
Abstract
Obesity is a major risk factor for many common diseases and has a substantial heritable component. To identify new genetic determinants, we performed exome-sequence analyses for adult body mass index (BMI) in up to 587,027 individuals. We identified rare loss-of-function variants in two genes (BSN and APBA1) with effects substantially larger than those of well-established obesity genes such as MC4R. In contrast to most other obesity-related genes, rare variants in BSN and APBA1 were not associated with normal variation in childhood adiposity. Furthermore, BSN protein-truncating variants (PTVs) magnified the influence of common genetic variants associated with BMI, with a common variant polygenic score exhibiting an effect twice as large in BSN PTV carriers than in noncarriers. Finally, we explored the plasma proteomic signatures of BSN PTV carriers as well as the functional consequences of BSN deletion in human induced pluripotent stem cell-derived hypothalamic neurons. Collectively, our findings implicate degenerative processes in synaptic function in the etiology of adult-onset obesity.
Similar content being viewed by others


Main
Over 1 billion people worldwide live with obesity, a global health challenge that is rapidly increasing in scale1,2. Obesity is the second leading cause of preventable death, increasing the risk of diseases such as type 2 diabetes (T2D), cardiovascular disease and cancer1,3. Understanding the full range of social, psychological and biological determinants of energy intake and expenditure will be key to tackling this epidemic. Early studies in mice highlighted the role of the leptin−melanocortin pathway in appetite and body weight regulation4, which led to candidate gene sequencing studies of individuals with severe early-onset obesity. These studies identified rare loss-of-function mutations in key components of this pathway as causes of severe early-onset obesity5, the most common of which affect the melanocortin 4 receptor (MC4R)6,7. In parallel, using a ‘hypothesis-free’ approach, large-scale population-based genome-wide association studies (GWAS) have identified hundreds of common genetic variants associated with body mass index (BMI) in adults8. These variants are mostly noncoding and are enriched near genes expressed in the brain9. Individually, the effect of each variant is small, and cumulatively, the ~1,000 common variants identified to date explain only ~6% of the population variance in BMI8.
The recent emergence of whole-exome sequencing (WES) data at the population scale has enabled exome-wide association studies (ExWAS), leading to a convergence of common and rare variant discoveries. In a landmark study, Akbari et al. used WES data from ~640,000 individuals to identify rare protein-coding variants in 16 genes associated with BMI10. These included genes with established roles in weight regulation (MC4R, GIPR and PCSK1) in addition to new targets, such as GPR75, in which loss-of-function mutations are protective against obesity in humans and mice10.
The current study was an ExWAS for BMI using WES data from 419,668 UK Biobank participants. Although this represents a subset of the exomes previously reported by Akbari et al.10, we were motivated by recent work demonstrating that, in the context of gene-burden analysis11, the various choices around how one defines a qualifying rare variant can highlight biologically relevant genes at exome-wide significance missed using alternative definitions12. Consistent with this, our approach identified new rare variant associations with BSN and APBA1, which we replicated in independent WES data from 167,359 individuals of predominantly non-European genetic ancestry. The rare protein-truncating variants (PTVs) detected in BSN and APBA1 have larger effects than other previously reported ExWAS genes10, and our findings collectively suggest emerging roles for neurodevelopment, neurogenesis and altered neuronal oxidative phosphorylation in the etiology of obesity.
Results
Exome-sequence analysis identifies rare alleles associated with BMI
To identify rare variants associated with adult BMI, we performed an ExWAS using genotype and phenotype data from 419,668 individuals of European ancestry from UK Biobank13. Individual gene-burden tests were performed by collapsing rare (minor allele frequency (MAF) < 0.1%) genetic variants across 18,658 protein-coding genes. We tested three categories of variants based on their predicted functional impact: high-confidence (HC) PTVs and two overlapping missense masks that used a REVEL14 score threshold of 0.5 or 0.7. This yielded a total of 37,691 gene tests with at least 30 informative rare allele carriers, corresponding to a multiple-test-corrected statistical significance threshold of P < 1.33 × 10−6 (0.05/37,691).
Genetic association testing was performed using BOLT-LMM15, which identified a total of nine genes that met the threshold for significant association with adult BMI (Supplementary Table 1). Our gene-burden ExWAS appeared to be statistically well calibrated, as indicated by low exome-wide test statistic inflation (λGC = 1.05−1.15) and by the absence of significant associations with any synonymous variant masks (Supplementary Figs. 1 and 2). Five of our identified associations were previously reported: PTVs in MC4R, UBR2, KIAA1109, SLTM and PCSK1 (ref. 10). At the other four genes, heterozygous PTVs conferred higher risk for increased adult BMI: BSN (effect = 3.05 kg m−2, standard error (s.e.) = 0.54, P = 2 × 10−8, carrier n = 65), TOX4 (effect = 3.61 kg m−2, s.e. = 0.71, P = 3.1 × 10−7, carrier n = 39), APBA1 (effect = 2.08 kg m−2, s.e. = 0.42, P = 6.1 × 10−7, carrier n = 111) and ATP13A1 (effect = 1.82 kg m−2, s.e.m. = 0.37, P = 1.1 × 10−6, carrier n = 139). For two of these genes, BSN and ATP13A1, we also found supporting evidence from common genetic variants at the same locus associated with BMI (Supplementary Fig. 3): noncoding alleles ~200 kb upstream of BSN (rs9843653, MAF = 0.49, β = −0.13 kg m−2, P = 9.5 × 10−46) and 400 kb upstream of ATP13A1 (rs72999063, MAF = 0.16, β = 0.09 kg m−2, P = 3.2 × 10−13; Supplementary Table 2). These GWAS signals were also associated with blood RNA expression levels of BSN and ATP13A1, respectively16 (Supplementary Table 2), and the BMI associations were replicated in independent GWAS data from the GIANT consortium9 (Supplementary Fig. 4 and Supplementary Table 2). We found no evidence of rare variant associations with BMI for any other genes at these GWAS loci (Supplementary Table 3).
We aimed to replicate our four new gene-burden rare variant associations in independent WES data from 167,359 individuals of predominantly non-European ancestry from the Mexico City Prospective Study (MCPS)17,18 and the Pakistan Genomic Resource (PGR) study (Fig. 1 and Supplementary Table 4). We observed supportive evidence for two of the four new genes identified above: for 32 BSN PTV carriers the mean BMI was 2.8 kg m−2 (s.e. = 0.84, P = 9.4 × 10−4) higher than for noncarriers, and for 20 APBA1 PTV carriers the mean BMI was 2.33 kg m−2 (s.e. = 1.05, P = 0.03) higher. Although the replication sample was smaller than the UK Biobank sample and evidence for replication at APBA1 was only nominally significant, these effect sizes were remarkably similar to those observed in UK Biobank (3.05 kg m−2 and 2.08 kg m−2 for BSN and APBA1, respectively).

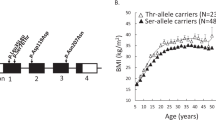
a, Discovery analyses were conducted in UK Biobank (n = 419,668) and replication was conducted in individuals from the MCPS and PGR study (n = 167,359). The means of the effect size estimates are presented with 95% CIs and were converted to kg m−2. Extended data can be found in Supplementary Tables 1 and 4. b, Variant-level results from the BOLT-LMM algorithm using a linear mixed model for association of HC PTVs in BSN and APBA1 with BMI. The y axis shows trait-increasing effects with −log10(P) and trait-decreasing effects with log10(P). The dashed lines denote a nominal significance threshold of P < 0.05. Statistics used to generate these plots are provided as source data.
The effect of BSN on BMI was larger than that of any previously reported ExWAS gene (Fig. 2) and substantially increased the risk of obesity (BMI > 30 kg m−2) in UK Biobank (BSN: odds ratio (OR) = 3.04 (95% confidence interval (CI), 1.87−4.94), P = 7.7 × 10−6, 49% case prevalence; APBA1: OR = 2.14 (1.46−3.13), P = 8.5 × 10−5, 41% case prevalence) and for BSN also increased the risk of severe obesity (BMI > 40 kg m−2) (OR = 6.61 (3.01−14.55), P = 2.6 × 10−6, 11% case prevalence) although this was not the case for APBA1 (OR = 1.91 (0.70−5.19), P = 0.20, 4% case prevalence; Fig. 3). Association statistics for individual variants in BSN and APBA1 in UK Biobank are shown in Fig. 1b and Supplementary Table 5. The gene-level associations of BSN and APBA1 with BMI were not driven by single HC PTVs (Supplementary Table 6), and carriers appeared to be geographically dispersed across the UK (Supplementary Fig. 5).

The means of the effect size estimates on BMI are presented with 95% CIs and are based on only UK Biobank participants (n = 419,668). The statistics used to generate this plot are provided as source data. pLOF, predicted loss of function.

The BMI categories appear according to guidance from the World Health Organization. The statistics used to generate this plot are provided as source data.
In a case−cohort study that included the Severe Childhood-Onset Obesity Project (SCOOP) and the INTERVAL Study (INTERVAL), we identified an excess of BSN PTV carriers among patients affected by severe early-onset obesity (3/927 cases; p.Arg1276*, p.Arg1787*, p.Arg2925*; Supplementary Table 4) compared to the control cohort (1/4,057; OR = 13 (1.05−686), Pexact = 0.02). Furthermore, the one PTV found among controls (p.Trp3926*) is located at the final amino acid of the BSN-encoded protein bassoon and is therefore unlikely to affect its function (Pexact = 0.006, when excluding p.Trp3926*).
Phenotypic characterization of BSN and APBA1 rare allele carriers
We next sought to understand the broader phenotypic profile of carriers of PTVs in BSN and APBA1. In UK Biobank, these genes showed diverse associations with body composition, with higher fat and lean mass across body compartments (Supplementary Table 7), but showed no association with adult height (P > 0.05) or waist-to-hip ratio adjusted for BMI (P > 0.05). In contrast to almost all previously reported obesity-associated genes, neither BSN nor APBA1 showed any association with childhood body size or puberty timing (P > 0.05), suggesting adult-onset effects on body weight based on the phenotypes available in UK Biobank. In UK Biobank, carriers of PTVs in BSN also had a higher risk of T2D (OR = 3.03, (1.60−5.76), P = 7.1 × 10−4, 18% case prevalence)—an effect size comparable to those of previously reported rare variant associations for T2D19,20. A broader phenome-wide analysis across 11,693 traits revealed a number of other associations (Supplementary Table 8); notably, BSN PTV carriers had a substantially higher risk of nonalcoholic fatty liver disease, as defined by a fatty liver index of ≥60 (ref. 21) or a hepatic steatosis index of >36 (ref. 22), compared to noncarriers (OR = 3.73 (2.26−6.16), P = 8.4 × 10−7, 45% case prevalence).
BSN carrier status magnifies the effect of common genetic variants
Previous studies have reported that common BMI-associated alleles increased the penetrance of obesity in rare allele carriers in an additive model10. To evaluate this for BSN and APBA1, we created a common variant polygenic score (PGS) in UK Biobank, using individual variant effect estimates obtained from independent GIANT consortium GWAS data9. By testing the multiplicative interaction between the PGS and rare variant carrier status on BMI in a linear regression model, we observed significant effect modification by BSN PTVs (interaction P = 0.01; Supplementary Fig. 6), but not APBA1 PTVs (P = 0.22). In carriers of BSN PTVs, the effect size of the PGS on BMI was double (0.6 s.d. increase in BMI per unit increase in PGS, equivalent to 2.9 kg m−2) that in noncarriers (0.3 s.d., equivalent to 1.4 kg m−2).
Evaluating the impact of BSN and APBA1 functions on the plasma proteome
To explore the putative biological mechanisms through which BSN and APBA1 might exert their effects, we first characterized the plasma proteomic signature of PTV carriers using Olink data on 1,463 circulating proteins available in ~50,000 UK Biobank participants23,24. Using the available proteomics data, we identified 6 and 17 PTV carriers for BSN and APBA1, respectively. No changes in plasma protein levels were associated with APBA1 carrier status after multiple-test correction (P < 3.42 × 10−5 (0.05/1,463)); however, BSN PTV carriers had higher levels of lymphotoxin alpha (LTα, previously known as TNFβ) than noncarriers (effect = 1.07, s.e. = 0.183, P = 5.3 × 10−9) (Supplementary Table 9). Furthermore, circulating LTα levels were positively associated with BMI (increase of 1.18 kg m−2 in BMI per 1 s.d. increase in LTα concentration, P = 7.6 × 10−122), and common genetic variants at the LTA locus were associated with BMI (rs3130048, MAF = 0.72, β = −0.10 kg m−2 per allele, P = 1.10 × 10−23). We repeated these analyses using the common BMI-associated variant (rs9843653) at BSN and identified 23 associated proteins, the most significant of which was semaphorin-3F (−0.03 s.d. per BMI-increasing allele, P = 6.7 × 10−45), a member of the semaphorin family that has been previously implicated in obesity etiology25. In total, 10 of the genes encoding these 24 proteins (including SEMA3F and LTA) were also implicated by common variant signals for BMI (Supplementary Table 10).
Differential gene expression in BSN +/− hypothalamic neurons
Finally, we explored the functional consequences of deleting BSN, which is highly expressed in the brain, by generating CRISPR−Cas9-edited human induced pluripotent stem cell-derived hypothalamic neurons heterozygous for the BSN p.Leu400Trpfs*114 PTV (BSN+/−) (Methods). On visual inspection, BSN+/− cells showed no obvious morphological effect on neuronal differentiation (Supplementary Fig. 7). To assess transcriptional differences between BSN+/− and wild-type cells, we performed single-nucleus RNA sequencing (snRNA-seq) in 61,016 hypothalamic neurons (32,198 BSN+/−, 28,818 wild type). We identified 18 distinct cell clusters, as shown via a uniform manifold approximation and projection plot (Supplementary Fig. 8; marker genes listed in Supplementary Table 11). Eight clusters were neurons (clusters 4, 5, 6, 9, 11, 13, 14 and 15; total n = 18,873) marked with RBFOX3 (NeuN), BSN and the bassoon binding partner PCLO (Supplementary Fig. 8). Because BSN is universally expressed in neurons, we combined expression data across all eight neuronal clusters in the differential gene expression analysis and performed pathway enrichment analyses to examine the possible global consequences of BSN+/−. Differential expression analyses revealed 778 genes (defined by P < 0.05 and log2(fold change (FC)) > 1 or < −1) (Supplementary Table 12), including downregulation of genes with reported roles in body weight regulation, such as SEMA3C25 and APOE26,27. The top enriched pathways included ‘neuroactive ligand-receptor interaction’ and ‘negative regulation of neurogenesis’, as well as ‘respiratory chain complex I (gamma subunit) mitochondrial’. Furthermore, when we examined the differential expression within individual clusters, NTNG1 was downregulated (log2(FC) = −0.66 to −0.93, P < 0.05) in four of eight BSN+/− populations (Supplementary Table 12). NTNG1 is closely associated with bassoon within the presynaptic active zone; it belongs to a class of synaptic adhesion molecules crucial for synaptic function28 and has a role in axon guidance in neurons29. Interestingly, common variants of NTNG1 are associated with BMI30,31. Differentially expressed genes within cluster 13 were also enriched for common variant associations with BMI (Supplementary Tables 13 and 14), including associations in APOE, DOC2A, COMT and GABPB2. Taken together, these results highlight dysregulation of neurodevelopment, neurogenesis and neuronal oxidative phosphorylation as possible underlying mechanisms linking BSN deficiency to obesity (Supplementary Table 15).
Discussion
We found that rare PTVs in APBA1 and BSN were associated with a substantial increase in adult BMI and higher risks of obesity and severe obesity in adults. Rare PTVs in BSN were also associated with higher risks for T2D and nonalcoholic fatty liver disease. The associations with adult BMI were confirmed in independent cohorts and were also supported by mapping of common variant signals to whole-blood expression quantitative trait loci for APBA1 and BSN. Rare PTVs in BSN were also found in three individuals with severe early-onset obesity; however, in UK Biobank, 65 BSN PTV carriers showed no difference in childhood adiposity-related traits compared to noncarriers. Therefore, APBA1 and BSN appear to be among the few genetic determinants of predominantly adult-onset obesity. The recalled childhood adiposity trait in UK Biobank shows a high genetic correlation with measured childhood BMI32; however, we acknowledge that it may still be an insensitive measure and longitudinal studies are needed.
APBA1 encodes a neuronal adaptor protein that interacts with amyloid precursor protein, encoded by the Alzheimer disease-associated APP gene. It has a putative role in signal transduction as a vesicular trafficking protein with the potential to couple synaptic vesicle exocytosis to neuronal cell adhesion33. BSN encodes bassoon, a scaffolding protein essential for organization of the presynaptic cytoskeleton and exocytosis-mediated neurotransmitter release34. Bsn knockout in mice reduces excitatory synaptic transmission because vesicles are unable to efficiently fuse with the synaptic membrane35. BSN is expressed primarily in the brain and is reportedly upregulated in the frontal lobes of patients with multiple system atrophy, a progressive neurodegenerative disease36. Furthermore, rare predicted-damaging missense mutations in BSN have been reported in four patients with progressive supranuclear palsy-like syndrome with features of multiple system atrophy and Alzheimer disease37. The links identified here with predominantly adult-onset obesity may be consistent with the putative roles of APBA1 and BSN in aging-related neurosecretory vesicle dysfunction and neurodegeneration. Therefore, we posit that adult obesity could result from some form of subtle age-dependent degeneration in primary appetitive regulatory pathways.
Previous studies have reported additive effects of common and rare susceptibility alleles on BMI10, but there is no evidence for epistatic interactions that are indicative of biological interactions. Notably, we found that carriers of rare PTVs in BSN showed enhanced susceptibility to the influence of a common variant PGS for adult BMI. The mechanistic basis for this statistical interaction is unclear. However, as the common genetic susceptibility to obesity is thought to act predominantly via central regulation of food intake9,38, we hypothesize that BSN may have widespread involvement in neurodevelopment and neurogenesis, with BSN variants leading to increased appetitive drive. We propose that future studies explore the impact of BSN PTVs on primary appetitive regulatory pathways across the life course.
The associations identified with rare PTVs in APBA1 and BSN were not highlighted in previous ExWAS analyses using overlapping data. We acknowledge the differences between such studies in relation to variant quality control and the thresholds used for in silico functional prediction. We posit that standardization in this field would be premature. Instead, studies should clearly detail their analytical approaches and seek replication and other forms of confirmation.
In conclusion, rare genetic disruptions of APBA1 and BSN have larger impacts on adult BMI and obesity risk than heterozygous disruptions of any previously described obesity risk gene. Rare PTVs in APBA1 and BSN appear to preferentially confer risk of adult-onset obesity, which we propose might be due to widespread dysregulation of neurodevelopment, neurogenesis and neuronal oxidative phosphorylation in neurons within the central feeding circuitry.
Methods
Ethics
Our research complies with all relevant ethical regulations. All studies included in this research were approved by the relevant board or committee. UK Biobank has approval from the North West Multi-centre Research Ethics Committee (REC reference 13/NW/0157) as a Research Tissue Bank (RTB) approval, and informed consent was provided by each participant. This approval means that researchers do not require separate ethical clearance and can operate under RTB approval. This RTB approval was granted initially in 2011 and is renewed every 5 years; hence, UK Biobank successfully renewed approval in 2016 and 2021. The MCPS was approved by the Mexican Ministry of Health, the Mexican National Council for Science and Technology and the University of Oxford. The PGR study was approved by the institutional review board at the Center for Non-Communicable Diseases (IRB: 00007048, IORG0005843, FWAS00014490) and all participants provided informed consent. The SCOOP cohort was approved by the Multi-regional Ethics Committee and the Cambridge Local Research Ethics Committee (MREC 97/21 and REC number 03/103). Participants (or parents for individuals <16 years old) provided written informed consent; minors provided oral consent. The INTERVAL study received ethics committee approval from the National Research Ethics Service Committee (11/EE/0538), and all participants provided informed consent before joining the study.
UK Biobank data processing and quality control
We used the same processing strategies as those outlined in our previous paper to analyze the WES data and perform quality control steps19. We queried WES data from 454,787 individuals in UK Biobank39, excluding those with excess heterozygosity, those with autosomal variant missingness on genotyping arrays of ≥5%, or those not included in the subset of phased samples as defined by Bycroft et al.13.
WES data were stored as population-level variant call format (VCF) files, aligned to GRCh38 and accessed through the UK Biobank Research Analysis Platform (RAP). In addition to the quality control measures already applied to the released data, as described by Backman et al.39, we conducted several additional quality control procedures. First, we used ‘bcftools v1.14 norm’40 to split the multiallelic sites and left-correct and normalize indels. Next, we filtered out variants that failed our quality control criteria, including those with: (1) read depth of <7; (2) genotype quality of <20; and (3) binomial test P value for alternative allele reads versus reference allele reads of ≤0.001 for heterozygous genotypes. For indel genotypes, we kept only variants with read depth of ≥10 and genotype quality of ≥20. Variants that failed quality control criteria were marked as missing (that is, ./.). After filtering, variants where more than 50% of the genotypes were missing were excluded from downstream analyses19.
The remaining variants underwent annotation using Ensembl Variant Effect Predictor (VEP v104)41 with the ‘-everything’ flag and additional plugins for REVEL14, CADD42 and LOFTEE43. For each variant, a single Ensembl transcript was prioritized on the basis of whether the annotated transcript was protein-coding, MANE select v0.97 (ref. 44) or the VEP canonical transcript. The individual consequence for each variant was then prioritized on the basis of severity as defined by VEP. Stop-gained, splice acceptor and splice donor variants were merged into a combined PTV category, while annotations for missense and synonymous variants were adopted directly from VEP. We included only variants on autosomes and the X chromosome that were within Ensembl protein-coding transcripts and transcripts included in the UK Biobank WES assay in our downstream analysis.
Our analyses focused primarily on individuals of European genetic ancestry, and we excluded those who withdrew consent from the study, resulting in a final cohort of 419,668 individuals.
Exome-wide gene-burden testing in UK Biobank
We used BOLT-LMM v2.3.6 (ref. 15) as our primary analytical tool to conduct the gene-burden test. To run BOLT-LMM, we first queried a set of genotypes with minor allele count (MAC) > 100, which was derived from the genotyping arrays for the individuals with the WES data to build the null model. To accommodate BOLT-LMM’s requirement for imputed genotyping data rather than per-gene carrier status, we developed dummy genotype files in which each gene was represented by a single variant. We then coded individuals with a qualifying variant within a gene as heterozygous, regardless of the total number of variants they carried in that gene. We then created dummy genotypes for the HC PTVs with MAF < 0.1% as defined by LOFTEE, missense variants with REVEL > 0.5 and missense variants with REVEL > 0.7. We then used BOLT-LMM to analyze phenotypes using default parameters, except for the inclusion of the ‘lmmInfOnly’ flag. In addition to the dummy genotypes, we included all individual markers in the WES data to generate association test statistics for individual variants. We used age, age2, sex and the first ten principal components (PCs) as calculated by Bycroft et al.13 and the WES release batch (50k, 200k, 450k) as covariates.
To check whether there was a single variant driving the association, we performed a leave-one-out analysis for BSN and APBA1 using linear regression in R v3.6.3 by dropping the HC PTVs contained in our analysis one by one. In addition, we also checked the geographic distribution of APBA1 and BSN HC PTV carriers.
Replication of findings in two independent non-European cohorts
We sought replication of our findings for the four new genes in two independent predominantly non-European exome-sequenced cohorts: the MCPS and the PGR study.
MCPS is a cohort study of 159,755 adults of predominantly admixed American ancestry. Participants aged 35 years or older were recruited between 1998 and 2004 from two adjacent urban districts of Mexico City. Phenotypic data were recorded during household visits, including height, weight, and waist and hip circumferences. Disease history was self-reported at baseline, and the participants were linked to Mexican national mortality records. The cohort has been described in detail elsewhere17,18.
The PGR study has been recruiting participants aged 15−100 years as cases or controls via clinical audits for specific conditions since 2005 from over 40 centers around Pakistan. Participants were recruited from clinics treating patients with cardiometabolic, inflammatory, respiratory or ophthalmological conditions. Information on lifestyle habits, medical and medication history, family history of diseases, exposure to smoking and tobacco consumption, physical activity, dietary habits, anthropometry, basic blood biochemistry and electrocardiogram traits was recorded during clinic visits. DNA, serum, plasma and whole blood samples were also collected from all study participants.
Exome sequencing data for 141,046 MCPS and 37,800 PGR participants were generated at the Regeneron Genetics Center and passed Regeneron’s initial quality control, which included identifying sex discordance, contamination, unresolved duplicate sequences and discordance with microarray genotype data for MCPS. Genomic DNA was subjected to paired-end 75-bp WES at Regeneron Pharmaceuticals using the IDT xGen v1 capture kit on the NovaSeq 6000 platform. Conversion of sequencing data in BCL format to FASTQ format and the assignments of paired-end sequence reads to samples were based on 10-base barcodes, using bcl2fastq v2.19.0.
These exome sequences were processed at AstraZeneca from their unaligned FASTQ state. A custom-built Amazon Web Services cloud computing platform running Illumina DRAGEN Bio-IT Platform Germline Pipeline v3.0.7 was used to align the reads to the GRCh38 genome reference and perform single-nucleotide variant (SNV) and insertion and deletion (indel) calling. SNVs and indels were annotated using SnpEff v4.3 (ref. 45) against Ensembl Build 38.92. All variants were additionally annotated with their gnomAD MAFs (gnomAD v2.1.1 mapped to GRCh38)43.
To further apply quality control to the sequence data, all MCPS and PGR exomes underwent a second screening using AstraZeneca’s bioinformatics pipeline, which has been described in detail previously46. Briefly, we excluded from the analysis sequences that had a VerifyBamID freemix (contamination) level of more than 4%, those for which inferred karyotypic sex did not match self-reported gender or those for which less than 94.5% of the consensus coding sequence (CCDS release 22) achieved a minimum tenfold read depth. We further removed one individual from every pair of genetic duplicates or monozygotic twins with a kinship coefficient of >0.45. Kinship coefficients were estimated from exome genotypes using the kinship function from KING v2.2.3 (ref. 47). For the MCPS, we additionally excluded sequences with an average CCDS read depth of at least 2 s.d. below the mean. After the above quality control steps, 139,603 (99.0%) MCPS and 37,727 (99.3%) PGR exomes remained.
For the MCPS, we predicted the genetic ancestry of participants using PEDDY v0.4.2 (ref. 48), with 1000 Genomes Project sequences as population ref. 49, and retained individuals with a predicted probability of admixed American ancestry of ≥0.95 who were within 4 s.d. of the means for the top four PCs. In the PGR study, we retained individuals with a predicted probability of South Asian ancestry of ≥0.95 who were within 4 s.d. of the means for the top four PCs. Following ancestry filtering, 137,059 (97.2%) MCPS and 36,280 (95.5%) PGR exomes remained.
We assessed the association of BMI and weight quantitative traits with genotype at the four proposed new genes of interest using a previously described gene-level collapsing analysis framework implementing a PTV collapsing analysis model46. We classified variants as PTVs if they had been annotated by SnpEff as follows: exon_loss_variant, frameshift_variant, start_lost, stop_gained, stop_lost, splice_acceptor_variant, splice_donor_variant, gene_fusion, bidirectional_gene_fusion, rare_amino_acid_variant and transcript_ablation.
We applied MAF filters to target rare variants: MAF < 0.001 in gnomAD (overall and every population except OTH) and leave-one-out MAF < 0.001 among our combined case and control test cohort. For variants to qualify, they had to also meet the following quality control filters: minimum site coverage of 10×; annotation in CCDS transcripts (release 22); at least 80% alternative reads in homozygous genotypes; a percentage of alternative reads for heterozygous variants of ≥0.25 and ≤0.8; a binomial test of alternative allele proportion departure from 50% in the heterozygous state result of P > 1 ×10−6; GQ of ≥20; FS of ≤200 (indels) or ≤60 (SNVs); MQ of ≥40; QUAL of ≥30; read position rank sum score of ≥−2; MQRS of ≥−8; DRAGEN variant status = PASS; and test cohort carrier quality control failure of < 0.5%. If the variant was observed in gnomAD exomes, we also applied the following filters: variant site achieved tenfold coverage in ≥25% of gnomAD exomes; variant site achieved exome z-score of ≥−2.0; exome MQ of ≥30; and random forest probability that the given variant is a true SNV or indel of >0.02 and >0.01, respectively50.
For the quantitative traits and for each gene, the difference in mean between the carriers and noncarriers of PTVs was determined by fitting a linear regression model, correcting for age and sex. In addition to calculating individual statistics for the MCPS and the PGR study, we also meta-analyzed the individual study effect sizes to generate a combined replication statistic using an inverse variance-weighted fixed-effect meta-analysis using the rma.uni() function from the metafor package v3.8-1 (ref. 51) in R v3.6.3.
BSN PTV carriers in the SCOOP−INTERVAL case−cohort study
To test whether there was an association between pLOF variants in the BSN gene and severe early-onset obesity, we studied 927 exomes from white British participants with severe early-onset obesity recruited to the Genetics of Obesity Study (GOOS) (SCOOP cohort) and 4,057 control exomes from the INTERVAL cohort of UK blood donors. SCOOP comprises UK patients with severe obesity (BMI more than 3 s.d. above the mean for age and sex) of early onset (<10 years) recruited to the GOOS. Exome sequencing in a subset of people of white British ancestry (the SCOOP cohort) was performed as described previously52,53,54. INTERVAL comprises predominantly healthy blood donors in the UK55 (https://www.intervalstudy.org.uk).
SCOOP and INTERVAL variants were joint-called and filtered for variant-level and sample-level quality control, as previously described52. A total of 927 cases (SCOOP) and 4,057 controls (INTERVAL) passed the quality control filters53. After splitting multiallelic variants and left normalizing, we annotated variants using VEP with Ensembl v96 (GRCh37) and identified high-impact variants (predicted protein-truncating, null or splice-disrupting) in the gene BSN (transcript ENST00000296452) using VEP IMPACT=‘HIGH’. This definition includes stop-gain variants (SNVs resulting in stop codons), frameshifts and splice donor/acceptor variants. We verified that the predicted consequences and stop codon positions were maintained in the latest minor version of the transcript (ENST00000296452.5, NM_003458.4) using VEP v110 after lifting over to GRCh38. Missense variants were detected in almost all BSN exons among SCOOP exomes (7/10 coding exons) and INTERVAL exomes (8/10 coding exons), suggesting that BSN stop-gain detection rates in cases and controls are unlikely to be driven by differential read coverage within the BSN gene.
The one PTV identified in INTERVAL (p.Trp3926*) is located at the final amino acid of the bassoon protein and is therefore unlikely to affect expression levels (note that the LOFTEE in silico stop-gain filter for low-confidence loss of function based on the ‘50-bp rule’ does not apply to the BSN gene because the termination codon is itself >55 bp from the final exon−exon boundary56). After excluding this variant on the basis of low confidence for loss of function, we performed a nested gene-burden analysis on the remaining three variants: n = 3 pLOF carriers in SCOOP and n = 0 carriers in INTERVAL controls (OR (95% CI) = inf (1.8−inf), P = 0.006, Fisher’s exact test; adding +0.5 to each cell, OR = 31). Studies in vitro are required to establish the effect of each stop-gain variant on bassoon protein expression levels and localization.
Phenome-wide analysis in UK Biobank
We included binary and quantitative traits made available in the June 2022 UK Biobank data release, harmonizing the phenotype data as previously described46. This resulted in 11,690 phenotypes for analysis, which are available on https://azphewas.com. On the basis of clinical relevance, we derived three additional phenotypes.
For UK Biobank phenome-wide analyses of the four putatively new genes, the same data generation and quality control processes described for the MCPS and PGR study were applied to UK Biobank exomes. Following the Regeneron and AstraZeneca quality control steps, 445,570 UK Biobank exomes remained. The phenome-wide analysis was performed in UK Biobank participants of predominantly European descent, whom we identified based on a PEDDY-derived predicted probability of European ancestry of ≥0.95 and were within 4 s.d. of the means for the top four PCs. On the basis of predicted ancestry pruning, 419,391 UK Biobank exomes were included in the phenome-wide analyses of the four prioritized genes.
As described previously, we assessed the association of the 11,693 phenotypes with genotypes at the four genes of interest, using a PTV collapsing analysis model46, and classifying variants as PTVs using the same SnpEff definitions as described for the MCPS and PGR analyses. For variants to qualify for inclusion in the model, we applied the same MAF and quality control filters used in the MCPS and PGR analyses, with the exception that due to the larger sample size of UK Biobank, only <0.01% of the test cohort carriers were permitted to fail quality control.
Association testing for other anthropometric phenotypes and protein expression levels
We ran association tests of APBA1 and BSN HC PTV carriers and carriers of a BMI-associated common variant (rs9843653) at the BSN locus with a list of anthropometric phenotypes available in UK Biobank using R v3.6.3 (Supplementary Table 5), including the same covariates we used in our exome-wide gene-burden tests. We acquired normalized protein expression data generated by the Olink platform from the UK Biobank RAP23,24. The detailed Olink proteomics assay, data processing and quality control were described by Sun et al.23. For the association tests of APBA1 and BSN PTV carriers and BMI-associated common variant (rs9843653) at the BSN locus carriers with expression levels for 1,463 proteins, we added age2, age × sex, age2 × sex, Olink batch, UK Biobank center, UK Biobank genetic array, number of proteins measured and the first 20 genetic PCs as covariates, as suggested by Sun et al.23. We chose the Bonferroni-corrected P value (P < 3.42 × 10−5 (0.05/1,463)) as the threshold for significance.
BMI GWAS lookup and downstream analyses
Identified genes were queried for proximal BMI GWAS signals, using data from UK Biobank, for signals within 500 kb upstream of the gene’s start site to 500 kb downstream of the gene’s end site. Such signals were further replicated in an independent BMI GWAS9.
We also performed colocalization tests, using the approximate Bayes factor method in R v4.0.2 using the package ‘coloc’ v5.1.0 and blood gene expression data from the eQTLGen study16. Genomic regions were defined as the regions ±500 kb around each gene, and loci exhibiting an H4 posterior probability of >0.5 were considered to show evidence of colocalization.
Finally, we used the GWAS data to calculate gene-level common variant associations, using MAGMA v1.09 (ref. 57). To do this, we used all common but nonsynonymous (coding) variants within a given gene. Gene-level scores were further collapsed into pathway-level associations where appropriate.
Interaction effect between the PGS and PTV carrier status
To examine whether there is an interaction effect between PTV carrier status for BSN and APBA1 and the PGS, we included an interaction term between the PGS and the carrier status for BSN and APBA1 PTVs in a linear regression model adjusted for sex, age and age2, and the first 10 PCs.
The PGS was constructed for 419,581 individuals of white European ancestry who had both genotype and exome sequencing data and a BMI record in UK Biobank. We used summary statistics of BMI from Locke et al.9, which included samples not in UK Biobank. Data were downloaded from the GIANT consortium. The summary statistics included 2,113,400 single-nucleotide polymorphisms (SNPs) with at least 500,000 samples in a cohort of 322,154 participants of European ancestry. For the genotype data of UK Biobank participants, a light quality check procedure was applied, where SNPs were removed if they had a MAF of <0.1%, Hardy−Weinberg equilibrium P < 1 × 10-6 or more than 10% missingness. In addition, SNPs that were mismatched with those in the summary statistics (with the same rsID but different chromosomes or positions) were excluded. We used the package ‘lassosum’ v4.0.5 (ref. 58) in R v3.6.0 to construct the PGS. The R2 of the model including the PGS regressed on rank-based inverse normal-transformed BMI and adjusted for sex, age and age2, and the first 10 PCs as covariates was 11%.
Cellular work and single-cell analyses
A detailed description of the methods used in cellular work and single-cell analyses can be found in the Supplementary Note.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The UK Biobank phenotype and WES data described here are publicly available to registered researchers through the UK Biobank data access protocol. Information about registration for access to the data is available at https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access. Data for this study were obtained under resource applications 26041 and 9905. The MCPS welcomes open-access and collaboration data requests from bona fide researchers. For more details on accessibility, the study’s data and sample sharing policy can be downloaded (in English or Spanish) from https://www.ctsu.ox.ac.uk/research/mcps. Available study data can be examined in detail through the study’s Data Showcase, available at https://datashare.ndph.ox.ac.uk/mexico/. SCOOP and INTERVAL WES data are accessible from the European Genome-phenome Archive with accession numbers EGAS00001000124 (SCOOP) and EGAS00001000825 (INTERVAL). snRNA-seq data are available from the NCBI Gene Expression Omnibus (GEO), under accession number: GSE243112. Source data are provided with this paper.
Code availability
The pipeline code for processing, filtering, annotating and burden testing UK Biobank WES data using the UK Biobank RAP is publicly available (https://github.com/mrcepid-rap)59. No custom code for analyzing the UK Biobank WES data was developed for this study. The analysis code for single-nucleus sequencing is available on GitHub (https://github.com/mariachukanova1/BSN_paper)60 and has been deposited on Zenodo at https://doi.org/10.5281/zenodo.10687754 (ref. 61).
References
Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
GBD 2015 Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017).
Di Cesare, M. et al. The epidemiological burden of obesity in childhood: a worldwide epidemic requiring urgent action. BMC Med. 17, 212 (2019).
Zhang, Y. et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 23, 120–133 (2022).
Vaisse, C., Clement, K., Guy-Grand, B. & Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 20, 113–114 (1998).
Yeo, G. S. H. et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. 20, 111–112 (1998).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Akbari, P. et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 373, eabf8683 (2021).
Povysil, G. et al. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat. Rev. Genet. 20, 747–759 (2019).
Stankovic, S. et al. Genetic susceptibility to earlier ovarian ageing increases de novo mutation rate in offspring. Preprint at medRxiv https://doi.org/10.1101/2022.06.23.22276698 (2022).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Loh, P. R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Tapia-Conyer, R. et al. Cohort profile: the Mexico City Prospective Study. Int. J. Epidemiol. 35, 243–249 (2006).
Ziyatdinov, A. et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature 622, 784–793 (2023).
Gardner, E. J. et al. Damaging missense variants in IGF1R implicate a role for IGF-1 resistance in the etiology of type 2 diabetes. Cell Genom. 2, 100208 (2022).
Zhao, Y. et al. GIGYF1 loss of function is associated with clonal mosaicism and adverse metabolic health. Nat. Commun. 12, 4178 (2021).
Bedogni, G. et al. The Fatty Liver Index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 6, 33 (2006).
Lee, J. H. et al. Hepatic steatosis index: a simple screening tool reflecting nonalcoholic fatty liver disease. Dig. Liver Dis. 42, 503–508 (2010).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Dhindsa, R. S. et al. Rare variant associations with plasma protein levels in the UK Biobank. Nature 622, 339–347 (2023).
van der Klaauw, A. A. et al. Human Semaphorin 3 variants link melanocortin circuit development and energy balance. Cell 176, 729–742 (2019).
Huang, J. et al. Genomics and phenomics of body mass index reveals a complex disease network. Nat. Commun. 13, 7973 (2022).
Chung, J. Y. et al. Identification of five genetic variants with differential effects on obesity-related traits based on age. Front. Genet. 13, 970657 (2022).
Seiradake, E. et al. Structural basis for cell surface patterning through NetrinG−NGL interactions. EMBO J. 30, 4479–4488 (2011).
Nakashiba, T. et al. Netrin-G1: a novel glycosyl phosphatidylinositol-linked mammalian netrin that is functionally divergent from classical netrins. J. Neurosci. 20, 6540–6550 (2000).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174 (2019).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019).
Richardson, T. G., Sanderson, E., Elsworth, B., Tilling, K. & Smith, G. D. Use of genetic variation to separate the effects of early and later life adiposity on disease risk: mendelian randomisation study. BMJ 369, m1203 (2020).
Butz, S., Okamoto, M. & Südhof, T. C. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94, 773–782 (1998).
Tom Dieck, S. et al. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J. Cell Biol. 142, 499–509 (1998).
Altrock, W. D. et al. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron 37, 787–800 (2003).
Hashida, H. et al. Cloning and mapping of ZNF231, a novel brain-specific gene encoding neuronal double zinc finger protein whose expression is enhanced in a neurodegenerative disorder, multiple system atrophy (MSA). Genomics 54, 50–58 (1998).
Yabe, I. et al. Mutations in bassoon in individuals with familial and sporadic progressive supranuclear palsy-like syndrome. Sci. Rep. 8, 819 (2018).
De Lauzon-Guillain, B. et al. Mediation and modification of genetic susceptibility to obesity by eating behaviors. Am. J. Clin. Nutr. 106, 996–1004 (2017).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532 (2021).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Pedersen, B. S. & Quinlan, A. R. Who’s who? Detecting and resolving sample anomalies in human DNA sequencing studies with Peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Singh, T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016).
Marenne, G. et al. Exome sequencing identifies genes and gene sets contributing to severe childhood obesity, linking PHIP variants to repressed POMC transcription. Cell Metab. 31, 1107–1119 (2020).
Hendricks, A. E. et al. Rare variant analysis of human and rodent obesity genes in individuals with severe childhood obesity. Sci. Rep. 7, 4394 (2017).
Moore, C. et al. The INTERVAL trial to determine whether intervals between blood donations can be safely and acceptably decreased to optimise blood supply: study protocol for a randomised controlled trial. Trials 15, 363 (2014).
Nagy, E. & Maquat, L. E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199 (1998).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Mak, T. S. H., Porsch, R. M., Choi, S. W., Zhou, X. & Sham, P. C. Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 41, 469–480 (2017).
mrcepid-rap. GitHub https://github.com/mrcepid-rap (2024).
mariachukanova1/BSN_paper. GitHub https://github.com/mariachukanova1/BSN_paper (2024).
Chukanova, M. snRNAseq analysis for “Protein-truncating variants in BSN are associated with severe adult-onset obesity, type 2 diabetes and fatty liver disease”. Zenodo https://doi.org/10.5281/zenodo.10687754 (2024).
Acknowledgements
We thank the participants and investigators in the UK Biobank study who made this work possible (resource application number 26041; 9905), the UK Biobank Exome Sequencing Consortium (UKB-ESC) members AbbVie, Alnylam Pharmaceuticals, AstraZeneca, Biogen, Bristol-Myers Squibb, Pfizer, Regeneron and Takeda for funding the generation of the data; the Regeneron Genetics Center for completing the sequencing and initial quality control of the exome sequencing data; and the AstraZeneca Centre for Genomics Research analytics and informatics team for processing and analyzing the sequencing and phenotype data. We thank the physicians who referred people to the GOOS and the participants and families for their involvement. Y.Z., K.A.K., R.Y.J., E.J.G., F.R.D., L.R.K., N.J.W., K.K.O. and J.R.B.P. are supported by the UK MRC (Unit Programmes MC_UU_00006/1 and MC_UU_00006/2). M.C. and A.M.S. are supported by a project grant from the MRC (MR/S026193/1). Y.-C.L.T., B.Y.H.L. and G.S.H.Y. are supported by the MRC Metabolic Diseases Unit (MC_UU_00014/1). G.K.C.D. is supported by the BBSRC Doctoral Training Programme. The MCPS has received funding from the Mexican Health Ministry, the National Council of Science and Technology for Mexico, the Wellcome Trust (058299/Z/99), Cancer Research UK, the British Heart Foundation and the UK MRC (MC_UU_00017/2). I.S.F. is supported by a Wellcome Principal Research Fellowship (207462/Z/17/Z), the National Institute for Health and Care Research (NIHR) Cambridge Biomedical Research Centre, the Botnar Foundation, the Bernard Wolfe Health Neuroscience Endowment and an NIHR Senior Investigator award. I.B. acknowledges funding from an ‘Expanding Excellence in England’ award from Research England. F.M. is a New York Stem Cell Foundation−Robertson Investigator (NYSCF-R-156) and is supported by the Wellcome Trust and Royal Society (211221/Z/18/Z) and a Ben Barres Early Career Acceleration Award from the Chan Zuckerberg Initiative (CZI NDCN 191942). This work was supported by the NIHR Exeter Biomedical Research Centre. Next-generation sequencing was performed at the Institute of Metabolic Science Genomics and Bioinformatics Core supported by the MRC (MC_UU_00014/5) and the Wellcome Trust (208363/Z/17/Z) and the Cancer Research UK Cambridge Institute Genomics Core. This study was supported by the NIHR Cambridge Biomedical Research Centre. These funding sources had no role in the design, conduct, or analysis of the study or in the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
All authors reviewed and contributed toward the drafting of the manuscript. J.R.B.P., G.S.H.Y., K.K.O. and S.O.R. designed the study. J.R.B.P., K.K.O., Y.Z., K.A.K., R.Y.J., E.J.G., F.R.D., L.R.K. and N.J.W. contributed toward the bioinformatics, genetic analyses and genotype–phenotype association testing of the UK Biobank data. Q.W., J.B.-C., P.K.-M., R.T.-C., J.A.-D., J.E., J.M.T., R.C., K.R.S., D.S., D.S.P., Z.F.-H. and S.P. contributed to statistical analyses and/or genotype/phenotype preparation of replication cohorts. K.L., I.B. and I.S.F. conducted the bioinformatic and genetic analyses on SCOOP and INTERVAL. M.C., A.M.S., G.K.C.D., Y.-C.L.T., B.Y.H.L., H.-J.C.C., F.M. and G.S.H.Y. designed and conducted the cellular work and single-cell analyses.
Corresponding author
Ethics declarations
Competing interests
Z.F.-H., Q.W., K.R.S., D.S.P. and S.P. are employees and/or stockholders of AstraZeneca. J.R.B.P. and E.J.G. are employees and shareholders of Insmed. J.R.B.P. receives research funding from GSK. Y.Z. is a UK University worker at GSK. I.S.F. has consulted for a number of companies developing weight loss drugs, including Eli Lilly, Novo Nordisk and Rhythm Pharmaceuticals. G.S.H.Y. receives grant funding from Novo Nordisk and consults for both Novo Nordisk and Eli Lilly. S.O.R. has undertaken remunerated consultancy work for Pfizer, Third Rock Ventures, AstraZeneca, NorthSea Therapeutics and Courage Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Timothy Frayling and Adam Locke for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information
Supplementary Notes and Figs. 1−8.
Supplementary Tables
Supplementary Tables 1−16.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Chukanova, M., Kentistou, K.A. et al. Protein-truncating variants in BSN are associated with severe adult-onset obesity, type 2 diabetes and fatty liver disease. Nat Genet 56, 579–584 (2024). https://doi.org/10.1038/s41588-024-01694-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-01694-x
This article is cited by
-
New genes associated with adult-onset obesity
Nature Reviews Endocrinology (2024)
