interference activity of the seven Cas13 proteins. By synthesizing the human-codon-optimized version of each protein, we generated mammalian expression plasmids carrying the catalytically active or inactive proteins by mutating RxxxxH motifs7,15 (Extended Data Fig. 2b). Each protein was then fused with both N- and C-terminal nuclear localization signals (NLSs). These VI-X and VI-Y proteins were paired with two distinct forms of guide RNAs, with either a 30-nucleotide (nt) spacer flanked by two 36-nt direct repeat (DR) sequences to mimic an unprocessed guide RNA (pre-crRNA) or a 36-nt DR with a 30-nt spacer (crRNA) predicted to mimic mature guide RNAs (Fig. 1c,d). To determine crRNA architecture, we first tested DR position at the 5ʹ or 3ʹ end of crRNA with a reporter inhibition assay (Extended Data Fig. 2c). The crRNA with a 3ʹ DR instead of a 5ʹ DR showed substantial suppression of reporter expression (Extended Data Fig. 2d), indicating that the crRNA accompanying Cas13X.1 shared a similar 3ʹ DR structure with that of previously reported Cas13b (ref. 16). We then assessed the abilities of different VI-X and VI-Y proteins to knock down the mCherry reporter level in HEK293T cells. At 2 d after transfection with the plasmids expressing each of the VI-X and VI-Y proteins and the corresponding single target-specific crRNA, we observed significant reduction of mCherry protein, with Cas13X.1 exhibiting the highest knockdown efficiency (Fig. 1d,e). In contrast, transfection with nontargeting crRNA together with each Cas13, or, alternatively, crRNA with inactive Cas13, had no significant effect on the mCherry level (Fig. 1d,e and Extended Data Fig. 2d), suggesting crRNA- and HEPN-dependent knockdown. We also found that both the single DR crRNA and pre-crRNA with dual DR could mediate potent knockdown, and NLS significantly improved knockdown activity of Cas13X.1 (Fig. 1d,e). To determine the optimal spacer length for efficient Cas13X.1 targeting, we targeted mCherry with crRNA-carrying spacers of different lengths ranging from 5 to 50 nt, varying with a step of 1 nt between 15 and 30 nt or with step of 5 nt for the rest of the lengths (Extended Data Fig. 2e). We found reporter inhibition activity to be robustly efficient at all three mCherry-targeting loci when using a 30-nt spacer; this length was consistent with the finding in the Cas13X-associated CRISPR array of the uncultured microorganism (Extended Data Fig. 2e). Furthermore, 15 nt was determined as the minimal length for the spacer to mediate detectable knockdown in HEK293T cells. Thus, crRNAs with a 30-nt spacer were used for the following RNA interference experiments unless otherwise indicated. To investigate any protospacer flanking sequence (PFS) requirements for Cas13X.1, we carried out the PFS screening analysis and found no PFS bias in Cas13X.1 for efficient RNA knockdown activity (Extended Data Fig. 2f).
We next sought to compare the knockdown efficiency of Cas13X.1 and Cas13Y.1 against that of previously identified Cas13 proteins, Cas13a (ref. 17), Cas13b (ref. 16) and Cas13d (ref. 15). Across three target loci in mCherry, Cas13X.1, Cas13Y.1 and RfxCas13d overall outperformed LwaCas13a and PspCas13b in HEK293T cells at 48 h after transfection (Fig. 2a and Extended Data Fig. 3c). To confirm that RNA interference by Cas13X.1 and Cas13Y.1 is broadly applicable as previously reported Cas13a/b/d, we selected a panel of 12 additional human genes with diverse roles in mammalian cells, using one crRNA per gene. Cas13X.1, Cas13Y.1 and RfxCas13d showed comparable or higher knockdown efficiency than LwaCas13a and PspCas13b (Fig. 2b and Extended Data Fig. 3d). Moreover, we designed two additional crRNAs on the same 12 genes for Cas13X.1 and found that Cas13X.1 consistently showed high-level knockdown activity for each gene, using any of the three crRNAs (Extended Data Fig. 3a,b), indicating the uniformity of the Cas13X.1 system for RNA interference. Because the Cas13 family is capable of processing its own CRISPR array15, we next leveraged this property for the delivery of pre-crRNA for multiple targeting with a simple single-vector system (Fig. 2c). We found that robust simultaneous knockdown of four RNA transcripts could be achieved by transfection of Cas13X.1 together with an array encoding four crRNAs, each tiling one mRNA (EZH2, HRAS or PPARG) and a nuclear-localized long noncoding RNA (MALAT1) (Fig. 2c). Furthermore, we performed transcriptome-wide RNA-sequencing (RNA-seq) analysis on Cas13X.1 targeting B4GALNT1 and EZH2 genes in HEK293T cells. It was found that Cas13X.1 induced a comparable number of differentially expressed genes with RfxCas13d after knockdown of B4GALNT1 and EZH2 (Fig. 2d). Among the 48 downregulated genes in the Cas13X-mediated EZH2 knockdown experiment, 9 affected downregulated genes were reported to be regulated by EZH2 (Supplementary Tables 7–10). We further made a genome-wide search for similar sequences with a crRNA target from both B4GALNT1 and EZH2 genes, and found that the most similar sequences had at least 6 mismatches with crRNA targets. All predicted off-target genes were also nonsignificantly regulated for Cas13X.1 (Extended Data Fig. 4). To examine the target-dependent collateral ribonuclease activity of Cas13X.1 with previously compared RfxCas13d and LwaCas13a, we used EGFP-transgenic HEK293T cells to monitor EGFP expression as an indicator of the potential collateral effect in mammalian cells (Extended Data Fig. 5a) when targeting transiently overexpressed mCherry and endogenously expressed genes. It was found that comparably low collateral activities for EGFP were found for LwaCas13a, RfxCas13d and Cas13X.1 when transiently overexpressing the mCherry target in HEK293T (Extended Data Fig. 5b); but, for endogenous RNA knockdown, collateral effects for three different Cas13 proteins were undetectable on three target genes by the reporter assay, which agrees with a previous study18 (Extended Data Fig. 5c–e). Furthermore, we conducted a fluorophore-quencher assay, in which cleavage of dye-labeled single-stranded RNA (ssRNA) generates a fluorescent signal, and found that Cas13X.1 exhibited lower in vitro collateral RNase activity than RfxCas13d and LwaCas13a (Extended Data Fig. 6a,b). Taken together, these results indicate that Cas13X.1 offers a compact RNA interference tool with relatively high efficiency and specificity.

a, Reporter inhibition assay results of comparing activity among Cas13X.1, Cas13Y.1, LwaCas13a, PspCas13b and RfxCas13d for three different mCherry-targeting crRNAs in HEK293T. Normalized MFI, mean fluorescence intensity relative to the nontargeting condition. b, Comparison of knockdown efficiency for 12 endogenous transcripts by Cas13X.1, Cas13Y.1, LwaCas13a, PspCas13b and RfxCas13d, each with one guide and a nontargeting crRNA in HEK293T cells. c, Arrays of four guides; each mediates target knockdown by Cas13X.1 in HEK293T cells via transient transfection. d, Differential gene expression analysis of RNA-seq for B4GALNT1 and EZH2 knockdown in HEK293T cells by Cas13X.1 and RfxCas13d (three biological replicates). Knockdown relative to an NT crRNA was determined by qPCR. DRG, downregulated genes; KD, knockdown; NT, nontargeting crRNA. All values shown are mean ± s.e.m (n = 3).
Antiviral activity of Cas13X.1 in mammalian cells
CRISPR–Cas13, as a naturally evolved antiviral system, could potentially be developed as an antivirus modality for combating human infections19,20,21. Therefore, we investigated whether Cas13X.1, with smaller size and comparable efficiency to previously identified RfxCas13d, could be used for prophylactic RNA virus inhibition to complement the current Cas13 toolbox. To create effective and specific crRNA sequences to target and cleave SARS-CoV-2, we first performed a bioinformatics analysis by aligning published SARS-CoV-2 genomes22 and selected 30 crRNAs targeting RNA sites coding for RdRP (RNA-dependent RNA polymerase) and E (envelope) proteins (with 15 crRNAs for each). Proof-of-concept experiments were performed on RdRP and E sequences which are conserved among SARS-CoV viruses (Fig. 3a). The RdRP protein is the antiviral target for remdesivir23 and the E protein is critical for SARS-CoV pathogenesis24. To evaluate whether Cas13X.1 is effective for degrading SARS-CoV-2 sequences, we created a reporter by fusing GFP with synthesized partial SARS-CoV-2 fragments of RdRP (genome coordinates 15,037–15,158 base pairs (bp)) and E (26,232–26,394 bp) (Fig. 3a). At 48 h after cotransfection of HEK293T cells with the reporter and Cas13X.1/crRNAs, we observed that nearly all RdRP- and E-targeting crRNAs tested (27 out of 30) were able to support the suppression of GFP fluorescence in the cells by about 70%, as compared with that found for control transfection with the nontargeting crRNA (Fig. 3a).

a, Top, comparison of sequence identity between SARS-CoV-2 and SARS-CoV-1 genomes, and alignment comparison of SARS-CoV-2 and all coronavirus genomes. Middle, schematic diagram of the reporter consists of EFS promoter, GFP and the synthesized RdRP and E fragment sequences. Bottom, GFP expression after cotransfection of the reporter and Cas13X.1/crRNA, as measured by flow cytometry. Mean GFP fluorescence intensity changes of the reporter caused by 30 different targeting crRNAs, relative to nontargeting (NT) crRNA. b, Left, crRNA nucleotide identity or mismatch types (single-nucleotide mismatch or two tandem-nucleotide mismatches). Right, changes of GFP fluorescence intensity caused by cotransfection of reporter, Cas13X.1, RfxCas13d or LwaCas13a together with each of 20 different versions of crRNA_1 with mismatched mutations, relative to NT crRNA. c, Procedure for testing Cas13X.1-mediated anti-H1N1 activity in H1N1 IAV-infected MDCK cells. MDCK cells were first transfected with Cas13X.1/crRNA vectors and later challenged with H1N1 IAV. Supernatant was collected to analyze IAV abundance after 48 h of IAV infection. d, IAV RNA knockdown efficiency resulting from transfection of nucleoprotein (NP)-targeting Cas13X.1/crRNA. Transcript levels are relative to NT crRNA control. e, Changes of IAV abundance following Cas13X.1/crRNA transfection, analyzed by absolute RT–qPCR of supernatant from infected cultures. All values in a, b, d and e are shown as mean ± s.e.m (n = 3). IAV, influenza A virus; M, membrane protein; N, nucleocapsid protein; S, spike protein.
We next examined the minimal number of crRNAs required to target the majority of known coronaviruses found in both humans and animals, using a similar strategy to that previously described19. From all known 3,137 coronavirus genomes, we identified approximately 7.1 million potential unique crRNA targets (Extended Data Fig. 7a). We estimated that only five 22-nt and six 30-nt crRNAs with zero mismatch were able to target over 90% of coronavirus genomes (Extended Data Fig. 7b,c). We next examined whether Cas13 could tolerate mismatches between the crRNA and the target viral RNA, enabling inhibition of more coronavirus variants without increasing crRNA number and prevention of virus escaping via mutagenesis. Thus, we assessed the knockdown activity for an example crRNA (SARS-CoV-2 crRNA_1) with one or two mismatches (Fig. 3b), and found that both Cas13X.1 and RfxCas13d could well tolerate a single-nucleotide mismatch at different positions on the example crRNA (Fig. 3b). Results on two tandem mismatches revealed a critical (seed) region between 16 and 30-nt of the crRNA for efficient Cas13X.1-induced knockdown (Fig. 3b). With the tolerance for single-nucleotide mismatch, we estimated that 3, 10 and 17 crRNAs could target 95.3%, 99.1% and 100% of all coronaviruses, respectively (Extended Data Fig. 7d).
Next, we applied the CRISPR–Cas13X.1 strategy for inhibiting influenza RNA virus H1N1 which has a tropism for respiratory tract epithelial cells similar to SARS-CoV-2. We directly designed four crRNAs targeting at the nucleoprotein segment of the H1N1 genome which is essential for viral replication and transcription20,24. To test the antiviral ability of Cas13X.1 in a setting that mimics virus infection, we used influenza H1N1 strain ‘A/Puerto Rico/8/1934’ (ref. 25) in the Madin–Darby canine kidney (MDCK) cell line (Fig. 3c). Compared with nontargeting crRNA, three out of four crRNAs showed high knockdown efficiency on the nucleoprotein transcript (Fig. 3d). Consistently, target-specific crRNAs significantly reduced the abundance of nucleoprotein-positive H1N1 virus found in the supernatant of infected cultures, indicating effective inhibition of viral growth (Fig. 3e). Together, these results showed that the Cas13X.1 system could be used to confer antiviral ability for mammalian cells.
Truncated dCas13X.1 with ADAR2dd variants for efficient RNA base conversions
Base editing at the RNA level by RNA-guided Cas13 enables reversible nucleotide exchange with broad applicability in biomedical research and treatment of genetic diseases. However, previous Cas13b or Cas13d was too large after fusion with ADAR2 (adenosine deaminase acting on RNA type 2) to be widely used for in vivo viral delivery. Therefore, we first fused dCas13X.1 with high-fidelity ADAR2dd (with E488Q/T375G, referred as ADAR2dd*) to generate A-to-I RNA base editors (termed ‘xABE’). To test the activity of xABE, we generated an RNA-editing reporter using a mutated mCherry with a nonsense mutation (W98X (UGG to UAG)), which could functionally be repaired to the wild-type codon through A-to-I editing, and mCherry fluorescence could be detected after editing (Extended Data Fig. 8a). We found that xABE effectively induced mCherry fluorescence in cells transfected with mutant mCherry transcripts, together with both xABE and 50-nt crRNA, but not with either alone (Fig. 4a). To reduce the size of dCas13X.1 for efficient in vivo delivery, we generated various base editors by fusing the truncated dCas13X.1 (using a structure-guided method) with ADAR2dd* (Fig. 4b and Extended Data Fig. 8b). To this end, we systematically screened the editing activity of a variety of fused base editors with different dCas13X.1 truncations at either or both N and C termini (Fig. 4b), and identified the miniature and functional editor (‘mini’) with 150-aa and 180-aa truncation at C and N termini, respectively (Fig. 4b), suitable for packaging into commonly used AAV. We then examined the effect of mismatched base position within a 50-nt spacer on A-to-I editing efficiency for both full-size (xABE) and mini xABE (mxABE) (Extended Data Fig. 9a), and found that mismatched base position from 15 to 25 nt on the crRNA sequence yielded higher editing efficiency than other positions (Extended Data Fig. 9b). Furthermore, we compared xABE (1,195 aa) and mxABE (865 aa) with dCas13b-ADAR2dd* (REPAIR, 1,388 aa) and CasRx-ADAR2dd* (1,375 aa) using crRNA with or without DR, and found only xABE/mxABE achieved efficient crRNA-guided editing (Fig. 4c). By contrast, both REPAIR and CasRx-ADAR2dd* generated substantial editing with DR-free guide sequence, indicating editing mostly via a crRNA-independent pathway (Fig. 4c). Next, the RNA-editing efficiency of the full-size and mini mxABE systems was further examined in mammalian cells for several endogenous transcripts in comparison with REPAIR. We found crRNA-dependent A-to-I conversions were efficiently achieved by xABE/mxABE editors but not REPAIR, confirming the results with reporter assay (Fig. 4d and Extended Data Fig. 9c). To extend the base-editing capability of the dCas13X.1 protein, we further generated a C-to-U base editor (‘xCBE’) by fusing full-length or truncated dCas13X.1 with RNA cytosine deaminase derived from evolved ADAR2 (ref. 26), and found both full-length and mini xCBE (mxCBE) could achieve more efficient C-to-U editing than the previously reported RESCUE-S (1,495 aa) system in HEK293T cells (Fig. 4e). Moreover, both truncated mxABE and mxCBE exhibited transcriptome-wide high-fidelity activity and reduced RNA off-target edits to the base level in contrast with those of full-length xABE/xCBE and REPAIR by RNA-seq analysis (Fig. 4f and Extended Data Fig. 10a,b). Therefore, mxABE and mxCBE demonstrated great potential as compact, efficient and safe RNA base editors to facilitate an AAV-based method for treating genetic diseases.

a, Restored expression of mutant reporter by xABE RNA base editor as measured with flow cytometry. b, The activity of a variety of truncated xABE variants was analyzed by reporter assay. The black bar on Cas13X.1 indicates the HEPN domain. c, Reporter assay shows A-to-I editing activity among various editors including mxABE, xABE, REPAIR_v2 and RfxCas13d-ADAR2dd*. d, A-to-I editing efficiency of xABE, mxABE and REPAIR_v2 on endogenous transcripts in HEK293T analyzed with deep sequencing. e, C-to-U editing efficiency of xCBE, mxCBE and RESCUE_S on endogenous transcripts in HEK293T analyzed with deep sequencing. f, Manhattan plots of transcriptome-wide off-target RNA editing analysis for GFP/mCherry (control), xABE, mxABE, xCBE and mxCBE transfection experiments in HEK293T cells (A-to-I editor targeting endogenous SMAD4 RNA; C-to-U editor targeting endogenous PPIB RNA). The x and y axes are proportionally enlarged with each Manhattan plot to make the axis legend clear. Non-DR, guide RNA without DRs; NT, nontargeting crRNA. All values are presented as mean ± s.e.m (n = 3).
Discussion
Here, we identified two families of compact CRISPR–Cas13 systems (type VI-X and VI-Y) by mining metagenomic sequence datasets of natural uncultivated microbes, highlighting the diversity of natural microbial CRISPR systems. Given the sequence and structural differences in Cas13 subtypes and their crRNA DR regions, Cas13 proteins might evolve to have different activity towards the same target. Thus, it is worthwhile to enrich the Cas13 toolbox by computational mining of more metagenomic sequences generated by samples from diverse environments. In addition, we found the identified Cas13s could induce collateral cleavage of RNA in cells by targeting exogenous overexpressed genes, which is consistent with previous studies7,15,16,27,28,29,30. Although collateral activity was undetectable for Cas13 proteins when transiently targeting endogenous genes, a more sensitive way to evaluate collateral effects in mammalian cells and further long-term safety evaluation are needed for future therapeutic applications. Notably, all known Cas13 orthologs from the type VI-X and VI-Y families originated from microorganisms living in hypersaline habitats, reducing the risk of preexisting immunity found in Cas9 and Cas12 orthologs identified from pathogenic bacteria samples closely related to human31. Furthermore, by structure-guided engineering, we successfully truncated Cas13X.1 from 775 to 445 aa to generate a minimal RNA base editor for A-to-I or C-to-U editing on various RNA loci in mammalian cells, overcoming the in vivo delivery obstacle of various large Cas13-based base editors. We envision that these RNA editors with the compact Cas13X.1 will be useful for in vivo RNA editing-based research and therapeutic applications27,28,29,30.
Methods
Computational identification of the CRISPR–Cas13 systems
Metagenome sequences were downloaded from DOE JGI Integrated Microbial Genomes9. A computational pipeline was used to produce an expanded database of class 2 CRISPR–Cas systems from metagenomic sources. CRISPR arrays were identified using Piler-CR35, with all default parameters. Proteins were predicted with Prodigal36 in anon mode on all contigs at least 5 kb in length, and de-duplicated (that is, removing identical protein sequences) to construct a database. Proteins with length between 400 and 900 residues were obtained. RNAfold (http://rna.tbi.univie.ac.at/) was used to predict the secondary structure of DR sequences. The NR database was used to remove proteins with significant similarity (E < 1 × 10–10) to proteins of known function, and Cas proteins in NCBI were used for functional characterization of the candidate Cas13 proteins. Multiple sequence alignment was then conducted for each candidate Cas effector protein using MAFFT37. MEGA38 was used to construct the phylogenetic tree. I-TASSER39 was used to perform the protein structure prediction.
Natural crRNA target analysis
To search for natural crRNA targets, all crRNA spacer sequences in the CRISPR loci were used as a query to find matched sequences in CRISPSRTarget40 in GenBank-phage, RefSeq-plasmid and IMG/VR databases. The ‘Mismatch-search.pl’ script deposited in our GitHub repository was used to search the natural target in the metagenomic datasets in which the Cas13 proteins were detected. The natural crRNA targets with no more than five mismatches with each spacer were retained.
Plasmid constructions
Retrieved coding sequences of Cas13X and Cas13Y were human-codon-optimized and synthesized for cloning into SalI/NotI-digested pCX539 backbone with the Gibson assembly method. Predicted DR sequences for each Cas13 variant were synthesized as oligos for cloning downstream of the human U6 promoter for expression in mammalian cells. A G > A amber mutation was introduced in the mCherry coding sequence to generate mutant fluorescence protein as the RNA base-editing reporter. All primers and Cas13 sequences used in this study are provided in Supplementary Tables 2 and 11.
Cell culture, transfection and flow cytometry analysis
Mammalian cell lines used in the study were HEK293T and N2A. Media for culturing cells were prepared by supplementing DMEM with 10% FBS, GlutMAX, sodium pyruvate and penicillin/streptomycin. Transfection of HEK293T and N2A cells was conducted with Lipofectamine 3000 following the manufacturer manual, and cells were analyzed by BD FACSAria II or sorted by MoFlo XDP at 48 h after transfection. Flow cytometry results were analyzed with FlowJo X (v.10.0.7).
RNA editing and sequencing analysis
To analyze A-to-I or C-to-U base-editing efficiency of dCas13X, successfully transfected cells were sorted for RNA extraction. RNA was extracted with RNA-easy Isolation Reagent according to the manufacturer protocol. The complementary DNAs were reverse-transcribed from RNAs by HiScript II One Step RT-PCR Kit, and crRNA target sites were amplified from cDNAs with Phanta Max Super-Fidelity DNA Polymerase for Sanger or deep sequencing methods. Deep sequencing libraries were prepared with Nextera XT DNA Library Prep Kit according to the manufacturer manual and sequenced on a HiSeq. Sequencing data were first de-multiplexed by Cutadapt (v.2.8)41 based on sample barcodes. The de-multiplexed reads were then processed by CRISPResso2 (ref. 42) for the quantification of A-to-I or C-to-U conversion efficiency at each target site. Sanger sequencing results were analyzed with EditR43 to quantify A-to-I or C-to-U conversion efficiency at each target site.
Quantitative PCR with reverse transcription, RNA-seq and analysis
To quantify RNA knockdown efficiency of Cas13 effectors, RNAs were extracted from successfully transfected cells and reverse-transcribed to cDNAs with HiScript II One Step RT-PCR Kit (Vazyme, Biotech). Quantitative PCR (qPCR) was performed with the cDNA for each sample on a Roche 480 II-A, using AceQ Universal SYBR qPCR Master Mix (Vazyme, Biotech). qPCR results were analyzed with the −ΔΔCT method, in which differences between average CT values of target genes and reference gene GAPDH for three biological replicates were used to calculate the relative expression level of the target gene and normalized by that of control groups.
To analyze the functional specificity of Cas13 effectors, RNAs were extracted with a TRIZOL (Ambion)-based method, fragmented and reverse-transcribed to cDNAs with a HiScript II One Step RT-PCR Kit according to the manufacturer protocol. An RNA-seq library was generated with a TruSeq Stranded Total RNA library preparation kit using the standard protocol. The transcriptome libraries were sequenced using a 150-bp paired-end Illumina Xten platform.
RNA-seq data were analyzed as previously described28 and presented as the mean of all repeats. After filtering the low-quality reads with SolexaQA (v.3.1.7.1)44, RNA-seq reads of RNA knockdown experiments were aligned to the hg38 reference genome with Hisat2 (v.2.0.4)45. All uniquely mapped reads were processed by HTSeq-count46 to generate a read count matrix. DESeq2 (ref. 47) was used to calculate differentially expressed genes. Genes with fold change >2 and false discovery rate (FDR) < 0.05 were treated as differentially expressed genes. A customized script, HTSeq2FPKM.pl, was used to calculate fragments per kilobase per million mapped fragments (FPKM) values from the read count matrix for plotting visualization.
RNA-seq reads of RNA base-editing experiments were aligned to the hg38 reference genome with Hisat2 (v.2.0.4)45. RNA editing sites were calculated using REDItools48 with the following parameters: -t 24 -e -d -l -U [AG or TC or CT or GA] -p -u -m20 -T6-0 -W -v 1 -n 0.0. dbSNP49 (v.146) database downloaded from NCBI was used to filter the sites overlapped with common single nucleotide variants (SNVs). The sites with less than ten mutated or nonmutated reads were further filtered.
Prediction of potential off-target sites
Potential off-target sites were identified by a sliding-window method with step size of 1 nt. The window-size (W) is the same with target length. The mismatches between potential off-target sites and target site were less than 0.3 × W. The ‘Mismatch-search.pl’ script deposited in our GitHub repository was used to predict the off-target sequences in the hg38 genome and transcriptome. For the genome searching method, if the number of mismatches between any sequence or the reverse complementary sequence of this sequence and the spacer was no more than eight, the sequence was retained as a potential off-target sequence. We further used the ‘OffTarget_gene.pl’ script in our GitHub repository to identify off-target sequence-associated genes with less than eight mismatches between the forward sequence of genes and the reverse complementary sequence of the spacer. For the transcriptome searching, if a forward sequence of a transcript had no more than eight mismatches with the reverse complementary sequence of the spacer, the transcript was retained. Based on the RNA-seq count matrix generated by ‘HTSeq-count’, the genes with more than five read counts in at least one sample were labeled as the genes expressed in HEK293 cell line. Finally, the genes obtained by the two methods were combined as suspicious genes with predicted off-target sequences, listed in Supplementary Tables 12–15.
Collateral activity analysis in cell culture
To examine the effect of collateral activity for Cas13X.1/crRNA targeting different genes, we used GFP fluorescence intensity change as the indicator of suspicious collateral activity in a constitutively expressed EGFP-transgenic HEK293T cell line. For endogenous gene knockdown, a plasmid encoding Cas13X.1/crRNA and mCherry was transfected into the cells with Lipofectamine 3000 (L3000008, Thermofisher). For mCherry knockdown, a plasmid encoding Cas13X.1/crRNA and mCherry and a plasmid encoding BFP were cotransfected into the cells. BFP was used for normalizing transfection efficiency difference with Lipofectamine 3000. At 48 h after transfection, cells were collected and analyzed by BD FACSAria II. Flow cytometry results were analyzed with FlowJo X (v.10.0.7).
PFS analysis
To analyze the PFS requirements for Cas13X.1 activity, target sequences having 16 types of PFS sequence15 surrounding the protospacer were designed and cloned upstream of the EGFP gene (designated as PFS-EGFP). Plasmids encoding Cas13X.1/crRNA, mCherry and PFS-EGFP variants were transfected into wild-type HEK293T cells. EGFP knockdown efficiency was analyzed 48 h after transfection by BD FACSAria II. Fluorescence of mCherry was used as an indicator for successful transfection. Flow cytometry results were analyzed with FlowJo X (v.10.0.7).
Cas13 protein purification
Cas13 protein purification was performed with the protocol previously described16. The human-codon-optimized gene for Cas13X/Y/a/d was synthesized (Huagene) and cloned into a bacterial expression vector (pC013-Twinstrep-SUMO-huLwCas13a from Dr. Feng Zhang’s laboratory, deposited in Addgene as Plasmid no. 90097) after the plasmid digestion by BamHI and NotI with NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs). The Cas13X/Y expression constructs were transformed into BL21 (DE3) (TIANGEN) cells. Next, 1 l of lysogeny broth (LB) growth medium (tryptone 10.0 g; yeast extract 5.0 g; NaCl 10.0 g, Sangon Biotech) was inoculated with 10 ml of culture grown for 12 h. Cells were then grown at 37 °C to a cell density of 0.6 OD600, and then SUMO-Cas13 expression was induced by supplementing with 500 mM isopropylthiogalactoside. The induced cells were grown at 16 °C for 16–18 h before collection by centrifuge (4,000 r.p.m., 20 min). The collected cells were resuspended in Buffer W (Strep-Tactin Purification Buffer Set, IBA) and lysed using an ultrasonic homogenizer (Scientz). Cell debris was removed by centrifugation and the clear lysate was loaded onto a Strep-Tactin Sepharose High Performance Column (StrepTrap HP, GE Healthcare). The nonspecific binding protein and contaminants were flowed through. The target proteins were eluted with elution buffer (Strep-Tactin Purification Buffer Set, IBA). The N-terminal 6xHis/Twinstrep-SUMO tag was removed by SUMO protease (4 °C, >20 h). Then, target proteins were subjected to a final polishing step by gel filtration (S200, GE Healthcare). The purity was assessed by SDS–PAGE.
Nuclease assay
A dye-labeled ssRNA reporter assay for Cas13 ribonuclease activity was performed and analyzed as previously described50,51. For on-target ribonuclease activity analysis, the assay was performed with 45 nM purified Cas13X/Y/a/d, 22.5 nM crRNA, 5 or 20 nM quenched cy5-labeled target ssRNA reporter (Sangon Biotech), 1 µl of murine RNase inhibitor (New England Biolabs), 100 ng of background total human RNA (purified from HEK293T cell culture) and varying amounts of input nucleic acid target, unless otherwise indicated, in nuclease assay buffer (40 mM Tris-HCl including 25 mM Tris-HCL, pH 7.5, and 25 mM Tris-HCL, pH 7.0, 60 mM NaCl, 6 mM MgCl2, pH 7.3). For collateral ribonuclease activity analysis, the assay was performed with 45 nM purified Cas13X/Y/a/d; 22.5 nM crRNA; 0, 5, or 20 target ssRNAs; 125 nM quenched FAM-labeled nontarget ssRNA reporter (Sangon Biotech); 1 µl of murine RNase inhibitor; 100 ng of background total human RNA (purified from HEK293T cell culture); and varying amounts of input nucleic acid target, unless otherwise indicated, in nuclease assay buffer. Reactions were allowed to proceed for 1–3 h at 37 °C on a fluorescence plate reader (Analytik Jena) with fluorescence kinetics measured every 5 min.
Antiviral experimental and analysis method
MDCK cells were seeded onto 96-well plates and incubated with DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin. The cells were further infected with influenza A virus H1N1 (A/Puerto Rico/8/1934)25 at 100 times doses of median tissue culture infective dose (TCID50). At 1 h post-infection, the medium was replaced with DMEM containing 0.1% BSA and 1 μg ml−1 of TPCK-trypsin. At 48 h post-infection, supernatant and cell lysate were collected for measuring viral titers. Total RNA was extracted from supernatants of virus-infected MDCK cells, and quantitative PCR with reverse transcription (RT–qPCR) was performed using influenza virus-specific primers for determination of relative levels of viral loads. All primers used in this study are provided in Supplementary Table 11.
Statistical analysis
All values are shown as mean ± s.e.m. Unpaired Student’s t-test (two-tailed) was used for comparisons and P < 0.05 was considered to be statistically significant. Details of statistical values are provided in Source Data Figs. 1–4 and Extended Data Figs. 1–10. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Material availability
All materials are available upon reasonable request.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the sequencing data have been deposited in the NCBI SRA under project accession number PRJNA680488. Meta-information on all raw NGS datasets is provided in Supplementary Table 16. Source data are provided with this paper.
Code availability
Bioinformatics codes were deposited in the GitHub repository (https://github.com/yszhou2016/Cas13).
Change history
02 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41592-021-01379-x
References
Makarova, K. S. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736 (2015).
Fonfara, I. et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–2590 (2014).
Harrington, L. B. et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018).
Pausch, P. et al. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369, 333–337 (2020).
Wang, D., Zhang, F. & Gao, G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell 181, 136–150 (2020).
Shmakov, S. et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell 60, 385–397 (2015).
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016).
Smargon, A. A., Shi, Y. J. & Yeo, G. W. RNA-targeting CRISPR systems from metagenomic discovery to transcriptomic engineering. Nat. Cell Biol. 22, 143–150 (2020).
Markowitz, V. M. et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122 (2012).
O’Connell, M. R. Molecular mechanisms of RNA targeting by Cas13-containing type VI CRISPR-Cas systems. J. Mol. Biol. 431, 66–87 (2019).
Makarova, K. S. et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020).
Lynch, K. H., Stothard, P. & Dennis, J. J. Genomic analysis and relatedness of P2-like phages of the Burkholderia cepacia complex. BMC Genomics 11, 599 (2010).
Saridaki, A. et al. Wolbachia prophage DNA adenine methyltransferase genes in different Drosophila-Wolbachia associations. PLoS ONE 6, e19708 (2011).
Sternberg, N. & Coulby, J. Cleavage of the bacteriophage P1 packaging site (pac) is regulated by adenine methylation. Proc. Natl Acad. Sci. USA 87, 8070–8074 (1990).
Konermann, S. et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676.e614 (2018).
Smargon, A. A. et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol. Cell 65, 618–630.e617 (2017).
Abudayyeh, O. O. et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284 (2017).
Wang, Q. et al. The CRISPR-Cas13a gene-editing system induces collateral cleavage of RNA in glioma cells. Adv. Sci. (Weinh.) 6, 1901299 (2019).
Abbott, T. R. et al. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell https://doi.org/10.1016/j.cell.2020.04.020 (2020).
Freije, C. A. et al. Programmable inhibition and detection of RNA viruses using Cas13. Mol. Cell 76, 826–837.e811 (2019).
Nguyen, T. M., Zhang, Y. & Pandolfi, P. P. Virus against virus: a potential treatment for 2019-nCov (SARS-CoV-2) and other RNA viruses. Cell Res. 30, 189–190 (2020).
Hulo, C. et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 39, D576–D582 (2011).
de Wit, E. et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl Acad. Sci. USA 117, 6771–6776 (2020).
Ruch, T. R. & Machamer, C. E. The coronavirus E protein: assembly and beyond. Viruses 4, 363–382 (2012).
Harding, A. T., Heaton, B. E., Dumm, R. E. & Heaton, N. S. Rationally designed influenza virus vaccines that are antigenically stable during growth in eggs. mBio https://doi.org/10.1128/mBio.00669-17 (2017).
Abudayyeh, O. O. et al. A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386 (2019).
Kushawah, G. et al. CRISPR-Cas13d induces efficient mRNA knockdown in animal embryos. Dev. Cell 54, 805–817 (2020).
He, B. et al. Modulation of metabolic functions through Cas13d-mediated gene knockdown in liver. Protein Cell https://doi.org/10.1007/s13238-020-00700-2 (2020).
Zhou, H. et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell https://doi.org/10.1016/j.cell.2020.03.024 (2020).
Zhou, C. et al. CasRx-mediated RNA targeting prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Natl Sci. Rev. https://doi.org/10.1093/nsr/nwaa033 (2020).
Mehta, A. & Merkel, O. M. Immunogenicity of Cas9 protein. J. Pharm. Sci. 109, 62–67 (2020).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
East-Seletsky, A., O’Connell, M. R., Burstein, D., Knott, G. J. & Doudna, J. A. RNA targeting by functionally orthogonal type VI-A CRISPR-Cas enzymes. Mol. Cell 66, 373–383.e373 (2017).
Cox, D. B. T. et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017).
Edgar, R. C. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8, 18 (2007).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evolution 30, 772–780 (2013).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evolution 33, 1870–1874 (2016).
Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008).
Biswas, A., Gagnon, J. N., Brouns, S. J., Fineran, P. C. & Brown, C. M. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. https://doi.org/10.14806/ej.17.1.200 (2011)
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Kluesner, M. G. et al. EditR: a method to quantify base editing from Sanger sequencing. CRISPR J. 1, 239–250 (2018).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11, 485 (2010).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Picardi, E. & Pesole, G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814 (2013).
Smigielski, E. M., Sirotkin, K., Ward, M. & Sherry, S. T. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28, 352–355 (2000).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017).
Tambe, A., East-Seletsky, A., Knott, G. J., Doudna, J. A. & O’Connell, M. R. RNA binding and HEPN-nuclease activation are decoupled in CRISPR-Cas13a. Cell Rep. 24, 1025–1036 (2018).
Acknowledgements
We thank M. Poo for discussions and comments on this manuscript; and the FACS facility, H. Wu and L. Quan at ION. This work was supported by the Basic Frontier Scientific Research Program of Chinese Academy of Sciences From 0 to 1 original innovation project (grant no. ZDBS-LY-SM001), the R&D Program of China (grant nos. 2017YFC1001300 and 2018YFC2000100), the CAS Strategic Priority Research Program (grant no. XDB32060000), the National Natural Science Foundation of China (grant nos. 31871502, 31925016, 91957122, 31901047, 82021001), the Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX05), the Shanghai City Committee of Science and Technology Project (grant nos. 18411953700, 18JC1410100, 19XD1424400, 19YF1455100) and the International Partnership Program of Chinese Academy of Sciences (grant no. 153D31KYSB20170059).
Author information
Authors and Affiliations
Contributions
C.X., Y.Z. and H.Y. conceived the project. C.X., Y.Z., Q.X., B.H. and G.G. designed and conducted experiments. Y.Z. and J.L. performed bioinformatics analysis. Z.W. and B.C. assisted with plasmids construction and RNA analysis. T.Y. assisted with cell experiments. X.W., D.Z. and X.H. assisted with virus experiments. H.Y. designed experiments and supervised the whole project. C.X., Y.Z. and H.Y. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
H.Y. is a founder of Hui-Gene Therapeutics. H.Y., C.X., Y.Z., and Q.X. are co-inventors on US patent application 16/864,982 relating to the Cas proteins described in this manuscript. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Lei Tang was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Computational pipeline for the identification of new CRISPR RNA-Targeting System Cas13 and protein schematics for Cas13 effectors.
a, Schematic describing a fast and classical computational pipeline for CRISPR system identification. In fast pipeline, a minimal definition for a putative class 2 CRISPR locus was used, requiring only a CRISPR repeat array and a nearby protein of 400 to 900 aa in length. b, Distribution of RNxxxH, RHxxxH, RQxxxH motifs among Cas13 proteins. All values are presented as percentage of motif occurrence (n = 180). c, Schematics describing protein-size and HEPN motif position of previously identified Cas13a/b/c/d and Cas13X/f proteins used in this study. The RxxxxH HEPN motif is highlighted. d, Similarity analysis between type E and F effectors by position-specific iterated BLAST. E value of >1e-10 defined as cutoff for non-significant similarity. e, HEPN domain alignment between type VI-X/Y and previously reported Cas13 effectors. Conserved residuals for Cas13X and Cas13Y were highlighted in pink and previously reported Cas13 in green.
Extended Data Fig. 2 Pre-crRNA processing mechanism of Cas13X.1 and effect of crRNA length and PFS on Cas13X.1 activity.
a, Procedure diagram of mCherry reporter inhibition assay for testing Cas13 activity in HEK293T. b, Schematics describing HEPN-active and -inactive Cas13X.1. The wild-type and inactivated mutant RxxxxH HEPN motifs are highlighted. c, Schematic diagram of potential mechanisms for pre-crRNA to mature crRNA processing by Cas13X.1. d, Reporter inhibition assay revealed ribonuclease-activity dependence on HEPN domains and crRNAs with 3’DR. e, Top, Schematic diagram showing the mCherry specific crRNAs with different spacer lengths. Bottom, Changes of mCherry fluorescence intensity for mCherry specific crRNAs of different lengths relative to non-targeting (NT) crRNA, as measured by flow cytometry. f, PFS analysis with crRNA targeting sequences flanked by different PFS. All values are presented as mean ± s.e.m (n = 3). P values are by two-sided unpaired t-test.
Extended Data Fig. 3 Comparison of knockdown activity between previously reported Cas13 and Cas13X.1/Cas13Y.1.
a, Relative target RNA knockdown by individual position-matched Cas13X.1 and RfxCasRx crRNAs. b, Cas13X.1 targeting efficiency of 12 endogenous transcripts, each with 3 guides and a non-targeting (NT) crRNA in HEK293T cells. c, Comparison of mCherry knockdown activity among Cas13X.1, Cas13Y.1, LwaCas13a, PspCas13b and RfxCas13d (n = 3 for each protein). d, Comparison of endogenous genes knockdown activity among Cas13X.1, Cas13Y.1, LwaCas13a, PspCas13b and RfxCas13d (n = 12 for each protein). *P < 0.05, ***P < 0.001, two-sided unpaired t-test. All values are presented as mean ± s.e.m (n = 3). P values are by two-sided unpaired t-test.
Extended Data Fig. 4 Genome-wide search for similar sequences with crRNA target from both B4GALNT1 and EZH2 gene predicted several potential off-target genes and volcano plot showed their differential expression significance.
Red and blue dot indicate significantly upregulated and downregulated off-target genes. Grey dot depicts non-significantly regulated off-target genes. Large blue dot indicates target gene, B4GALNT1 and EZH2.
Extended Data Fig. 5 Collateral activity comparison between Cas13X.1, RfxCas13d and LwaCas13a in HEK293T cells.
a, Schematics describing collateral activity detection system based on EGFP-transgenic HEK293T cells. b-e, Target knockdown and collateral activity for Cas13X.1, RfxCas13d and LwaCas13a with mCherry, endogenous ANXA4, B4GALNT1 and EZH2-targeting crRNAs. All values are presented as mean ± s.e.m (n = 3). P values are by two-sided unpaired t-test.
Extended Data Fig. 6 Biochemical characterization of on-target and collateral ribonuclease activity.
a, On-target ribonuclease activity comparison among LwaCas13a, RfxCas13d, Cas13X.1 and Cas13Y.1. b, Collateral ribonuclease activity comparison among LwaCas13a, RfxCas13d, Cas13X.1 and Cas13Y.1. AU, arbitrary unit. All Values shown are mean (n = 3).
Extended Data Fig. 7 Bioinformatics analysis for identifying minimal number of coronaviruses-targeting crRNAs.
a, Schematic describing a computational pipeline for identifying the minimal number of crRNAs targeting all coronaviruses by analyzing available coronavirus genomes. b,c, Histograms showing the predicted minimal number of 22-nt and 30-nt crRNAs with zero mismatch to target all sequenced 3,137 coronavirus genomes. d, Seventeen 30-nt crRNAs with single-nt mismatch in the minimal pool that targets all coronaviruses and their similarity with human transcriptome.
Extended Data Fig. 8 Verification of eABE function with mCherry* reporter system and predicted Cas13X.1 protein structure.
a, Schematic procedure of testing eABE with the mCherry* reporter system. b, Predicted protein structure of Cas13X.1. Yellow denote N terminal; green indicate C terminal; red represent HEPN motif.
Extended Data Fig. 9 Effect of mismatched base position on xABE activity and Sanger sequencing results of editing human endogenous transcripts by xABE.
a, Mismatch positions in different crRNAs targeting the mCherry amber mutation. b, Effect of mismatched base position with 50-nt spacer on A-to-I editing efficiency by both full and mini xABE editors. All values are presented as mean ± s.e.m (n = 3). c, Sanger sequencing results showing representative A-to-I conversion on endogenous transcripts by full and mini xABE editors. Red triangles indicate mutation sites.
Extended Data Fig. 10 Off-target RNA editing effect for xABE, mxABE, xCBE, mxCBE and REPAIR system.
a, Transcriptome-wide off-target sites numbers for GFP/mCherry (control), xABE, mxABE, xCBE and mxCBE transfection experiments in HEK293T cells. All values are presented as mean ± s.e.m (n = 3). b, Manhattan plots of transcriptome-wide off-target RNA editing analysis for REPAIR transfection experiments in HEK293T cells (A-to-I editor targeting endogenous SMAD4 RNA). The x and y axis are proportionally enlarged with each Manhattan plot to make the axis legend clear. Non-DR, guide RNA without direct repeats. NT, non-targeting crRNA.
Supplementary information
Supplementary Information
Supplementary gating strategy and experimental protocol.
Supplementary Table 1
Genomic positions and habitat information of Cas13X.1-X.2 and Cas13Y.1-Y5
Supplementary Table 2
Cas13X.1-X.2 and Cas13Y.1-Y.5 protein sequences used in the study
Supplementary Table 3
Type VI-X and VI-Y classification criteria
Supplementary Table 4
HEPN domains sequence alignment of type VI-A to VI-Y Cas13 proteins
Supplementary Table 5
Spacer sequences in Cas13X and Cas13Y associated CRISPR arrays
Supplementary Table 6
Natural crRNA targets
Supplementary Table 7
Significant differentially downregulated and upregulated genes by EZH2 knockdown with Cas13X.1
Supplementary Table 8
Significant differentially downregulated and upregulated genes by EZH2 knockdown with RfxCas13d
Supplementary Table 9
Significant differentially downregulated and upregulated genes by B4GALNT1 knockdown with Cas13X.1
Supplementary Table 10
Significant differentially downregulated and upregulated genes by B4GALNT1 knockdown with RfxCas13d
Supplementary Table 11
Primers used in the study
Supplementary Table 12
Predicted off-target sequences from hg38 genome for B4GALNT1 crRNA
Supplementary Table 13
Predicted off-target sequences from hg38 transcriptome for B4GALNT1 crRNA
Supplementary Table 14
Predicted off-target sequences from hg38 genome for EZH2 crRNA
Supplementary Table 15
Predicted off-target sequences from hg38 transcriptome for EZH2 crRNA
Supplementary Table 16
RNA-seq datasets information
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
About this article

Cite this article
Xu, C., Zhou, Y., Xiao, Q. et al. Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes. Nat Methods 18, 499–506 (2021). https://doi.org/10.1038/s41592-021-01124-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-021-01124-4
This article is cited by
-
Programmable RNA base editing via targeted modifications
Nature Chemical Biology (2024)
-
The CRISPR-Cas13a Gemini System for noncontiguous target RNA activation
Nature Communications (2024)
-
Repurposing CRISPR-Cas13 systems for robust mRNA trans-splicing
Nature Communications (2024)
-
Tracking endogenous proteins based on RNA editing-mediated genetic code expansion
Nature Chemical Biology (2024)
-
Near-cognate tRNAs increase the efficiency and precision of pseudouridine-mediated readthrough of premature termination codons
Nature Biotechnology (2024)
