
Abstract
The predominant approach for antibody generation remains animal immunization, which can yield exceptionally selective and potent antibody clones owing to the powerful evolutionary process of somatic hypermutation. However, animal immunization is inherently slow, not always accessible and poorly compatible with many antigens. Here, we describe ‘autonomous hypermutation yeast surface display’ (AHEAD), a synthetic recombinant antibody generation technology that imitates somatic hypermutation inside engineered yeast. By encoding antibody fragments on an error-prone orthogonal DNA replication system, surface-displayed antibody repertoires continuously mutate through simple cycles of yeast culturing and enrichment for antigen binding to produce high-affinity clones in as little as two weeks. We applied AHEAD to generate potent nanobodies against the SARS-CoV-2 S glycoprotein, a G-protein-coupled receptor and other targets, offering a template for streamlined antibody generation at large.

Similar content being viewed by others
Main
It is hard to overstate the importance of antibodies in the life sciences. Because of their remarkable ability to specifically recognize biomolecules, antibodies are critical in basic research (western blotting, cytometry, imaging and structural biology), diagnostics (pathogen detection and histology), therapy (antibody drugs) and public health (epidemic response)1,2,3,4. Indeed, the versatility of antibodies has been on display in the current COVID-19 pandemic, where patient-derived5, animal-derived6 and synthetic7 antibodies have been fast-tracked for development into diagnostic tools and therapeutics.
Unfortunately, existing techniques for antibody discovery are typically slow, difficult to scale and often unreliable. For example, the main approach for generating custom antibodies through animal immunization suffers from fundamental challenges such as tolerance to self-antigens, immunodominance and incompatibility with membrane proteins that require detergent solubilization. Compounding these fundamental challenges that limit the scope of addressable targets are practical challenges, primarily the lengthy timelines and high cost associated with animal immunization, as well as ethical challenges in animal welfare8. In vitro display technologies that involve the selection of high-affinity antibodies from antibody libraries expressed on the surface of phage or cells have been developed to overcome the problems with animal immunization, but forfeit a key advantage of animal immune systems—the seamless transformation of low-affinity germline antibodies9 into high-affinity clones through the evolutionary process of affinity maturation by somatic hypermutation10,11. As a result, in vitro display technologies necessitate strategies that introduce their own hurdles in speed, cost and scalability. These include the execution of affinity maturation campaigns requiring technically complex rounds of antibody gene diversification, transformation and selection12 or the design and construction of massive (often proprietary) libraries13 that only partially compensate for the loss of dynamic sequence search during affinity maturation. An additional overarching challenge with animal immunization and in vitro antibody discovery technologies is that both techniques are specialized and not readily accessible to many researchers. This creates inefficiencies where research decisions—for example, which protein from a screen to follow up—are influenced by the availability of commercial antibodies rather than the merits of the research alone, research reproducibility is eroded by overdependence on unreliable external antibody sources14,15, and the extent and speed of antibody discovery efforts responding to urgent crises such as COVID-19 become constrained. The difficulty of generating high-quality antibodies therefore remains an important problem in biomedical research.
In this Article, we describe ‘autonomous hypermutation yeast surface display’ (AHEAD), a highly accessible animal-free antibody generation technology that mimics the process of vertebrate somatic hypermutation using yeast. AHEAD provides exceptional speed, simplicity and effectiveness in the generation of potent antibodies for the life sciences and is an initial step towards a future antibody engineering landscape that will require minimal human effort.
Results
Design of AHEAD
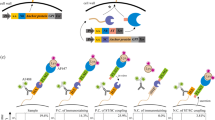
AHEAD pairs orthogonal DNA replication (OrthoRep) with yeast surface display (YSD) to achieve rapid antibody evolution through the simple cultivation and sorting of yeast cells. In OrthoRep, an orthogonal error-prone DNA polymerase replicates a special cytosolic plasmid (p1) that stably propagates in Saccharomyces cerevisiae without elevating genomic mutation rates16,17. This results in the durable continuous hypermutation of p1-encoded genes at a mutation rate of 10−5 substitutions per base, which is 100,000-fold higher than yeast’s genomic mutation rate of 10−1 substitutions per base. When antibody fragments are encoded on p1, yeast cells self-diversify their displayed antibodies, resulting in the autonomous exploration of sequence space. When subjected to sequential rounds of sorting for antigen binding, the continuously diversifying antibodies rapidly improve to yield high-affinity, high-quality antibody clones in a short period of time (Fig. 1a).

a, Scheme for the rapid evolution of high-affinity binding using AHEAD. Ab, antibody fragment; DNAP, DNA polymerase; HA, hemagglutinin tag. b, Cytometry plot showing detection of a functionally surface-displayed single-chain antibody fragment (scFv) and a functionally surface-displayed nanobody (Nb) encoded on the p1 orthogonal plasmid, replicated by an associated orthogonal DNAP. The orthogonal DNAP used in this case was the wt TP-DNAP1 (Methods) rather than the error-prone TP-DNAP1-4-2 variant that was used for all subsequent AHEAD evolution experiments. Cognate antigens for 4-4-20 (fluorescein) and AT110 (AT1R) were labeled with biotin and FLAG tag, respectively, and detected with AF647-conjugated streptavidin and allophycocyanin-conjugated anti-FLAG, respectively. The HA tag was detected with mouse anti-HA and a goat anti-mouse AF488-conjugated secondary antibody.
We first tested whether two known antibody fragments could be encoded on p1 for cell surface display. Specifically, we tested a single-chain variable fragment called 4-4-20 that binds fluorescein18 and a camelid single-domain antibody fragment (‘nanobody’) called AT110 that recognizes a prototypical G-protein-coupled receptor (GPCR)19 (Fig. 1b). Constitutive expression of 4-4-20 and AT110 fused to the mating adhesion receptor, Aga2p, along with induced expression of the genome-encoded cell wall anchoring subunit, Aga1p (ref. 18), resulted in functional display on the yeast surface as measured by flow cytometry (Fig. 1b). This set the stage for cycles of yeast culture and fluorescence activated cell sorting (FACS) on target binding to effect the rapid evolution of high-affinity antibodies, akin to affinity maturation in vertebrate immune systems9,20.
Evolution of antibody fragments with AHEAD
To test whether AHEAD could generate high-affinity antibodies through cycles of yeast culture and FACS, we sought to evolve the nanobody, AT110. Nanobodies are single-domain antibody fragments derived from the VHH domains of heavy-chain antibodies in llamas, camels and their relatives21 (Extended Data Fig. 1). These unique antibody fragments are capable of stabilizing distinct functional conformations of GPCRs and other dynamic proteins by virtue of their propensity to bind concave epitopes22. This has made the discovery of high-affinity nanobodies a mainstay of GPCR structural biology and pharmacology23. Nanobodies are also particularly appropriate for animal-free antibody generation approaches, as the alternate of immunizing camelids carries logistical challenges of large animal husbandry and associated animal welfare issues24. AT110 is a low-affinity nanobody that binds selectively to the active-state conformation of the angiotensin II receptor type 1 (AT1R), discovered via in vitro selection from a naïve synthetic nanobody library. However, AT110 required several rounds of manual error-prone polymerase chain reaction (PCR) diversification, selection and engineering to reach the affinity necessary for co-crystallization and structural studies19,25. Using AHEAD and starting from AT110, we carried out iterative cycles of yeast culture and FACS to improve AT110 (Fig. 2a). This experiment yielded nanobody AT110i103 (Supplementary Table 1), with an allosteric modulation potency of 2.5 nM for enhancing agonist binding to the AT1R GPCR (Fig. 2b), representing a roughly 20-fold functional affinity increase over the parent clone. Notably, some mutations (for example, I98V and a non-synonymous change at Y113) were identified both by AHEAD and previous efforts19, while other affinity-enhancing mutations (for example, R45C and R66H) were only identified by AHEAD (Extended Data Fig. 2). Interestingly, Y113H and I98V were synergistic in their ability to modulate agonist-bound AT1R (Fig. 2b), showing that AHEAD could find complex functional outcomes. Overall, this experiment demonstrated that AHEAD could produce high-affinity antibodies in a much more streamlined manner than conventional approaches.

a, Enrichment of affinity-increasing mutations in anti-AT1R nanobodies through cycles of AHEAD, as determined by NGS of the p1-encoded nanobody population in each cycle of AHEAD. ‘Pre’ indicates the population composition before the first cycle of AHEAD. b, Nanobody potency was assessed in a radioligand allosteric shift assay (Methods). This measures the ability of each nanobody to enhance agonist affinity by stabilizing an active-state receptor conformation, serving as an indirect measure of nanobody binding affinity. Error bars represent the s.e.m. from three independent experiments performed as single replicates. NA, not applicable.
An improved second-generation AHEAD system
Although successful, lessons learned during AT110 nanobody evolution motivated us to redesign AHEAD’s nanobody display constructs (Extended Data Fig. 3). In particular, we found that the average level of AT110 display was low, which complicated the FACS selection process in cycles of AHEAD, as only a small fraction of cells expressed AT110 at the levels needed to detect antigen binding. We temporarily overcame this problem by using magnetic activated cell sorting (MACS) before each FACS round to enrich the subpopulations of cells with high AT110 display. Because OrthoRep maintains p1 in multiple copies16, copy number fluctuations create subpopulations that express substantially more nanobody than average, allowing effective MACS enrichment of these higher expressers when large numbers of cells are used. However, the requirement for MACS steps substantially increased the time and effort needed for antibody evolution, counter to AHEAD’s potential as a rapid and streamlined antibody generation system.
To permanently address this challenge, we engineered four changes into AHEAD’s antibody expression cassette (Extended Data Fig. 3a). First, we adopted an improved display architecture26 that places the nanobody at the N terminus of the Aga2p fusion polypeptide (Extended Data Figs. 3a and 4a). Second, we introduced a new p1-specific promoter, called pGA, that contains two expression-enhancing mutations discovered in an unrelated OrthoRep-based protein engineering evolution project from our laboratory. Third, we engineered a stronger secretory leader using directed evolution from the strongest known secretory leader, app8 (ref. 27), resulting in secretory leader app8i1 (Extended Data Fig. 4b). Finally, we incorporated a polyadenosine tail downstream of the nanobody gene for AHEAD, which was previously found to increase the expression of p1-encoded genes28 (Extended Data Fig. 3a). These features together increased the surface display level of nanobodies by ~25-fold, allowing the typical cell in a population to display enough nanobody for direct antigen-binding selection by FACS, even when the binding was weak (Extended Data Fig. 3b). This second-generation system (AHEAD 2.0) supports our intended process of cycling directly between yeast culture and FACS to generate antibodies (Fig. 1a).
To validate AHEAD 2.0, we ran pilot antibody fragment evolution experiments against model antigens including human serum albumin (HSA) and green fluorescent protein (GFP). High-affinity nanobodies against HSA are useful as domains that can be fused to therapeutic proteins to increase their serum half-life29. High-affinity nanobodies against GFP are valuable for immunostaining in GFP-expressing tissue samples where conventional antibodies take too long to penetrate because of their high molecular weight (for example, cleared brain)30,31. Starting from moderate-affinity nanobodies targeting HSA and GFP, namely Nb.b201 (ref. 32) and Lag42 (ref. 33), respectively, we successfully evolved higher-affinity clones through four to six cycles of AHEAD (Extended Data Fig. 4c–e and Supplementary Table 1).
Evolution of SARS-CoV-2 antibodies using AHEAD 2.0
AHEAD is distinguished by its coincident speed and parallelizability for antibody maturation. These properties are particularly valuable for an outbreak response, where urgency demands rapid discovery (speed) of high-affinity antibodies through many independent antibody generation campaigns that collectively maximize the probability of success (parallelizability). In light of the current COVID-19 pandemic, we asked whether AHEAD 2.0 would be capable of generating collections of potent nanobodies against the novel coronavirus, SARS-CoV-234. Starting from an open-source naïve nanobody YSD library32, we selected eight clones that bound the receptor-binding domain (RBD) of the SARS-CoV-2 spike (S) protein (Extended Data Fig. 5a and Supplementary Table 1). Each of these distinct clones was then independently encoded on p1 for streamlined evolution (that is, affinity maturation). The parallelizability of AHEAD allowed us to simultaneously run eight independent affinity maturation experiments, one for each of the parental clones, to avoid competition among parents and initial lineages. We reasoned that this separation of lineages would (1) prevent the early loss of weak parents that have the potential to reach high affinity late in the affinity maturation process and (2) maintain the functional diversity in binding modes potentially represented across parents (Extended Data Fig. 5a). After three to eight cycles of AHEAD where the uninterrupted cycle time defined by yeast culturing and FACS was only three days, all eight experiments produced multi-mutation nanobodies (Fig. 3a,b and Supplementary Table 1) with higher affinity for RBD than their parents (Fig. 3c, Table 1, Supplementary Data 1 and Extended Data Fig. 6). Notably, nanobodies RBD1i13, RBD3i17, RBD6id, RBD10i10, RBD10i14 and RBD11i12 exhibited monovalent RBD-binding affinity improvements up to ~580-fold over the course of affinity maturation (Fig. 3 and Table 1), and one nanobody, RBD10i14, reached a subnanomolar monovalent Kd of 0.72 nM (Fig. 3c and Table 1). These levels of evolutionary improvement from naïve nanobodies, achieved through a fast and parallelizable process, confirm the power of AHEAD.

a, Sequential FACS plots showing affinity maturation of an anti-SARS-CoV-2 nanobody (parent = RBD10). Red polygons correspond to gates used for sorting. (See Extended Data Fig. 5b for similar plots showing affinity maturation from all parents.) b, Locations of nanobody mutations fixed in eight independent AHEAD experiments starting from different parental clones. (See Supplementary Data 1 for exact mutations.) c, SPR traces and associated monovalent affinities for select anti-SARS-CoV-2 nanobodies evolved using AHEAD. (See Extended Data Fig. 6 for affinity measurements on additional nanobodies.) Each nanobody was tested as an immobilized Fc fusion over which listed concentrations of RBD were flowed. RU, response unit. d, Neutralization activities of select anti-SARS-CoV-2 nanobodies on pseudotyped SARS-CoV-2 virus. Each nanobody concentration (x axis) was tested in replicate. n = 6, error bars represent ±s.d. (See Extended Data Fig. 7a for neutralization activities of additional nanobodies.) e, Biolayer interferometry traces measuring ACE2 competition for RBD binding in the presence of select anti-SARS-CoV-2 nanobodies evolved using AHEAD. (See Extended Data Fig. 7b for ACE2 competition activities of additional nanobodies and control antibodies.) f, Affinity and neutralization potency improvements of nanobodies isolated from different cycles of AHEAD during each parent nanobody’s affinity maturation. Each filled circle represents a nanobody’s affinity and the open circle of identical color represents the nanobody’s neutralization potency. The number within each circle designates the AHEAD cycle from which the nanobody was isolated.
Anti-RBD nanobodies neutralize SARS-CoV-2 pseudovirus
Anti-RBD monoclonal antibodies have emerged as promising therapeutics for the treatment of COVID-1935,36. These antibodies act by inhibiting the interaction between SARS-CoV-2 and its receptor, angiotensin-converting enzyme 2 (ACE2), thereby blocking entry into cells. To probe the potential of our anti-RBD nanobodies as therapeutic candidates, we genetically fused the nanobodies to the Fc region of the human immunoglobulin G1 (IgG1) antibody isotype and carried out neutralization assays against SARS-CoV-2-pseudotyped virus. We found that many of the evolved anti-RBD nanobodies had exceptional neutralization potencies representing up to ~925-fold improvements over their parent sequences (Fig. 3d, Table 1 and Extended Data Fig. 7a). For example, nanobodies RBD1i13, RBD3i17, RBD6id, RBD10i10, RBD10i14 and RBD11i12 exhibited low-nanomolar or subnanomolar half-maximal inhibitory concentration (IC50) values of 0.66, 1.51, 0.72, 2.44, 5.38 and 0.52 nM, respectively (Fig. 3d, Table 1, Supplementary Data 1 and Extended Data Fig. 7a). Interestingly, nanobodies RBD1i13 and RBD11i12, which had the strongest viral neutralization potencies among all evolved variants, descended from parents that were relatively poor neutralizers (Table 1 and Supplementary Data 1). This highlights the value of the experimental parallelizability available with AHEAD: through evolution experiments that kept each parent clone’s lineages separate (Extended Data Fig. 5a), early high achievers such as RBD6 could not outcompete the initially low-performing lineages that ultimately gave rise to the most potent neutralizers.
Anti-RBD nanobodies exhibit diversity in inhibition modes
To understand how evolved anti-RBD nanobodies inhibit SARS-CoV-2 pseudovirus infection, we tested potent neutralizers for their ability to compete with ACE2 in binding to RBD. Nanobodies RBD1i13, RBD6id and RBD11i12 strongly or moderately competed with ACE2, whereas a fourth clone, RBD10i10, did not (Fig. 3e, Table 1 and Extended Data Fig. 7b). This suggests that different nanobodies bind RBD at different locations, which may translate to potency against diverse SARS-CoV-2 variants. We probed this finding further by using a recently developed deep mutational scanning assay to reveal single mutations in RBD that escape nanobody binding37,38. In this assay, a library of yeast-displayed RBD mutants representing every single amino acid change was first sorted for those that maintain binding to soluble human ACE2, then labeled with each nanobody under investigation, and finally sorted for low nanobody labeling. This pipeline results in the enrichment of functional RBD mutants that escape nanobody binding, which can be measured by next-generation sequencing (NGS; Fig. 4a). The RBD escape mutations roughly define the epitope that the nanobody under investigation binds, and predicts which SARS-CoV-2 variants of concern may be addressable with that nanobody. This mutational scanning assay elucidated why we saw different degrees of ACE2 competition by nanobodies RBD1i13, RBD10i10 and RBD11i12. Specifically, RBD mutations that escape binding by RBD1i13’s parent nanobody, RBD1i1, are immediately adjacent to the ACE2 binding site when mapped to the structure of the RBD–ACE2 complex39, while the RBD mutations that escape nanobody RBD10i10 are not (Fig. 4b). RBD mutations that escape nanobody RBD11i12 are physically closer to ACE2 than those that escape nanobody RBD10i10 but more distal to ACE2 than those that escape nanobody RBD1i13 (Fig. 4b), consistent with our observation that RBD11i12 competes with ACE2 binding to RBD more modestly than RBD1i13 (Fig. 3e and Extended Data Fig. 7b). Notably, mutations in RBD capable of escaping nanobodies RBD1i13 and RBD10i10 do not include the concerning E484K and N501Y RBD mutants driving recent surges of COVID-1940, although all three nanobodies have reduced binding to the L452 RBD mutants that characterize more recent SARS-CoV-2 variants of concern41 (Fig. 4). AHEAD evolution experiments directly targeted to L452 RBD variants are under way in our laboratories.

a, Logo plots showing the enrichment of RBD mutations that escape binding by each nanobody for each of the libraries, as determined by NGS. Libraries 1 and 2 are biological replicates using independent RBD mutational scanning libraries to ensure consistency in the escape mutations identified. Following Greaney et al.37, enrichment is plotted as the ‘escape fraction’ for each mutation shown and is defined as the fraction of cells with a given RBD mutation sorted into the low nanobody labeling gate. b, Structural mapping of each nanobody’s binding site using escape profile information. The escape mutation positions are highlighted in red magenta and yellow for RBD1i1, RBD10i10 and RBD11i12, respectively. The images were prepared using the structure of the RBD–ACE2 complex (PDB: 6M17). RBD is colored in blue and ACE2 in green.
A naïve nanobody library can be encoded on AHEAD
In the experiments described above, AHEAD was used to rapidly evolve high-affinity nanobodies starting from isolated individual clones encoded on p1. It should also be possible to encode diverse naïve antibody libraries directly on the AHEAD system. The resulting yeast populations would act as off-the-shelf libraries that enable AHEAD experiments to encompass both initial clone discovery and affinity maturation in an end-to-end process for antibody generation against arbitrary targets. To test the feasibility of this approach, we computationally designed and synthesized a 200,000-member naïve nanobody library capturing key features of camelid immune repertoires42, encoded the library in AHEAD strains with 50-fold coverage, and subjected to selection for binding GFP as a test target. After three cycles of AHEAD, a single nanobody, NbG1, dominated the population. After two additional cycles, a C96Y mutation that increased GFP binding (half-maximum effective concentration, EC50) by 4.4-fold arose and fixed as NbG1i1 (Extended Data Fig. 8 and Supplementary Table 1). This shows that AHEAD can emulate the entire process of somatic recombination, clonal expansion and somatic hypermutation in the immune system. Although the gain in affinity achieved by NbG1i1 was rather modest in this proof-of-principle experiment, we expect that larger libraries or computationally designed nanobody libraries optimized for stability and evolvability will yield superior outcomes in future integrated nanobody discovery and maturation campaigns.
Discussion
In this work, we have engineered a streamlined system for rapid antibody evolution, called AHEAD, that leverages OrthoRep to autonomously diversify YSD nanobodies simply as cells are cultured. Cycles of yeast growth and FACS-based selection for binding result in the efficient affinity maturation of nanobodies in a process mimicking somatic hypermutation by animal immune systems, which allowed us to quickly generate potent nanobodies against SARS-CoV-2, as well as high-affinity nanobodies against a GPCR and other targets. Because AHEAD operates through an animal-free process involving only culturing and FACS, it compares favorably to immunization technologies in speed, ease and parallelizability, which should promote its widespread adoption as an antibody-generation system.
A notable difference between AHEAD-based diversification and natural somatic hypermutation (SHM) is the lack of mutational hotspots in AHEAD, because the entire nanobody sequence on p1 is hypermutated by OrthoRep. In natural SHM, the entire antibody sequence is also hypermutated, but there exist sequence motifs within complementarity-determining regions (CDRs) that promote enhanced hypermutation to generate mutational hotspots43. Although evidence is emerging that mutations outside of CDRs are important for antibody folding/solubility and antigen recognition44, including in our experiments where mutations within framework regions are commonly enriched and in some cases improve antigen binding (Fig. 3b and Supplementary Data 1), CDR mutational hotspots are generally accepted as a valuable feature of SHM because CDRs have an outsized influence on antigen binding. Future versions of AHEAD may be engineered to mimic mutational hotspots, for example by utilizing fusions of cytidine deaminases (AID) to clustered regularly interspaced short palindromic repeats (CRISPR) to deliberately boost mutation rates in CDRs45. Future versions may also include large libraries, new antibody scaffolds beyond nanobodies, developments in the underlying OrthoRep system (for example, higher-mutation-rate orthogonal DNAPs), as well as new ways of selecting for target binding and modulation that do not require FACS.
The application of AHEAD to the rapid evolution of anti-SARS-CoV-2 nanobodies merits further discussion. In this experiment, eight independent antibody-generation campaigns, starting from weak-affinity nanobody clones isolated from a naïve library, were carried out in an uninterrupted time of only 1.5–3 weeks. During this time, we saw major improvements in both affinity and neutralization potencies owing to the sequential fixation of affinity-enhancing mutations (Fig. 3f and Supplementary Data 1). This synthetic affinity maturation process may serve as a template for future outbreak response. Because we started from naïve synthetic nanobody libraries, our experiments did not depend on the prior discovery of antibodies from patients or animals, demonstrating capacity for immediate response once a molecular target is identified. Because we ran multiple independent evolution campaigns simultaneously, we readily obtained a collection of functionally diverse nanobody sequences, important for hedging against biological uncertainty and antibody developability in the face of future novel coronaviruses and SARS-CoV-2 mutants going forward. Running independent evolution campaigns also prevented the early loss of weak parents that are capable of evolving into superior descendants, akin to ‘demes’ in natural evolution46,47. Finally, because we relied only on simple cycles of yeast culturing and FACS to evolve potent nanobodies, our experiments should be broadly accessible and may enable wider and more distributed antibody development efforts in future pandemics. In the particular case of anticipating novel coronavirus outbreaks, our collection of SARS-CoV-2 nanobodies, already encoded on AHEAD, should be privileged starting points for rapid response, as additional evolutionary cycles may be able to direct our current nanobodies to become specific for new spike variants. In fact, we estimate that, given a new SARS-CoV-2 RBD variant37, we will be able to quickly discover multiple high-affinity binders. Overall, we believe that AHEAD’s core functionality for rapid and parallelizable antibody generation through autonomous hypermutation, along with the growing ecosystem of autonomous hypermutation systems in synthetic biology48,49, has the potential to broadly upgrade antibody generation, supporting all areas of biomedicine.
Methods
AHEAD base yeast display strain construction
We generated strain yAW301, which is essentially the EBY100 YSD strain18 harboring a ‘landing pad’ version of p1 along with p2 (ref. 50). yAW301 served as the base strain for all AHEAD experiments where (1) antibody fragment display cassettes were integrated onto the landing pad p1 and (2) a nuclear plasmid, pAR-633-Leu2 (Supplementary Table 2), encoding an error-prone orthogonal DNAP replicating p1, was transformed into the strain to drive continuous hypermutation of p1 and the antibody fragment display cassette encoded therein. To make strain yAW301, we first generated F102-2 (ref. 50) cells containing p1 and p2 but lacking the MET17 gene from the genome. To delete MET17, F102-2 cells were transformed with a linear DNA fragment encoding a G418 resistance cassette flanked on both sides by 45-bp sequences homologous to the surrounding regions of MET17 (SG ID S000004294). Following selection on solid media with G418, colonies were isolated and verified for the deletion of MET17 by PCR. In addition, the colonies’ inability to grow without the supplementation of methionine and cysteine was verified.
F102-2 cells harbor the unmodified cytosolic p1 and p2 plasmids. However, we wished to engineer a version of p1 designed for ease of integration and selection of antibody yeast display cassettes. To generate such a ‘landing pad’ p1, a DNA fragment was designed to recombine with F102-2’s p1 plasmid (8.9 kb) to replace positions 3201–8400 of p1 with MET17 driven by the p1-specific promoter, p2O5. This integration resulted in the desired landing pad p1 (5.3 kb) harbored in F102-2. The shifted size of p1 was validated by gel electrophoresis of total DNA treated with proteinase K (Thermo Fisher cat. no. E00491), as previously described50 and sequence-verified. This landing pad p1 could subsequently be transported from F102-2 to other strains through protoplast fusion, as previously described50. In particular, EBY100 YSD cells18 that had their genomic MET17 deleted using the same strategy for deleting MET17 in F102-2 were fused with F102-2 cells containing the landing pad p1. Selection for prototrophies uniquely encoded by nuclear genes in EBY100 (and not in F102-2) as well as selection for MET17 uniquely encoded on p1 resulted in strain yAW292 (Supplementary Table 3). Finally, plasmid pAR-633-Leu2, which encodes the error-prone orthogonal DNAP, TP-DNAP1-4-2 (ref. 17), was transformed into yAW292 so that the p1-encoded nanobody expression cassette would be replicated by the error-prone orthogonal DNAP to drive hypermutation. The resulting strain was dubbed yAW301 (Supplementary Table 3).
All strains used in this study are listed in Supplementary Table 3. All genetic modifications that were made during strain construction were verified by sequencing and phenotyping.
Cloning nanobodies into AHEAD
pAW24 or pAW240, plasmids that encode the linear cassette needed for integration of nanobodies into the landing pad p1, were designed and constructed for nanobody expression from p1 (Extended Data Fig. 3 and Supplementary Table 2). In pAW24 (corresponding to AHEAD 1.0; Extended Data Fig. 3), the nanobody gene within the integration cassette is driven by a previously reported p1-specific promoter called p10B228 and fused to a standard alpha mating factor secretory leader sequence18. In pAW240 (corresponding to AHEAD 2.0; Extended Data Fig. 3), the nanobody gene within the integration cassette is driven by a new p1-specific promoter (pGA) that was discovered in an unrelated OrthoRep-based protein evolution project (pGA is the same sequence as p10B2 with two G→A mutations at positions −5 and −34 upstream from the start codon). The nanobody gene within the integration cassette of pAW240 also includes a mutated leader sequence, app8i1, which was selected for higher-efficiency display (Extended Data Figs. 3 and 4b). Finally, the nanobody expression cassette in pAW240 contains a hard-coded poly-A tail28 to maximize expression. Single nanobodies or libraries were amplified with PCR using primers Nb_P240_F and Nb_P240_R (Supplementary Table 4), gel-purified and assembled into BseR1-digested pAW24 or pAW240 with a Gibson reaction. For pAW240, it is important to use BseR1-digested pAW240 as the ‘backbone’ for Gibson assembly, as opposed to a PCR-amplified ‘backbone’ of pAW240, because PCR causes truncation of the hard-coded poly-A tail sequence, which results in lower expression and a drop in antibody display levels. Once pAW24 or pAW240 was properly assembled to contain the desired nanobodies, the resulting plasmids were linearized with ScaI to expose flanking regions homologous to the landing pad p1 to direct their homologous recombination into p1 in yAW292 or yAW301. The ScaI linearized pAW24 or pAW40 plasmids containing desired nanobodies were then transformed into yAW301 by a standard yeast chemical transformation protocol51. Cells that successfully integrated the nanobody expression cassette onto p1 were selected on Synthetic Complete without histidine, leucine, uracil, tryptophan, methionine and cysteine (US Biological, EU Commodity code 38210000) with 2% glucose (SC-HLUWMC) agar plates. Although MET17 in the landing pad p1 is replaced by the nanobody expression cassette, we found that the exclusion of methionine and cysteine was appropriate for selection, because some copies of the landing pad p1 would still be present in cells containing the p1 integration product. We empirically found that selection for cells still containing the unmodified landing pad p1 increased the success rate of integration, possibly because the unmodified landing pad p1 also encodes an additional source of TP-DNAP1, allowing for a higher overall p1 copy number to aid the replication of both the unmodified landing pad p1 and the modified nanobody-expressing p1 integration product. Proper integration of the nanobody expression cassette into the landing pad p1 was varied by gel electrophoresis on total DNA, PCR using primers unique to the integration product, and DNA sequencing. For further cultivation, cells were grown in SC-HLUW (Synthetic Complete without histidine, leucine, uracil and tryptophan with 2% glucose, US Biological cat. no. D9540) liquid dropout medium. AHEAD experiments were started after this point.
Functional display of AT110 and 4-4-20 from AHEAD 1.0
When we first examined whether functional surface display was feasible from p1, by testing the 4-4-20 scFv and the AT110 nanobody, we used the wild-type DNAP for replicating p1 (Fig. 1b). However, when we transitioned to using the error-prone polymerase, TP-DNAP1-4-2, the functional display levels dropped substantially (Extended Data Fig. 3b), resulting from a decline in p1’s copy number as described previously16,17,28. The weak binding signal limited FACS selections, so we overcame these limitations with MACS, and later by designing AHEAD 2.0, as described in the following.
Affinity maturation of anti-AT1R nanobodies using the first-generation AHEAD 1.0 system
Yeast strain yAW301, harboring pAR-633-Leu2 encoding the error-prone orthogonal DNAP and an AT110 expression cassette (linearized plasmid pAW24) integrated onto p1, was grown and passaged in SC-HLUW. In preparation for each FACS selection step, display of AT110 was induced by transferring yeast to SC-HLUW, but with glucose replaced with 2% galactose as the sole sugar source. This results in the induction of Aga1p expression and subsequent surface display of the AT110 nanobody. Because of the low expression of nanobodies in the first-generation AHEAD 1.0 system (Extended Data Fig. 3b), cells expressing the highest levels of AT110 nanobodies were deliberately enriched before each AHEAD cycle. Accordingly, the following process defined each AHEAD 1.0 cycle, which is longer than the three-day AHEAD 2.0 cycle used for the majority of evolution experiments in this work. Between 2 × 109 and 1 × 1010 induced yeast cells were pelleted and resuspended in 2.5–5 ml of AT1R staining buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% BSA, 3 mM CaCl2, 0.1% maltose-neopentyl glycol, 0.01% cholesteryl hemisuccinate, 0.2% maltose, 20 µM angiotensin II) with 1 µM mouse anti-HA (hemagglutinin) antibody that was fluorescently labeled, alternately, with AlexaFluor (AF) 647 (Thermo Fisher cat. no. 26183-A647) or FITC (Abcam cat. no. ab6785). After incubation at 4 °C for 30 min, cells were pelleted and resuspended in 2.5–5 ml of AT1R staining buffer, followed by the addition of 250–500 µl anti-647 or anti-FITC microbeads (Miltenyi cat. nos. 130-091-395 and 130-048-701). Yeast cells were incubated with microbeads for 20 min at 4 °C and then pelleted, resuspended in staining buffer, and added to two or three LS columns (Miltenyi). The columns were then washed with staining buffer and bound yeast cells were eluted in 5 ml of staining buffer. From these elutions, ~1 × 108 cells were pelleted for FACS and stained with FLAG-tagged angiotensin II type 1 receptor19 for 45 min at 4 °C. Following this, yeast cells were again pelleted and stained with fluorescently labeled anti-FLAG antibody (AF647, Thermo Fisher cat. no. MA1-142-A647; FITC, Abcam cat. no. ab2492; AF488, Thermo Fisher cat. no. MA1-142-A488) and fluorescently labeled anti-HA antibody for 20 min at 4 °C, alternating between FITC, 647- and 488-labeled antibody for each AHEAD cycle to avoid selection for dye binding. FACS was performed with a Sony SH800 cell sorter using a 100-µm Sony sorting chip. Over the course of nanobody AT110 affinity maturation, cells were grown for a total of ~400 divisions. During that time, eight cycles of AHEAD were performed. Gating for singlets is shown in Extended Data Fig. 9.
In preparation for NGS, p1 plasmid was extracted, as previously described20, from yeast cultures after the FACS step of each AHEAD cycle. PCRs were performed with Q5 Master Mix (New England Biolabs cat. no. M0492S) and primers NGS_p1_F and NGS_p1_R. Following PCR reactions, samples were PCR-purified. Amplicon sequencing was performed by the MGH CCIB DNA Core and analyzed using Geneious Prime software version 2019.1.
Radioligand binding assay for anti-AT1R nanobodies
Nanobodies were purified from the periplasm of Escherichia coli by Ni-NTA affinity chromatography (Qiagen cat. no. 30210) and dialyzed into buffer consisting of 20 mM HEPES pH 7.4, 100 mM NaCl. Because AT110 and its derivatives allosterically increase the affinity of agonists for the AT1R, the effect of serially diluted nanobodies on the binding of the inverse agonist [3H]-olmesartan (American Radiolabeled Chemicals cat. no. ART1976) was assessed in the absence versus the presence of an ~IC20 concentration of the low-affinity agonist, TRV055 (Genescript). Purified wild-type AT1R (75 ng)19 was added to 2.5 nM [3H]-olmesartan, the indicated concentration of nanobody, and either assay buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.01% lauryl maltose neopentyl glycol and 0.1% BSA) or 1 μM TRV055. The final reaction volumes were 200 μl, with single replicates for each condition. The assay buffer consisted of 20 mM HEPES pH 7.4, 100 mM NaCl, 0.01% lauryl maltose neopentyl glycol and 0.1% BSA. After a 90-min incubation at room temperature, reactions were collected onto GF/B glass-fiber filter paper using a 96-well harvester (Brandel) and quickly washed three times with cold 20 mM HEPES pH 7.4, 100 mM NaCl. The fraction of [3H]-olmesartan bound in the presence versus the absence of TRV055 was determined at each nanobody concentration. Data from three independent experiments were fit to a one-site model in GraphPad Prism.
Engineering a stronger secretory leader sequence for the second-generation AHEAD 2.0 system
One of the parts responsible for higher nanobody display levels in AHEAD 2.0 is a mutated app8 secretory leader, which we selected from an error-prone PCR library of app8 (Extended Data Fig. 4b). Nanobody AT110 was fused to an app8 secretory leader27 and cloned into a nuclear centromere/autonomously replicating sequence (CEN/ARS) plasmid containing either a pREV1, pSAC6, pRPL18B or a pTDDH3 promoter52. Nanobody display from the pSAC6 promoter was determined to be most similar to expression from p1 with the pGA promoter. This plasmid, dubbed pAW258, was then used as template for preparing a library of mutated app8 secretory leader sequences. The app8 was amplified by error-prone PCR (Agilent cat. no. 200550) using primers AW_Sac6_mut_F and AW_Sac6_mut_R and cloned back into pAW258 using Gibson assembly. The library size was determined to be 2 × 107 by counting colonies on serially diluted antibiotic selection plates. Twelve single clones were picked and sequenced. All tested clones had intact reading frames, and the average number of mutations per clone was 1.4. The library was then transformed into EBY100 cells, induced through growth in 2% galactose as the sole sugar source for 1–2 days, and subjected to three rounds of FACS selection for strong HA tag display, as the HA tag is fused to AT110 and acts as a surrogate for AT110 display level. In each round, the top 0.05% expressing cells as determined by anti-HA signal were sorted (Extended Data Fig. 4b). After the third round, the cell population was plated, and 48 single colonies were picked and screened for nanobody display. The 12 clones that displayed the highest levels of nanobody expression were sequenced. Three different mutations were discovered, namely V10A, F48V and D28G. These mutations were reintroduced to plasmid p258 and assayed for their effect on AT110 display level. Although mutations F48V and D28G did not confer any increase in display levels, mutation V10A increased nanobody display by ~90% (Extended Data Fig. 4b). The app8 with V10A was dubbed app8i1 and used in the second-generation AHEAD 2.0, along with other modifications as described above.
Specific considerations for gating during FACS in AHEAD experiments
In yeast display antibody evolution, one typically gates for target binding, normalized to the nanobody display level. To measure nanobody display levels, we used a human influenza HA tag fused to the nanobody. We then gated on the ratio of [target (that is, RBD) binding]:[HA tag binding by a fluorescently labeled anti-HA antibody]. In other words, during selection for better target binding, we gate along a slope on the FACS plot where the y axis is HA level and the x axis is target-binding level (Extended Data Fig. 5b provides examples). However, because AHEAD hypermutates the entire content of p1, it is possible to obtain mutations in the HA tag that disable it, creating cells that have seemingly strong target binding per nanobody displayed if sorted exclusively for cells with a high [target binding]:[HA signal] ratio. This can lead to the gradual fixation of cheaters that bind the target weakly but were selected because they also have disabled HA tags. To overcome this issue, it is important to gate with a strict floor on HA signal rather than solely on the [target binding]:[HA signal] ratio. In our experiments, we found that if the floor for the HA signal was set to include only the top ~20% of cells on the HA signal axis (y axis), we could maintain selection for target-binding throughout rounds without carrying over cheaters that mutated the HA tag.
Affinity maturation of Nb.b201 and Lag42 using the second-generation AHEAD 2.0 system
HSA (Sigma cat. no. A3782) was directly labeled with AF647 (Thermo Fisher cat. no. A20000). 6XHis tagged GFP was expressed in E. coli and purified using Ni-NTA agarose (Qiagen cat. no. 30210). Nb.b201 was amplified using PCR from pYDS-Nb.b201 (ref. 32) and cloned into pAW240. Lag42 (ref. 33) was synthesized as a gBlock (IDT DNA Technologies) and cloned into pAW240. The resulting plasmids were linearized using ScaI, transformed into yAW301, and plated as described above (section ‘Cloning nanobodies into AHEAD’). A single colony was picked into SC-HLUW, grown to saturation, pelleted, induced in SC-HLUW with 2% galactose replacing glucose as the sole sugar source for ~24 h, and labeled with 50 nM HSA-AF647 (for Nb.b201 cells) or 200 nM GFP-AF647 (for Lag42 cells) and 1 µM mouse anti-HA antibody (Thermo Fisher cat. no. 26183) for 1 h at 4 °C in binding buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% BSA, 0.2% maltose). Cells were washed with binding buffer and incubated with 0.5 µM polyclonal goat anti-mouse AF488-labeled antibody (Thermo Fisher cat. no. A32723) for 15 min. Cells were washed again with binding buffer and sorted (Sony SH800) for increased affinity for HSA or GFP (Extended Data Fig. 4c,d) into a tube containing 2 ml of SC-HLUW. Cells were incubated with shaking for 1–2 days at 30 °C and subjected to the next AHEAD cycle.
Over four cycles of AHEAD (for Nb.b201) or six cycles of AHEAD (for Lag42), the selection stringency was increased by reducing the concentration of HSA or GFP as indicated (Extended Data Fig. 4c,d). During each FACS step, ~2 × 107 cells were used for sorting out 200–1,000 cells. After AHEAD, nanobodies were amplified from p1 and either sequenced directly or subcloned into a plasmid to isolate individual clones for sequencing and further characterization.
Isolation of anti-RBD nanobody parents
To isolate RBD-binding nanobodies, two initial rounds of MACS were performed using a synthetic yeast-displayed library of nanobodies32 followed by two rounds of FACS. S. cerevisiae containing the library were grown in tryptophan dropout medium (US Biological) with 2% glucose for one day, then expression and display were induced in tryptophan dropout with 2% galactose for two days. For the first round, 1 × 1010 yeast cells were centrifuged and resuspended in a ‘pre-clearing’ solution of 4.5 ml of binding buffer, 500 µl of anti-PE microbeads (Miltenyi) and 200 nM streptavidin-PE (BioLegend cat. no. 405203). After incubation for 40 min at 4 °C, yeast cells were passed through an LD column (Miltenyi) to remove cells interacting with microbeads or streptavidin. Yeast cells that flowed through the column were collected, centrifuged, resuspended in a ‘staining solution’ consisting of 2 ml of binding buffer with 1 µM SARS-CoV-2 RBD and 250 nM streptavidin-PE and incubated for 1 h at 4 °C. After incubation, yeast cells were centrifuged, resuspended in a ‘secondary solution’ of 4.5 ml of binding buffer and 500 µl anti-PE microbeads (Miltenyi cat. no. 130-048-80) and incubated for an additional 15 min at 4 °C. These yeast cells were then centrifuged, washed with binding buffer and passed into an LS column (Miltenyi). The LS column was washed with 7 ml of binding buffer and the remaining yeast cells were eluted in 5 ml of binding buffer, centrifuged and resuspended in 5 ml of tryptophan dropout medium for expansion. The second round of MACS was performed similarly to the first but starting with 1 × 109 yeast cells and substituting PE-labeled streptavidin with FITC-labeled streptavidin and anti-PE microbeads with anti-FITC microbeads. Additionally, volumes of the pre-clearing and secondary solutions were reduced fivefold and the staining solution by twofold.
For the first round of FACS, 1 × 108 induced cells were stained with 1 µM directly AF488-labeled SARS-CoV-2 RBD and 0.5 µM anti-HA AF647 (Cell Signaling Technology cat. no. 3444 S) antibody, to visualize expression, for 1 h at 4 °C. These cells were then centrifuged, resuspended in 2 ml of binding buffer and sorted. In total, 35,000 cells from 11,431,000 were collected and expanded. The second round of FACS was performed with similar conditions to the first; however, the RBD was labeled with AF647, anti-HA with AF488, and the concentration of RBD was reduced to 150 nM. For the second round, 104,000 cells were collected from 2,330,000 sorted. These cells were expanded in culture and then plated on tryptophan dropout medium to isolate single clones. Twenty-four colonies were picked, cultured and induced. Each culture was screened for binding by staining ~1 × 106 cells with 200 nM 647- and 488-labeled RBD along with 488- and 647-labeled anti-HA antibody, respectively, and binding reactions were evaluated using a flow cytometer. Promising clones were selected as parents for AHEAD experiments.
FACS selection for improved RBD binders using the improved second-generation AHEAD system
After cloning the RBD-binding parent nanobodies into AHEAD (section ‘Cloning nanobodies into AHEAD’), initial cultures (50 ml of SC-HLUW) were grown to saturation and optionally passaged once or twice into 50 ml of SC-HLUW at a ratio of 1:1,000 to prolong diversification of the nanobodies before the first AHEAD cycle. Upon final saturation (1–2 days), cells were pelleted and resuspended in SC-HLUW with 2% galactose replacing glucose as the sole sugar source for induction. Induction was performed for 24 h at room temperature with shaking at 250 r.p.m. Cells were collected, washed in binding buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% BSA, 0.2% maltose) and incubated with unlabeled 1 µM mouse anti-HA antibody (Thermo Fisher cat. no. 26183) and SARS-CoV-2 RBD directly labeled with AF647. Cells were then washed and incubated with 0.5 µM goat anti-mouse AF488-labeled antibody (Thermo Fisher cat. no. A32723) for 15 min. The cells were subjected to FACS (Sony SH800), whereby 200–500 cells were collected into a culture tube containing 3 ml of SC-HLUW, out of ~2 × 107 sorted cells. Each subsequent cycle of AHEAD involved growing the 3 ml of sorted cells with shaking at 30 °C until saturation (1–2 days), induction with 2% galactose as the sole sugar source for ~24 h at room temperature with shaking at 250 r.p.m., washing of cells in binding buffer, incubation with anti-HA and labeled RBD, washing steps to remove unbound RBD, and FACS sorting into 3 ml of SC-HLUW. RBD concentrations were diminished from cycle to cycle while the stringency of washes increased. After several rounds of AHEAD, nanobodies were amplified from p1 and either sequenced directly or subcloned into a plasmid to isolate individual clones for sequencing and further characterization.
Nanobody–Fc fusion purification
Nanobodies targeting RBD were expressed and secreted as Fc fusions by cloning into pFUSE-hlgG1-Fc2 (InvivoGen) using the NcoI and EcoRI restriction sites or by Gibson assembly. For each nanobody–Fc fusion, 100 ml of Expi293 cells (Thermo Fisher cat. no. A14527) were transfected with 90–150 µg of plasmid. After one day, cells were enhanced with 3 mM valproic acid and 0.45% glucose. Cell supernatants were collected four days after transfection. Before purification, supernatants were treated with benzonase nuclease and protease inhibitor, then passed through a 0.22-µm filter. Nanobody–Fc fusion supernatants were passed over a column with 4-ml protein G resin (Thermo Fisher cat. no. 20399), which was then washed with 40 ml of HBS, eluted with 100 mM citrate (pH 3) and then neutralized to pH 7 with concentrated HEPES (pH 8). Nanobody–Fc fusions were then dialyzed twice with HBS (pH 7.5).
On-yeast EC50 measurements
To determine nanobody affinities for their targets as surface-displayed proteins, individual nanobody sequences were cloned into plasmid p253, a plasmid for galactose-inducible expression of nanobodies for surface display in EBY100 cells. Plasmids were transformed into EBY100, induced in appropriate dropout medium with 2% galactose as the sole sugar source for ~24 h at room temperature, washed with binding buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% BSA, 0.2% maltose) and labeled with biotinylated antigen across a range of concentrations as well as with 1 μM mouse anti-HA antibody for 1 h at 4 °C. Cells were then washed and incubated 0.5 μM with goat anti-mouse AF488-labeled antibody (Thermo Fisher cat. no. A32723) and streptavidin-conjugated PE (BioLegend cat. no. 405203) for 15 min. After additional washing, fluorescence was measured using an Attune flow cytometer (Life Technologies). Antigen binding (PE signal) was recorded only for cells that express the nanobody, that is, cells in populations showing anti-HA staining signal. The average PE signal at each antigen concentration was determined and used to fit a one-site model in GraphPad Prism to determine the EC50. In all cases, volumes and number of cells used were chosen to accommodate doing assays in 96-well format and to avoid ligand depletion. Binding was measured in triplicate for each antigen concentration.
Surface plasmon resonance
Surface plasmon resonance (SPR) was performed using a Biacore T200 system (Cytiva). Nanobody–Fc fusion proteins were immobilized to a protein A sensor chip (Cytiva cat. no. 29127557) at a capture level of approximately 85–255 response units (RUs). Binding experiments with dilutions of RBD were performed in running buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 0.05% Tween). RBD dilutions were injected at a flow rate of 30 µl min−1 with a contact time of either 160 s or 280 s and a dissociation time of 600 s or 900 s. After each cycle, the protein A sensor chip was regenerated with 10 mM glycine-HCl pH 1.5. Kinetic data were double reference subtracted and fitted to a 1:1 binding model. For samples in which the on or off rates could not be determined, data were fitted to a steady-state affinity model.
SARS-CoV-2 and VSV-G lentivirus production
To generate lentivirus pseudotypes, the SARS-CoV-2 virus spike protein with the last 27 amino acids deleted (GenBank ID: QJR84873.1, residues 1–1246) was cloned into a pCAGGS vector and modified to include at its C-terminal tail the eight most membrane-adjacent residues of the cytoplasmic domain of the HIV-1 envelope glycoprotein (NRVRQGYS). Pseudotypes were packaged by transfecting HEK293T cells (ATCC CRL-11268) using Lipofectamine 3000 (Invitrogen cat. no. L3000001) with SARS-CoV-2 S in pCAGGS or VSV-G in pCAGGS (as previously described53), in addition to a packaging vector containing HIV Gag, Pol, Rev and Tat (psPAX2, provided by D. Trono, Addgene 12260), and a pLenti transfer vector containing GFP (pLenti-EF1a-Backbone, a gift from F. Zhang54, Addgene plasmid 27963). After 18 h, the transfection medium was removed from cells and replaced with DMEM containing 2% (vol/vol) FBS and 50 mM HEPES. Cells were incubated at 34 °C, before the supernatant was then collected at 48 and 72 h, centrifuged at 3,000g and filtered through a 0.45-µm filter. The filtered supernatant was then concentrated by layering on a 10% (vol/vol) sucrose cushion in Tris buffered saline (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.5 mM EDTA) and spun at 10,000g for 4 h at 4 °C. The viral pellet was resuspended in Opti-MEM (Thermo Fisher cat. no. 31985062) containing 5% (vol/vol) FBS and stored at −80 °C.
Pseudotype neutralization experiments
Nanobodies or PBS alone were pre-incubated with SARS-CoV-2 or VSV-G lentivirus for 60 min at 37 °C in a mixture that also contained 0.5 µg ml−1 polybrene (Sigma cat. no. TR-1003). Mixtures were then added to HEK293T cells overexpressing human ACE2 (a gift from H. Mou and M. Farzan, Scripps Research). After 24 h, the virus medium was removed and replaced with DMEM containing 10% (vol/vol) FBS, 5% (vol/vol) pen/strep and 1 µg ml−1 puromycin. The percentage of GFP-positive cells was determined by flow cytometry with an iQue Screener PLUS (Intellicyt) 48 h after initial infection. Percent relative entry was calculated using the following equation: relative entry (%) = (% GFP-positive cells in nanobody well)/(%GFP-positive cells in PBS-alone well). Percent neutralization was calculated using the following equation: neutralization (%) = 1 − (% GFP-positive cells in nanobody well)/(% GFP-positive cells in PBS-alone well). All experiments were performed twice in triplicate.
ACE2 competition assays using biolayer interferometry
Biolayer interferometry experiments for ACE2 competition assays were performed with an Octet RED96e system (Sartorius). For ACE2 competition assays, biotinylated SARS-CoV-2 RBD was loaded onto streptavidin sensors (ForteBio cat. no. 18-5019) at 1.5 µg ml−1 for 80 s. After a baseline measurement was obtained, nanobodies were associated at 250 nM for 300 s, followed by an association with ACE2–Fc at 250 nM for 300 s. Representative results of two replicates for each experiment are shown.
Deep mutational scanning assay to detect RBD mutants that escape nanobody binding and map the binding site of nanobodies
Deep mutational scanning was performed for nanobodies RBD1i1, RBD10i10 and RBD11i12 as described in refs. 37,38. Briefly, the deep mutational scanning plasmid libraries encoding RBD mutants were transformed into EBY100 cells, induced by incubating cultures in SC-W medium containing 2% galactose instead of glucose for 48 h, and labeled for ACE2 binding and RBD expression using biotin-ACE2 (Acro cat. no. H82F9) and the MYC epitope, respectively, using FITC-conjugated anti-Myc (Immunology Consultants Lab cat. no. CYMC-45F). Cells selected for ACE2 binding using FACS were recovered and subjected to a round of selection for nanobody escape by incubating with 10 nM Fc fused nanobody where the nanobody was labeled with a goat anti-human IgG dye-AF647-conjugated antibody (Thermo Fisher cat. no. A-21445). In this escape selection, cells encoding RBD variants that could not be bound by the nanobody were recovered by FACS. These recovered cells were expanded and subjected to another round of escape selection. Their DNA was extracted and the barcode region corresponding to RBD mutants was amplified by PCR and subjected to NGS and analyzed using the published computational pipeline37,38 for matching enriched barcodes to escape mutations in RBD. The results of this experiment, showing which mutations in RBD enrich when selected for escape from nanobody binding, are plotted in Fig. 4. (The raw sequencing files can be obtained from https://www.ncbi.nlm.nih.gov/sra?term=SRP320370, with identifier biosample accession numbers SAMN19242322, SAMN19242323, SAMN19242324, SAMN19242325, SAMN19242326, SAMN19242327 and SAMN19242328.)
Recombinant RBD expression and purification
Two preparations of the RBD of SARS-CoV-2 S (GenBank ID QHD43416.1, residues 319–541) were used in this study. For ACE2 competition assays, RBD was cloned into the pHLsec vector (PubMed ID 17001101). The construct contains an N-terminal 6xHis tag, a TEV protease site, and a BirA ligase site followed by a seven-residue linker. RBD was produced by transfecting HEK293T cells grown in suspension and collected after five days and was purified by reverse nickel affinity purification. The RBD was then biotinylated with BirA ligase (Creative Biomart cat. no. birA-339E) and again purified using reverse nickel affinity purification to remove the BirA ligase, followed by size exclusion purification on a Superdex 200 Increase column (GE Healthcare). For all other experiments including AHEAD evolution of anti-RBD nanobodies and their characterization, a pVRC8400 plasmid containing RBD was used for RBD expression55. The construct (pVRC8400-RBD) has RBD fused to a C-terminal HRV C3 protease cleavage site followed by an 8xHis tag and a streptavidin binding peptide (SBP). 0.5 mg of plasmid was transfected into HEK293T cells and grown in suspension for three days. The culture medium was then dialyzed against PBS overnight and RBD was purified using Ni-NTA agarose. The eluted His-tagged RBD was then incubated with biotin-tagged human rhinovirus (HRV) C3 protease (Sigma cat. no. SAE0110) and passed through a streptavidin-agarose column to deplete the protease and the 8xHis-SBP peptide. The eluate was collected and analyzed on a denaturing sodium dodecyl SDS-PAGE gel to confirm the purity of the RBD.
Recombinant ACE2–Fc fusion protein expression and purification
To generate a recombinant ACE2–Fc fusion protein, we cloned the ectodomain of human ACE2 (GenBank ID BAB40370.1 residues 18–740) with a C-terminal Fc tag into the pVRC8400 vector containing the human IgG1 Fc. We transfected the construct (pVRC8400-hACE2) into Expi293F cells using an ExpiFectamine transfection kit (Thermo Fisher cat. no. A14525) according to the manufacturer’s protocol. The supernatant was collected after five days and purified using a MabSelect SuRE Resin (GE Healthcare cat. no. GE17-5438-01) followed by size exclusion purification on a Superose 6 Increase column (GE Healthcare). The supernatant was collected five days after transfection and purified with a CaptureSelect KappaXL Affinity Matrix (Thermo Fisher cat. no. 194321005) followed by size exclusion chromatography on a Superdex 200 Increase column (GE Healthcare).
Cloning a 200,000-member nanobody library into AHEAD 2.0
A computationally designed 200,000-member naïve nanobody CDR3 library was synthesized as an oligonucleotide pool of CDR3 sequences by Agilent, as previously reported42. To clone this CDR3 library into AHEAD, we first made plasmid pAW240-NbCM, which encodes the nanobody scaffold with fixed CDR1 and CDR2 sequences but with CDR3 replaced with a NotI restriction site. The CDR3 oligo library was then inserted into pAW240-NbCM using the NotI site and ligation. Transformation of the ligated plasmid products into E. coli resulted in ~108 transformants. 200 µg of plasmid DNA was then prepared, linearized with ScaI (NEB) and transformed into strain yAW301 by scaling up the process described in the section ‘Cloning nanobodies into AHEAD’ by 100-fold. The total transformation resulted in ~107 transformants such that the 200,000-member library was covered 50 times.
Selection and affinity maturation of a GFP-binding nanobody from a computationally designed naïve nanobody library encoded on AHEAD 2.0
A 10-ml saturated culture of yAW301 cells expressing the computationally designed 200,000-member naïve nanobody library was induced in SC-HLUW with 2% galactose replacing glucose as the sole sugar source. Induction was performed for 24 h at room temperature with shaking at 250 r.p.m. Cells were collected, washed in binding buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% BSA, 0.2% maltose) and first subjected to negative selection against streptavidin binders by MACS. Specifically, cells were incubated for 1 h at 4 °C with 0.5 µM streptavidin-conjugated FITC, washed and incubated with anti-FITC microbeads (Miltenyi). Cells were washed again and passed through an LD column to deplete streptavidin binders. Recovered cells eluted from the column were incubated with 200 nM GFP–biotin and 1 µM mouse anti-HA antibody. Cells were washed and incubated for 15 min in binding buffer containing 0.5 μM goat anti-mouse AF488-labeled antibody (Thermo Fisher) and streptavidin-conjugated AF647. Cells were washed again and subjected to the first cycle of FACS for GFP binders. In the following cycles of AHEAD, cells were incubated with GFP directly labeled with AF647 (Extended Data Fig. 8). These cycles of AHEAD followed the same process for nanobody evolution using AHEAD 2.0 described above.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this Article.
Data availability
All data generated for the present study are available upon request to the corresponding authors. pAW240 and its sequence are available at Addgene (plasmid 170791). NGS data are available at NCBI’s SRA website https://www.ncbi.nlm.nih.gov/sra?term=SRP320370 (identifier biosample accession numbers SAMN19242322, SAMN19242323, SAMN19242324, SAMN19242325, SAMN19242326, SAMN19242327 and SAMN19242328).
References
Lu, R. M. et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27, 1 (2020).
Gravbrot et al. Therapeutic monoclonal antibodies targeting immune checkpoints for the treatment of solid tumors. Antibodies 8, 51 (2019).
Czajka, T. F., Vance, D. J. & Mantis, N. J. Slaying SARS-CoV-2 one (single-domain) antibody at a time. Trends Microbiol. 29, 195–203 (2021).
Byrne, B., Stack, E., Gilmartin, N. & O’Kennedy, R. Antibody-based sensors: principles, problems and potential for detection of pathogens and associated toxins. Sensors 9, 4407–4445 (2009).
Yao, H. et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 6, 76 (2020).
Hanke, L. et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 11, 4420 (2020).
Schoof, M. et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive spike. Science 370, 1473–1479 (2020).
Gray, A. et al. Animal-free alternatives and the antibody iceberg. Nat. Biotechnol. 38, 1234–1239 (2020).
Rajewsky, K. Clonal selection and learning in the antibody system. Nature 381, 751–758 (1996).
Mishra, A. K. & Mariuzza, R. A. Insights into the structural basis of antibody affinity maturation from next-generation sequencing. Front. Immunol. 9, 117 (2018).
Teng, G. & Papavasiliou, F. N. Immunoglobulin somatic hypermutation. Annu. Rev. Genet. 41, 107–120 (2007).
Boder, E. T., Raeeszadeh-Sarmazdeh, M. & Price, J. V. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 526, 99–106 (2012).
Almagro, J. C., Pedraza-Escalona, M., Arrieta, H. I. & Pérez-Tapia, S. M. Phage display libraries for antibody therapeutic discovery and development. Antibodies 8, 44 (2019).
Baker, M. Reproducibility crisis: blame it on the antibodies. Nature 521, 274–276 (2015).
Voskuil, J. L. A. The challenges with the validation of research antibodies. F1000Res. 6, 161 (2017).
Ravikumar, A., Arrieta, A. & Liu, C. C. An orthogonal DNA replication system in yeast. Nat. Chem. Biol. 10, 175–177 (2014).
Ravikumar, A., Arzumanyan, G. A., Obadi, M. K. A., Javanpour, A. A. & Liu, C. C. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 175, 1946–1957 (2018).
Boder, E. T. & Wittrup, K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 (1997).
Wingler, L. M., McMahon, C., Staus, D. P., Lefkowitz, R. J. & Kruse, A. C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176, 479–490 (2019).
Neuberger, M. Antibodies: a paradigm for the evolution of molecular recognition. Biochem. Soc. Trans. 30, 341–350 (2002).
Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 (2013).
Zavrtanik, U., Lukan, J., Loris, R., Lah, J. & Hadži, S. Structural basis of epitope recognition by heavy-chain camelid antibodies. J. Mol. Biol. 430, 4369–4386 (2018).
Manglik, A., Kobilka, B. K. & Steyaert, J. Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 57, 19–37 (2017).
Gray, A. C., Sidhu, S. S., Chandrasekera, P. C., Hendriksen, C. F. M. & Borrebaeck, C. A. K. Animal-based antibodies: obsolete. Science 353, 452–453 (2016).
Wingler, L. M. et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367, 888–892 (2020).
Wang, Z., Mathias, A., Stavrou, S. & Neville, D. M. A new yeast display vector permitting free scFv amino termini can augment ligand binding affinities. Protein Eng. Des. Sel. 18, 337–343 (2005).
Rakestraw, J. A., Sazinsky, S. L., Piatesi, A., Antipov, E. & Wittrup, K. D. Directed evolution of a secretory leader for the improved expression of heterologous proteins and full-length antibodies in Saccharomyces cerevisiae. Biotechnol. Bioeng. 103, 1192–1201 (2009).
Zhong, Z., Ravikumar, A. & Liu, C. C. Tunable expression systems for orthogonal DNA replication. ACS Synth. Biol. 7, 2930–2934 (2018).
Makrides, S. C. et al. Extended in vivo half-life of human soluble complement receptor type 1 fused to a serum albumin-binding receptor. J. Pharmacol. Exp. Ther. 277, 534–542 (1996).
Renier, N. et al. IDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014).
Chung, K. et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Fridy, P. C. et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 11, 1253–1260 (2014).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Cohen, J. ‘Provocative results’ boost hopes of antibody treatment for COVID-19. Science https://doi.org/10.1126/science.abf0591 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014 (2020).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57 (2020).
Starr, T. N. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Tang, J. W., Toovey, O. T. R., Harvey, K. N. & Hui, D. D. S. Introduction of the South African SARS-CoV-2 variant 501Y.V2 into the UK. J. Infect. 82, e8–e10 (2021).
Deng, X. et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell https://doi.org/10.1016/j.cell.2021.04.025 (2021).
Shin, J. E. et al. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 12, 2403 (2021).
Wei, L. et al. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc. Natl Acad. Sci. USA 112, E728–E737 (2015).
Ovchinnikov, V., Louveau, J. E., Barton, J. P., Karplus, M. & Chakraborty, A. K. Role of framework mutations and antibody flexibility in the evolution of broadly neutralizing antibodies. eLife 7, e33038 (2018).
Hess, G. T. et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016).
Wright, S. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc. Sixth Int. Congr. Genet. 1, 356–366 (1932).
Rix, G. et al. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun. 11, 5644 (2020).
Rix, G. & Liu, C. C. Systems for in vivo hypermutation: a quest for scale and depth in directed evolution. Curr. Opin. Chem. Biol. 64, 20–26 (2021).
Wang, T., Badran, A. H., Huang, T. P. & Liu, D. R. Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol. 14, 972–980 (2018).
Gunge, N. & Sakaguchi, K. Intergeneric transfer of deoxyribonucleic acid killer plasmids, pGKl1 and pGKl2, from Kluyveromyces lactis into Saccharomyces cerevisiae by cell fusion. J. Bacteriol. 147, 155–160 (1981).
Gietz, R. D. & Schiestl, R. H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007).
Lee, M. E., DeLoache, W. C., Cervantes, B. & Dueber, J. E. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 4, 975–986 (2015).
Radoshitzky, S. R. et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446, 92–96 (2007).
Zhang, F. et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 29, 149–153 (2011).
Iyer, A. S. et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci. Immunol. 5, eabe0367 (2020).
Acknowledgements
We thank W. Capel for assistance with nanobody purifications, Z. Zhong, C. Carlson, T. Loveless, A. Banks and other members of the Liu and Kruse groups for experimental assistance, materials and thoughtful discussions, and G. Arzumanyan for the pGA promoter mutations discovered in his OrthoRep continuous protein evolution experiments (unrelated to this study). We thank D. Trono (EPFL), F. Zhang (Broad Institute), H. Mou (Scripps Research) and M. Farzan (Scripps Research) for the gift of plasmids and cells used in our study. We also acknowledge the support of the Center for Macromolecular Interactions at Harvard Medical School. This work was funded by NIH 1DP2GM119163 (C.C.L.), NIH NIGMS 1R35GM136297 (C.C.L.), the Moore Inventor Fellowship (C.C.L.), the UCI COVID-19 Basic, Translational and Clinical Research Fund (C.C.L.), NIH DP5OD021345 (A.C.K.), a Vallee Scholars Award (A.C.K.), NIH NIAID R01AI146779 (A.G.S.), a Massachusetts Consortium on Pathogenesis Readiness (MassCPR; A.G.S.), training grants NIGMS T32GM007753 (B.M.H. and T.M.C.) and T32AI007245 (J.F.) and NIH NCI 1R01CA260415 (C.C.L., A.C.K. and D.S.M.).
Author information
Authors and Affiliations
Contributions
All authors contributed to experimental design and data analysis. A.W., C.M., A.C.K. and C.C.L. were responsible for the conception of AHEAD. A.W., M.H.H., V.J.H. and K.M.N. carried out experiments establishing the first generation of AHEAD and made improvements to reach the second-generation AHEAD system. C.M. carried out AHEAD experiments for the evolution of anti-AT1R nanobodies and selected parent anti-SARS-CoV-2 for evolution using AHEAD. A.W., J.R.C. and M.H.H. carried out AHEAD experiments for the evolution of anti-GFP, anti-HSA and anti-SARS-CoV-2 nanobodies. A.W., C.M., M.S.A.G., S.C. and L.M.W. characterized the activities of evolved nanobodies in binding assays (A.W., C.M. and L.M.W.), SPR measurements (C.M. and M.S.A.G.), neutralization assays (S.C.) and ACE2 competition assays (S.C.). J.F., B.M.H., T.M.C. and A.W. were responsible for the expression of RBD used throughout this study. A.W. and V.J.H. were responsible for the RBD mutational scanning experiments and NGS data analysis that mapped target epitopes and RBD escape mutations for anti-RBD nanobodies. J.-E.S. and D.S.M. were responsible for computational design aspects for the naïve ~200,000-member nanobody library and A.W. inserted that library into AHEAD. A.C.K. and C.C.L. oversaw all aspects of the project, D.S.M. supervised computational nanobody library design, J.A. supervised neutralization and ACE2 competition assays, and A.G.S. supervised the preparation of RBD. A.W. carried out the deep mutational scanning analysis. A.W., C.M., A.C.K. and C.C.L. wrote the manuscript, with input and contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
Provisional patents (US Patent Application No. 63/123,558 and US Patent Application No. 63/111,860) have been filed on this work. A.C.K. is a co-founder and advisor of Tectonic Therapeutic, Inc., and of the Institute for Protein Innovation. C.C.L. is a co-founder of K2 Biotechnologies, Inc., which focuses on the use of continuous evolution technologies applied to antibody engineering.
Additional information
Peer review information Nature Chemical Biology thanks Theam Soon Lim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Antibody fragments.
Single-chain variable fragments and nanobodies are displayed on the surface of yeast in this study. Their relationships to conventional antibodies are depicted.
Extended Data Fig. 2 Evolution of anti-AT1R nanobodies by AHEAD.
a, Contributions of individual mutations fixed during the evolution of AT110 by AHEAD. Affinity (EC50) of each nanobody for AT1R was determined by measuring binding of yeast-displayed nanobodies to each concentration of AT1R-angiotensin II complex (X-axis) in a single replicate and fitting the resulting binding curve. b, Amino acid sequence of AT110 and evolved variants. Mutations that were discovered using AHEAD are underlined in bold. Mutations that were discovered in a previous AT110 evolution experiment using a standard error prone PCR library approach19 are highlighted in yellow.
Extended Data Fig. 3 Optimization of antibody display in AHEAD.
a, Maps of orthogonal p1 plasmids containing OrthoRep parts driving expression of nanobodies in the first-generation AHEAD 1.0 and improved second-generation AHEAD 2.0 systems. Nb = nanobody, tAHD1 = ADH1 terminator, polyA = polyadenosine tail. b, Increased functional expression of nanobody AT110 using all AHEAD 2.0 parts as determined by FACS. The induced population in AHEAD 2.0 shows an ~25-fold increase in nanobody display levels (determined by mean fluorescence intensity of the cell population) compared to AHEAD 1.0.
Extended Data Fig. 4 Optimization of antibody display in AHEAD and evolution of anti-GFP and anti-HSA antibodies using the optimized second-generation AHEAD 2.0 system.
a, Architectures for nanobody display in the first-generation AHEAD 1.0 and improved second-generation AHEAD 2.0 systems. b, Selection of a new leader sequence for higher nanobody display. FACS plots showing the progressive enrichment of higher efficiency leader sequences across 3 rounds of selection (left panel). Nanobody display level using app8 compared to the selected app8i1 variant (right panel). n = 6, error bars represent ± s.d. c, Selected FACS plots showing affinity maturation of Nb.b201 through AHEAD cycles. d, Selected FACS plots showing affinity maturation of Lag42 through AHEAD cycles. e, (left) Affinities (EC50) of improved high-affinity anti-HSA nanobodies evolved using AHEAD. Binding of yeast-displayed nanobodies by each concentration of HSA was measured in replicate (n = 3, error bars represent ± s.d.) and EC50s were determined by fitting each binding curve. (right) Affinities (EC50) of improved high-affinity anti-GFP nanobodies evolved using AHEAD. Binding of yeast-displayed nanobodies by each concentration of GFP was measured in replicate (n = 3, error bars represent ± s.d.) and EC50s were determined by fitting each binding curve.
Extended Data Fig. 5 Evolution of anti-RBD nanobodies.
a, Isolation of parent anti-RBD nanobodies. (left) FACS plot showing enrichment of initial anti-RBD nanobody clones from a naïve nanobody library32. The green polygon corresponds to the gate used for sorting. (right) Schematic showing the separation of parent clones into different AHEAD experiments in order to minimize competition among parents and their lineages, avoiding early loss of weak parents that have the potential to yield superior descendants later during affinity maturation. b, Selected FACS plots showing anti-RBD affinity maturation by cycles of AHEAD in 8 independent experiments, each starting from one of the 8 parent clones identified from the naïve nanobody library (see Extended Data Fig. 5a). Red polygons correspond to the gates used for sorting.
Extended Data Fig. 6 Affinities of anti-RBD nanobodies determined by surface plasmon resonance (SPR) or EC50 measurements.
SPR or EC50 binding curves are shown for each anti-RBD nanobody characterized in this study. For SPR measurements (Y-axis = Response), kinetic fits are shown where available and steady-state affinity fits are shown for nanobodies for which the on and off rates could not be determined. For EC50 affinities (Y-axis = Normalized Fluorescence), binding of yeast-displayed nanobodies by each concentration of RBD was determined in biological triplicate (n = 3, error bars represent ± s.d.) and EC50s were determined by fitting each binding curve.
Extended Data Fig. 7 Neutralization assays and ACE2 competition assays for anti-RBD nanobodies evolved with AHEAD.
a, Neutralization plots for all anti-RBD nanobodies characterized in this study. Each nanobody concentration (X-axis) was tested in replicate. n = 6, error bars represent ± s.d. b, Biolayer interferometry (BLI) traces measuring ACE2 competition for anti-RBD nanobodies. CR3022 is an anti-RBD antibody that does not compete with ACE2 binding (no competition control) whereas SC1A-B12 is an anti-RBD antibody that competes strongly with RBD binding.
Extended Data Fig. 8 Evolution of an anti-GFP nanobody from a computationally-designed 200,000-member naïve nanobody library encoded on AHEAD.
a, Representative FACS plots showing enrichment of a GFP-binding clone from the nanobody library and subsequent emergence and fixation of a mutation that increases GFP binding across AHEAD cycles. b, Affinity (EC50) of the AHEAD-evolved anti-GFP nanobody, NbG1i1, isolated from AHEAD cycle 6 as compared to its parent, NbG1, that fixed in AHEAD cycle 3. Binding of yeast-displayed nanobodies by each concentration of GFP was determined in relicate (n = 3, error bars represent ± s.d.) and EC50s were determined by fitting each binding curve.
Extended Data Fig. 9 Gating strategy for singlets in all FACS experiments.
(left) Forward scatter (horizontal axes) versus side scatter (vertical axes) of a representative population of yeast cells. Red circle represents cells passing the gate. (right) Forward scatter area (horizontal axes) vs. forward scatter height (vertical axes) gating of cells that passed through the previous gate. Green boundary represents cells passing the gate. For all FACS experiments, only cells sorted through both gates were used in nanobody expression and binding gates and.
Supplementary information
Supplementary Information
Supplementary Tables 1–4.
Supplementary Data 1
Information and activities for all anti-SARS-CoV-2 nanobodies characterized in this study.
Rights and permissions
About this article

Cite this article
Wellner, A., McMahon, C., Gilman, M.S.A. et al. Rapid generation of potent antibodies by autonomous hypermutation in yeast. Nat Chem Biol 17, 1057–1064 (2021). https://doi.org/10.1038/s41589-021-00832-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-021-00832-4
This article is cited by
-
Automated in vivo enzyme engineering accelerates biocatalyst optimization
Nature Communications (2024)
-
Discovery of VH domains that allosterically inhibit ENPP1
Nature Chemical Biology (2024)
-
Efficient evolution of human antibodies from general protein language models
Nature Biotechnology (2024)
-
Machine learning for functional protein design
Nature Biotechnology (2024)
-
Engineered bacterial orthogonal DNA replication system for continuous evolution
Nature Chemical Biology (2023)
