
Abstract
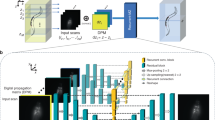
We demonstrate that a deep neural network can be trained to virtually refocus a two-dimensional fluorescence image onto user-defined three-dimensional (3D) surfaces within the sample. Using this method, termed Deep-Z, we imaged the neuronal activity of a Caenorhabditis elegans worm in 3D using a time sequence of fluorescence images acquired at a single focal plane, digitally increasing the depth-of-field by 20-fold without any axial scanning, additional hardware or a trade-off of imaging resolution and speed. Furthermore, we demonstrate that this approach can correct for sample drift, tilt and other aberrations, all digitally performed after the acquisition of a single fluorescence image. This framework also cross-connects different imaging modalities to each other, enabling 3D refocusing of a single wide-field fluorescence image to match confocal microscopy images acquired at different sample planes. Deep-Z has the potential to improve volumetric imaging speed while reducing challenges relating to sample drift, aberration and defocusing that are associated with standard 3D fluorescence microscopy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
We declare that all the data supporting the findings of this work are available within the manuscript and its supplementary information.
Code availability
Deep learning models reported in this work used standard libraries and scripts that are publicly available in TensorFlow. Through a custom-written Fiji-based plugin, we provided our trained network models (together with some sample test images) for the following objective lenses: Leica HC PL APO 20×/0.80 NA DRY (two different network models trained on TxRd and FITC channels), Leica HC PL APO 40×/1.30 NA oil (trained on TxRd channel), Olympus UPLSAPO20X 0.75 NA (trained on TxRd channel). We made this custom-written plugin and our models publicly available through the following links: http://bit.ly/deep-z-git and http://bit.ly/deep-z.
References
Nguyen, J. P. et al. Whole-brain calcium imaging with cellular resolution in freely behaving Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 113, E1074–E1081 (2016).
Schrödel, T., Prevedel, R., Aumayr, K., Zimmer, M. & Vaziri, A. Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat. Methods 10, 1013–1020 (2013).
Tomer, R. et al. SPED light sheet microscopy: fast mapping of biological system structure and function. Cell 163, 1796–1806 (2015).
Siedentopf, H. & Zsigmondy, R. Uber sichtbarmachung und größenbestimmung ultramikoskopischer teilchen, mit besonderer anwendung auf goldrubingläser. Ann. Phys. 315, 1–39 (1902).
Lerner, T. N. et al. Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162, 635–647 (2015).
Hell, S. W. & Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 (1994).
Hell, S. W. Far-field optical nanoscopy. Science 316, 1153–1158 (2007).
Henriques, R. et al. QuickPALM: 3D real-time photoactivation nanoscopy image processing in Image. J. Nat. Methods 7, 339–340 (2010).
Abraham, A. V., Ram, S., Chao, J., Ward, E. S. & Ober, R. J. Quantitative study of single molecule location estimation techniques. Opt. Express 17, 23352–23373 (2009).
Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M. & Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 8, 1027–1036 (2011).
Juette, M. F. et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5, 527–529 (2008).
Pavani, S. R. P. et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl Acad. Sci. USA 106, 2995–2999 (2009).
Prevedel, R. et al. Simultaneous whole-animal 3D imaging of neuronal activity using light-field microscopy. Nat. Methods 11, 727–730 (2014).
Levoy, M., Ng, R., Adams, A., Footer, M. & Horowitz, M. Light Field Microscopy. In ACM SIGGRAPH 2006 Papers 924–934 (ACM, 2006).
Pégard, N. C. et al. Compressive light-field microscopy for 3D neural activity recording. Optica 3, 517–524 (2016).
Broxton, M. et al. Wave optics theory and 3-D deconvolution for the light field microscope. Opt. Express 21, 25418–25439 (2013).
Cohen, N. et al. Enhancing the performance of the light field microscope using wavefront coding. Opt. Express 22, 24817–24839 (2014).
Wagner, N. et al. Instantaneous isotropic volumetric imaging of fast biological processes. Nat. Methods 16, 497–500 (2019).
Rosen, J. & Brooker, G. Non-scanning motionless fluorescence three-dimensional holographic microscopy. Nat. Photonics 2, 190–195 (2008).
Brooker, G. et al. In-line FINCH super resolution digital holographic fluorescence microscopy using a high efficiency transmission liquid crystal GRIN lens. Opt. Lett. 38, 5264–5267 (2013).
Siegel, N., Lupashin, V., Storrie, B. & Brooker, G. High-magnification super-resolution FINCH microscopy using birefringent crystal lens interferometers. Nat. Photonics 10, 802–808 (2016).
Abrahamsson, S. et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods 10, 60–63 (2013).
Abrahamsson, S. et al. MultiFocus polarization microscope (MF-PolScope) for 3D polarization imaging of up to 25 focal planes simultaneously. Opt. Express 23, 7734–7754 (2015).
Belthangady, C. & Royer, L. A. Applications, promises, and pitfalls of deep learning for fluorescence image reconstruction. Nat. Methods https://doi.org/10.1038/s41592-019-0458-z (2019).
Rivenson, Y. et al. Deep learning microscopy. Optica 4, 1437–1443 (2017).
Ouyang, W., Aristov, A., Lelek, M., Hao, X. & Zimmer, C. Deep learning massively accelerates super-resolution localization microscopy. Nat. Biotechnol. 36, 460–468 (2018).
Nehme, E., Weiss, L. E., Michaeli, T. & Shechtman, Y. Deep-STORM: super-resolution single-molecule microscopy by deep learning. Optica 5, 458–464 (2018).
Rivenson, Y. et al. Deep learning enhanced mobile-phone microscopy. ACS Photonics 5, 2354–2364 (2018).
Haan, K., de, Ballard, Z. S., Rivenson, Y., Wu, Y. & Ozcan, A. Resolution enhancement in scanning electron microscopy using deep learning. Sci. Rep. 9, 1–7 (2019).
Wang, H. et al. Deep learning enables cross-modality super-resolution in fluorescence microscopy. Nat. Methods 16, 103– (2019).
Weigert, M. et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat. Methods 15, 1090– (2018).
Zhang, X. et al. Deep learning optical-sectioning method. Opt. Express 26, 30762–30772 (2018).
Wu, Y. et al. Bright-field holography: cross-modality deep learning enables snapshot 3D imaging with bright-field contrast using a single hologram. Light Sci. Appl. 8, 25 (2019).
Barbastathis, G., Ozcan, A. & Situ, G. On the use of deep learning for computational imaging. Optica 6, 921–943 (2019).
Goodfellow, I. et al. Generative Adversarial Nets. Adv. Neural Inf. Process. Syst. 27, 2672–2680 (2014).
Mirza, M. & Osindero, S. Conditional Generative Adversarial Nets. Preprint at arXiv https://arxiv.org/abs/1411.1784 (2014).
Shaw, P. J. & Rawlins, D. J. The point-spread function of a confocal microscope: its measurement and use in deconvolution of 3-D data. J. Microsc. 163, 151–165 (1991).
Kirshner, H., Aguet, F., Sage, D. & Unser, M. 3-D PSF fitting for fluorescence microscopy: implementation and localization application. J. Microsc. 249, 13–25 (2013).
Nguyen, J. P., Linder, A. N., Plummer, G. S., Shaevitz, J. W. & Leifer, A. M. Automatically tracking neurons in a moving and deforming brain. PLoS Comput. Biol. 13, e1005517 (2017).
Gonzalez, R. C., Woods, R. E. & Eddins, S. L. Digital Image Processing Using MATLAB (McGraw-Hill, 2004).
Tinevez, J.-Y. et al. TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017).
Kato, S. et al. Global brain dynamics embed the motor command sequence of Caenorhabditis elegans. Cell 163, 656–669 (2015).
Nagy, S., Huang, Y.-C., Alkema, M. J. & Biron, D. Caenorhabditis elegans exhibit a coupling between the defecation motor program and directed locomotion. Sci. Rep. 5, 17174 (2015).
Toyoshima, Y. et al. Accurate automatic detection of densely distributed cell nuclei in 3D space. PLoS Comput. Biol. 12, e1004970 (2016).
Huang, B., Wang, W., Bates, M. & Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008).
Antipa, N. et al. DiffuserCam: lensless single-exposure 3D imaging. Optica 5, 1–9 (2018).
Shechtman, Y., Sahl, S. J., Backer, A. S. & Moerner, W. E. Optimal point spread function design for 3D imaging. Phys. Rev. Lett. 113, 133902 (2014).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Strange, K. (Ed.) C. elegans: Methods and Applications (Humana Press, 2006).
Thevenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998).
Forster, B., Van de Ville, D., Berent, J., Sage, D. & Unser, M. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc. Res. Tech. 65, 33–42 (2004).
Zack, G. W., Rogers, W. E. & Latt, S. A. Automatic measurement of sister chromatid exchange frequency. J. Histochem. Cytochem. 25, 741–753 (1977).
Mao, X. et al. Least squares generative adversarial networks. In Proc. 2017 IEEE International Conference on Computer Vision 2813–2821 (IEEE, 2017).
Ronneberger, O., Fischer, P. & Brox, T. U-Net: convolutional networks for biomedical image segmentation. In Medical Image Computing and Computer-Assisted Intervention 2015 (eds Navab, N. et al.) 234–241 (Springer, 2015).
Wu, Y. et al. Extended depth-of-field in holographic imaging using deep-learning-based autofocusing and phase recovery. Optica 5, 704–710 (2018).
Glorot, X. & Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. In Proc. Thirteenth International Conference on Artificial Intelligence and Statistics 249–256 (2010).
Abadi, M. et al. TensorFlow: a system for large-scale machine learning. In Proc. 12th USENIX Symposium on Operating Systems Design and Implementation 265–283 (USENIX, 2016).
Wang, Z., Bovik, A. C., Sheikh, H. R. & Simoncelli, E. P. Image quality assessment: from error visibility to structural similarity. IEEE Trans. Image Process. 13, 600–612 (2004).
Shi, J. & Malik, J. Normalized cuts and image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 22, 888–905 (2000).
von Luxburg, U. A tutorial on spectral clustering. Stat. Comput. 17, 395–416 (2007).
Acknowledgements
The authors acknowledge Y. Luo, X. Tong, T. Liu, H. C. Koydemir and Z. S. Ballard of University of California, Los Angeles (UCLA), as well as Leica Microsystems for their help with some of the experiments. The Ozcan group at UCLA acknowledges the support of Koc Group, National Science Foundation and the Howard Hughes Medical Institute. Y.W. also acknowledges the support of a SPIE John Kiel scholarship. Some of the reported optical microscopy experiments were performed at the Advanced Light Microscopy/Spectroscopy Laboratory and the Leica Microsystems Center of Excellence at the California NanoSystems Institute at UCLA with funding support from National Institutes of Health Shared Instrumentation grant S10OD025017 and National Science Foundation Major Research Instrumentation grant CHE-0722519. We also thank Double Helix Optics for providing their SPINDLE system and DH-PSF phase mask, which was used for engineered PSF data capture, and acknowledge X. Yang and M.P. Lake for their assistance with these engineered PSF experiments and related analysis.
Author information
Authors and Affiliations
Contributions
A.O., Y.W. and Y.R. initiated the research. Y.W., Y.R. and H.W. performed the experiments. E.B.-D. cultured and prepared C. elegans samples. Y.W. and Y.L. processed the data. L.A.B. and C.P. helped with the experiments and analysis. A.O., Y.W. and Y.R. prepared the manuscript. A.O. supervised the research.
Corresponding author
Ethics declarations
Competing interests
A.O., Y.W. and Y.R. have a pending patent application on the presented framework.
Additional information
Peer review information Rita Strack was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Quantification of SNR improvement through Deep-Z.
Top row: an input image of a 300 nm fluorescent bead was digitally refocused to a plane 2 µm above it using Deep-Z, where the ground truth was the mechanically scanned fluorescence image acquired at this plane. Bottom row: same images as the first row, but saturated to a dynamic range of [0, 10] to highlight the background. The SNR values were calculated by first taking a Gaussian fit (see the Methods section) on the pixel values of each image to find the peak signal strength. Then the pixels in the region of interest (ROI) that were 10σ away (where σ2 is the variance of the fitted Gaussian) were regarded as the background (marked by the region outside the red dotted circle in each image) and the standard deviation of these pixel values was calculated as the background noise. The Deep-Z network rejects background noise and improves the output image SNR by ~ 40 dB, compared to the mechanical scan ground truth image. Analysis was performed on a randomly selected particle from a group of 96 images with similar results.
Supplementary Figure 2 Digital refocusing of fluorescence images of C. elegans worms.
(a, k) Measured fluorescence images (Deep-Z input). (b, d, l, n) Deep-Z output at different target heights (z). (c, e, m, o) Ground truth (GT) images, captured using a mechanical axial scanning microscope at the same heights as the Deep-Z outputs. (f, p) overlay of Deep-Z output images in magenta and GT images in green. (g, i, q, s) Absolute difference images of Deep-Z output images and the corresponding GT images at the same heights. (h, j, r, t) Absolute difference images of Deep-Z input and the corresponding GT images. Structural similarity index (SSIM) and root mean square error (RMSE) were calculated for the output vs. GT and the input vs. GT for each region, displayed in (g, i, q, s) and (h, j, r, t), respectively. Scale bar: 25 µm. Experiments were repeated with 20 images, achieving similar results.
Supplementary Figure 3 Structural similarity (SSIM) index and correlation coefficient (Corr. Coeff.) analysis for digital refocusing of fluorescence images from an input plane at zinput to a target plane at ztarget.
We created a scanned fluorescence z-stack of a C. elegans sample, within an axial range of -20 µm to 20 µm, with 1 µm spacing. First column: each scanned image at zinput in this stack was compared against the image at ztarget, forming cross-correlated SSIM and Corr. Coeff. matrices. Both the SSIM and Corr. Coeff. fall rapidly off the diagonal entries. Second column: A Deep-Z network trained with fluorescence image data corresponding to +/- 7.5 µm propagation range (marked by the cyan diamond in each panel) was used to digitally refocus images from zinput to ztarget. The output images were compared against the ground truth images at ztarget using SSIM and Corr. Coeff. Third column: same as the second column, except the training fluorescence image data included up to +/- 10 µm axial propagation (marked by the cyan diamond that is now enlarged compared to the second column). These results confirm that Deep-Z learned the digital propagation of fluorescence, but it is limited to the axial range that it was trained for (determined by the training image dataset). Outside the training range (defined by the cyan diamonds), both the SSIM and Corr. Coeff. values considerably decrease.
Supplementary Figure 4 3D imaging of C. elegans head neuron nuclei using Deep-Z.
The input and ground truth images were acquired by a scanning fluorescence microscope with a 40×/1.4NA objective. A single fluorescence image acquired at z=0 µm focal plane (marked by dashed yellow rectangle) was used as the input image to Deep-Z and was digitally refocused to different planes within the sample volume, spanning around -4 to 4 µm; the resulting images provide a good match to the corresponding ground truth images. Scale bar: 25 µm. Experiments were repeated with 12 images, achieving similar results.
Supplementary Figure 5 Digital refocusing of fluorescence microscopy images of BPAEC using Deep-Z.
The input image was captured using a 20×/0.75 NA objective lens, using the Texas Red and FITC filter sets, occupying the red and green channels of the image, for the mitochondria and F-actin structures, respectively. Using Deep-Z, the input image was digitally refocused to 1 µm above the focal plane, where the mitochondrial structures in the green channel are in focus, matching the features on the mechanically-scanned image (obtained directly at this depth). The same conclusion applies for the Deep-Z output at z = 2 µm, where the F-actin structures in the red channel come into focus. After 3 µm above the image plane, the details of the image content get blurred. The absolute difference images of the input and output with respect to the corresponding ground truth images are also provided, with SSIM and RMSE values, quantifying the performance of Deep-Z. Scale bar: 20 µm. Experiments were repeated with 180 images, achieving similar results.
Supplementary Figure 6 Photobleaching analysis.
(a) Fluorescence signal of nanobeads imaged in 3D, for 180 times of repeated axial scans, containing 41 planes, spanning +/- 10 µm with a step size 0.5 µm. The accumulated scanning time is ~30 min. (b) The corresponding scan for a single plane, which is used by Deep-Z to generate a virtual image stack, spanning the same axial depth within the sample (+/- 10 µm). The accumulated scanning time for Deep-Z is ~ 15 seconds. The center line represents the mean and the shaded region represents the standard deviation of the normalized intensity for 681 and 597 individual nanobeads (for (a) and (b), respectively) inside the sample volume.
Supplementary Figure 7 Refocusing capability of Deep-Z under lower image exposure.
(a) Virtual refocusing of images containing two microbeads under different exposure times from defocused distances of -5, 3 and 4.5 µm, using two Deep-Z models trained with images captured at 10 ms and 100 ms exposure times, respectively. (b) Median FWHM values of 91 microbeads imaged inside a sample FOV after the virtual refocusing of an input image across a defocus range of -10 µm to 10 µm by the Deep-Z (100 ms) network model. The test images have different exposure times spanning 3 ms to 300 ms. (c) Same as (b), but plotted for the Deep-Z (10 ms) model.
Supplementary Figure 8 Deep-Z based virtual refocusing of a different sample type and transfer learning results.
(a) The input image records the neuron activities of a C. elegans that is labeled with GFP; the image is captured using a 20×/0.8NA objective under the FITC channel. The input image was virtually refocused using both the optimal worm strain model (denoted as: same model, functional GFP) as well as a different model (denoted as: different model, structural tagRFP); we also report here the results of a transfer learning model which used the different model as its initialization and functional GFP image dataset to refine it after ~ 500 iterations (~30 min of training). (b) A different C. elegans sample is shown. The input image records the neuron nuclei labeled with tagRFP imaged using a 20×/0.75NA objective under the Texas Red channel. The input image was virtually refocused using both the exact worm strain model (same model, structural, tagRFP) as well as a different model (different model, 300 nm red beads); we also report here the results of a transfer learning model which used the different model as its initialization and structural ragRFP image dataset to refine it after ~ 4,000 iterations (~6 hours training). Image correlation coefficient (r) is shown at the lower right corner of each image, in reference to the ground truth mechanical scan performed at the corresponding microscope system (Leica and Olympus, respectively). The transfer learning was performed using 20% of the training data and 50% of the validation data, randomly selected from the original data set.
Supplementary Figure 9 Virtual refocusing of a different microscope system and transfer learning results.
The input image records the C. elegans neuronal nuclei labeled with tag GFP, imaged using a Leica SP8 microscope with a 20×/0.8NA objective. The input image was virtually focused using both the exact model (Leica SP8 20×/0.8NA) as well as a different model (denoted as: different model, Olympus 20×/0.75NA); we also report here the results of a transfer learning model using the different model as its initialization and Leica SP8 image dataset to refine it after ~ 2,000 iterations (~40 min training). Image correlation coefficient (r) is shown at the lower right corner of each image, in reference to the ground truth mechanical scan performed at the corresponding microscope system. The transfer learning was performed using 20% of the training data and 50% of the validation data, randomly selected from the original data set.
Supplementary Figure 10 Time-modulated signal reconstruction using Deep-Z.
(a) A time-modulated illumination source was used to excite the fluorescence signal of microbeads (300 nm diameter). Time-lapse sequence of the sample was captured under this modulated illumination at the in-focus plane (z = 0 µm) as well as at various defocused planes (z = 2–10 µm) and refocused using Deep-Z to digitally reach z = 0 µm. Intensity variations of 297 individual beads inside the FOV (after refocusing) were tracked for each sequence. (b) Based on the video captured in (a), we took every other frame to form an image sequence with twice the frame-rate and modulation frequency, and added it back onto the original sequence with a lateral shift. These defocused and super-imposed images were virtually refocused using Deep-Z to digitally reach z=0 µm, in-focus plane. Group 1 contained 297 individual beads inside the FOV with 1 Hz modulation. Group 2 contained the signals of the other (new) beads that are super-imposed on the same FOV with 2 Hz modulation frequency. Each intensity curve was normalized, and the mean (plotted as curve center) and the standard deviation (plotted as error bar) of the 297 curves were plotted for each time-lapse sequence. Virtually-refocused Deep-Z output tracks the sinusoidal illumination, very closely following the in-focus reference time-modulation reported in target (z = 0 µm). Also see the Supplementary Video 5, related to this figure.
Supplementary Figure 11 Deep-Z based virtual refocusing of a laterally shifted weaker fluorescent object next to a stronger object.
(a) A defocused experimental image (left bead) at plane z was shifted laterally by d pixels to the right and digitally weakened by a pre-determined ratio (right bead), which was then added back to the original image, used as the input image to Deep-Z. Scale bar: 5 μm. (b) An example of the generated bead pair with an intensity ratio of 0.2; we show in-focus plane, defocused planes of 4 and 10 µm, and the corresponding virtually-refocused images by Deep-Z. (c-h) Average intensity ratio of the shifted and weakened bead signal with respect to the original bead signal for 144 bead pairs inside a FOV, calculated at the virtually refocused plane using different axial defocus distances (z). The crosses “x” in each figure mark the corresponding lateral shift distance, below which the two beads cannot be distinguished from each other, color-coded to represent bead signal intensity ratio (spanning 0.2–1.0).
Supplementary Figure 12 Impact of axial occlusions on Deep-Z virtual refocusing performance.
(a) 3D virtual refocusing of two beads that have identical lateral positions but are separated axially by 8 µm; Deep-Z, as usual, used a single 2D input image corresponding to the defocused image of the overlapping beads. The virtual refocusing calculated by Deep-Z exhibits two maxima representing the two beads along the z-axis, matching the simulated ground truth image stack. (b) Simulation schematic: two defocused images in the same bead image stack with a spacing of d was added together, with the higher stack located at a depth of z=8 µm. A single image in the merged image stack was used as the input to Deep-Z for virtual refocusing. (c-d) report the average and the standard deviation (represented by transparent colors) of the intensity ratio of the top (i.e., the dimmer) bead signal with respect to the bead intensity in the original stack, calculated for 144 bead pairs inside a FOV, for z = 8 µm with different axial separations (d) and bead intensity ratios (spanning 0.2–1.0).
Supplementary Figure 13 Deep-Z inference results as a function of 3D fluorescent sample density.
(a) Comparison of Deep-Z inference against a mechanically-scanned ground truth image stack over an axial depth of +/- 10 µm with increasing fluorescent bead concentration. The measured bead concentration resulting from the Deep-Z output (using a single input image) as well as the mechanically-scanned ground truth (which includes 41 axial images acquired at a scanning step size of 0.5 µm) is shown on the top left corner of each image. MIP: maximal intensity projection along the axial direction. Scale bar: 30 µm. (b-e) Comparison of Deep-Z output against the ground truth results as a function of the increasing bead concentration. The red line is a 2nd order polynomial fit to all the data points. The black dotted line represents y=x, shown for reference. These particle concentrations were calculated/measured over a FOV of 1536×1536 pixels (500×500 µm2), i.e. 15-times larger than the specific regions shown in (a).
Supplementary Figure 14 C. elegans neuron segmentation comparison.
(a, d) The fluorescence image used as input to Deep-Z. (b, e) Segmentation results based on (a, d), respectively. (c, f) Segmentation results based on the virtual image stack (-10 to 10 µm) generated by Deep-Z using the input images in (a, d), respectively. (g) An additional fluorescence image, captured at a different axial plane (z = 4 µm). (h) Segmentation results on the merged virtual stack (-10 to 10 µm). The merged image stack was generated by blending the two virtual stacks generated by Deep-Z using the input images (d) and (g). (i) Segmentation results based on the mechanically-scanned image stack used as ground truth (acquired at 41 depths with 0.5 µm axial spacing). Each neuron was represented by a small sphere in the segmentation map and the depth information of each neuron was color-coded. (j-l) The detected neuron positions in (e, f, h) were compared with the positions in (i) (see the Supplementary Note 7 for details), and the axial displacement histograms between the Deep-Z results and the mechanically-scanned ground truth results were plotted.
Supplementary Figure 15 C. elegans neuron activity tracking and clustering.
(a) Max intensity projection (MIP) along the axial direction of the median-intensity image over time. The red channel (Texas red) labels neuron nuclei and the green channel (FITC) labels neuron calcium activity. A total of 155 neurons were identified in the 3D stack, as labeled here. Scale bar: 25 µm. Scale bar for the zoom-in regions: 10 µm. (b) The intensity of the neuron calcium activity, ΔF(t), of these 155 neurons is reported over a period of ~ 35 s at ~3.6Hz. Based on a threshold on the standard deviation of each ΔF(t), we separate neurons to be active (right-top, 70 neurons) and less active (right-bottom, 85 neurons). (c) The similarity matrix of the calcium activity patterns of the top 70 active neurons. (d) The top 40 eigen values of the similarity matrix. An eigen-gap is shown at k=3, which was chosen as the number of clusters according to eigen-gap heuristic (i.e. choose up to the largest eigenvalue before the eigenvalue gap, where the eigenvalues increase significantly). (e) Normalized activity ΔF(t)/ F0 for the k=3 clusters after the spectral clustering on the 70 active neurons. (f) Similarity matrix after spectral clustering. The spectral clustering rearranged the row and column ordering of the similarity matrix (c) to be block diagonal in (f), which represents three individual clusters of calcium activity patterns.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15 and Supplementary Notes 1–10
Supplementary Video 1
Deep-Z inference comparison against the images captured using a mechanical axial scan of a C. elegans sample. Left, a single fluorescence image of the C. elegans worm was captured at the reference plane (z = 0 µm), which was used as the Deep-Z input. Middle, the input image was appended with different DPMs and passed through the trained Deep-Z network to digitally refocus the input image to a series of planes, from −10 µm to 10 µm (with a step size of 0.5 µm) with respect to the reference plane. Right, mechanical axial scan of the same C. elegans worm at the same series of planes, used for comparison.
Supplementary Video 2
3D visualization of a C. elegans using Deep-Z inference. Left, a single fluorescence image of a C. elegans was captured and used as Deep-Z input. Middle, the input fluorescence image was digitally refocused using Deep-Z to a series of planes from −10 µm to 10 µm (with an axial step size of 0.5 µm) to generate a 3D stack. This 3D stack was rotated around the vertical axis of the input image, spanning 360° with a step size of 2°. Maximum intensity projection of the 3D volume at each rotated angle is shown in the video, which was generated using the ImageJ plugin ‘Volume Viewer’. Right, the same 3D stack was deconvolved using Lucy-Richardson deconvolution regularized by total variation in ImageJ plugin ‘DeconvolutionLab2’. The deconvolved 3D stack was rotated and displayed in the same way as the middle video.
Supplementary Video 3
Deep-Z 3D inference from a 2D video containing four moving C. elegans. A fluorescence video containing four C. elegans was recorded at a single plane (z = 0 µm) at 10 frames per second for 10 s. Each frame was digitally refocused using Deep-Z to a series of planes at z = −6, −4, −2, 2 and 4 µm away from the input plane, generating virtual videos at these different depths in 3D. The input video and the Deep-Z-generated videos were played simultaneously at one frame per second, that is, they were slowed down tenfold.
Supplementary Video 4
Deep-Z 3D inference from a 2D video containing a defocused moving C. elegans. A fluorescence video was captured at a single focal plane (z = 0 µm) at 3 frames per second for a duration of 18 s. Each frame was digitally refocused using Deep-Z to a series of planes at z = 2, 4, 6, 8 and 10 µm away from the input focal plane, generating virtual videos at these different depths in 3D. In the input video, the worm was mostly defocused owing to sample drift and motion. Using Deep-Z, neurons are rapidly refocused at these virtual planes in 3D.
Supplementary Video 5
Deep-Z-based refocusing of spatiotemporally modulated bead images. Videos contain two groups of 300-nm bead emitters (sinusoidally modulated at 1 Hz and 2 Hz, respectively). Deep-Z was used to digitally refocus the defocused videos to virtually reach the z = 0 µm plane. An example region of interest containing six pairs of such emitters was cropped and shown in this video (also see Supplementary Figure 10).
Supplementary Video 6
Tracking of neuron calcium activity events in 3D from a single 2D fluorescence video. A fluorescence video containing a fixed C. elegans was recorded at a single focal plane (z = 0 µm) at ~3.6 frames per second for ~35 s. The video contained two color channels: the red channel represents the Texas Red fluorescence targeting the red fluorescent protein (RFP)-tagged neuron nuclei and the green channel represents the FITC fluorescence targeting the green fluorescent protein (GFP)-tagged neuron activities. Each frame was digitally refocused using Deep-Z to a series of planes from −10 to 10 µm with a step size of 0.5 µm, for each of the fluorescence channels. MIP was applied along the axial direction to generate an extended depth of field image for each frame. Expanded views (2×) of the head and tail regions are shown for better visualization. The input video and the Deep-Z-generated MIP video were played simultaneously at ~3.6 frames per second.
Supplementary Video 7
Locations of the detected neurons in 3D. Individual neuron locations were isolated from the Deep-Z-inferred 3D stack of the neuron calcium activity video. The isolated neuron locations were plotted in 3D. A rough shape of the worm was also plotted, which was generated by thresholding the autofluorescence of the worm in the Deep-Z-generated 3D stack. The 3D plot was rotated around the z-axis at 1° steps for 360° and viewed at 15° tilt. Each neuron was color-coded according to its depth (z) location, as indicated by the color-bar on the right.
Supplementary Video 8
Deep-Z-based 3D structural imaging of C. elegans at 100 Hz. A C. elegans worm was imaged using a 20×/0.8 NA objective lens under the Texas Red channel to capture its tagRFP signal, labeling the neuron nuclei. A fluorescence video was captured at a single focal plane (z = 0 µm) at 100 full frames per second for a duration of 10 s using the stream mode of the camera. Each frame was digitally refocused using Deep-Z to a series of planes at z = −2, 2, 4, 6, 8 and 10 µm away from the input focal plane, generating virtual videos at these different depths in 3D, as well as a corresponding MIP video (over an axial depth range of ±10 µm).
Supplementary Video 9
Deep-Z-based 3D functional imaging of C. elegans at 100 Hz. A C. elegans worm was imaged using a 20×/0.8 NA objective lens under the FITC channel to capture its GFP signal that labels its calcium activity. A fluorescence video was captured at a single focal plane (z = 0 µm) at 100 full frames per second for a duration of 10 s using the stream mode of the camera. Each frame was digitally refocused using Deep-Z to a series of planes at z = −2, 2, 4, 6, 8 and 10 µm away from the input focal plane, generating virtual videos at these different depths in 3D, as well as a corresponding MIP video (over an axial depth range of ±10 µm).
Supplementary Video 10
Deep-Z-based refocusing of microscopic objects imaged with a 3D-engineered PSF. Left, a single fluorescence image of 300-nm red fluorescence beads with a 3D-engineered double-helix PSF was captured at the reference plane (z = 0 µm), which was used as the Deep-Z input. Middle, the input image on the left was appended with different DPMs and passed through a trained Deep-Z network to digitally refocus the input image to a series of planes, from −13 µm to 10 µm (with a step size of 0.2 µm) with respect to the reference plane. Right, mechanical axial scan of the same sample at the same corresponding planes, used for comparison.
Rights and permissions
About this article

Cite this article
Wu, Y., Rivenson, Y., Wang, H. et al. Three-dimensional virtual refocusing of fluorescence microscopy images using deep learning. Nat Methods 16, 1323–1331 (2019). https://doi.org/10.1038/s41592-019-0622-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-019-0622-5
This article is cited by
-
Visualizing cortical blood perfusion after photothrombotic stroke in vivo by needle-shaped beam optical coherence tomography angiography
PhotoniX (2024)
-
Quantitative phase imaging by gradient retardance optical microscopy
Scientific Reports (2024)
-
Establishing a reference focal plane using convolutional neural networks and beads for brightfield imaging
Scientific Reports (2024)
-
Signal Processing and Artificial Intelligence for Dual-Detection Confocal Probes
International Journal of Precision Engineering and Manufacturing (2024)
-
Optical-resolution photoacoustic microscopy with a needle-shaped beam
Nature Photonics (2023)
