
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with a lifetime risk of one in 350 people and an unmet need for disease-modifying therapies. We conducted a cross-ancestry genome-wide association study (GWAS) including 29,612 patients with ALS and 122,656 controls, which identified 15 risk loci. When combined with 8,953 individuals with whole-genome sequencing (6,538 patients, 2,415 controls) and a large cortex-derived expression quantitative trait locus (eQTL) dataset (MetaBrain), analyses revealed locus-specific genetic architectures in which we prioritized genes either through rare variants, short tandem repeats or regulatory effects. ALS-associated risk loci were shared with multiple traits within the neurodegenerative spectrum but with distinct enrichment patterns across brain regions and cell types. Of the environmental and lifestyle risk factors obtained from the literature, Mendelian randomization analyses indicated a causal role for high cholesterol levels. The combination of all ALS-associated signals reveals a role for perturbations in vesicle-mediated transport and autophagy and provides evidence for cell-autonomous disease initiation in glutamatergic neurons.
Similar content being viewed by others

Main
ALS is a fatal neurodegenerative disease affecting one in 350 individuals. Due to degeneration of both upper and lower motor neurons, patients suffer from progressive paralysis, ultimately leading to respiratory failure within 3–5 years after disease onset1. In ~10% of patients with ALS, there is a clear family history for ALS, suggesting a strong genetic predisposition, and currently a pathogenic mutation can be found in more than half of these cases2. On the other hand, apparently sporadic ALS is considered a complex trait for which heritability is estimated at 40–50% (refs. 3,4). There is no widely accepted definition of familial or sporadic ALS5, and they are likely to represent the ends of a spectrum with overlapping genetic architectures for which the same genes have been implicated in both familial and sporadic disease6,7,8,9,10,11. To date, partially overlapping GWASs have identified up to six genome-wide significant loci, explaining a small proportion of the genetic susceptibility to ALS11,12,13,14,15,16. Indeed, some of these loci found in GWASs harbor rare variants with large effects also present in familial cases (for example, C9orf72 and TBK1)6,17,18. For other loci, the role of rare variants remains unknown.
While ALS is referred to as a motor neuron disease, cognitive and behavioral changes are observed in up to 50% of patients, sometimes leading to frontotemporal dementia (FTD). The overlap with FTD is clearly illustrated by the pathogenic hexanucleotide repeat expansion in C9orf72, which causes familial ALS and/or FTD17,18 and the genome-wide genetic correlation between ALS and FTD19. Further expanding the ALS–FTD spectrum, a genetic correlation with progressive supranuclear palsy (PSP) has been described20. Shared pathogenic mechanisms between ALS and other neurodegenerative diseases, including common diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), can further reveal ALS pathophysiology and inform new therapeutic strategies.
Here, we combine new and existing individual-level genotype data in the largest GWAS of ALS to date. We present a comprehensive screen for pathogenic rare variants and short tandem repeat (STR) expansions as well as regulatory effects observed in brain cortex-derived RNA sequencing (RNA-seq) and methylation datasets to prioritize causal genes within ALS-risk loci. Furthermore, we reveal similarities and differences between ALS and other neurodegenerative diseases as well as the biological processes in disease-relevant tissues and cell types that affect ALS risk.
Results
Cross-ancestry meta-analysis reveals 15 risk loci for ALS
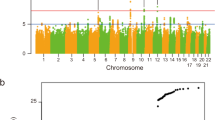
To generate the largest GWAS of ALS to date, we merged individual-level genotype data from 117 cohorts into six strata matched by genotyping platform. A total of 27,205 patients with ALS and 110,881 control participants of European ancestries passed quality control (including 6,374 newly genotyped cases and 22,526 control participants; Methods and Supplementary Tables 1 and 2). Patients were not selected for a family history of ALS. Through meta-analysis of these six strata, we obtained association statistics for 10,461,755 variants down to a minor allele frequency (MAF) of 0.1% in the Haplotype Reference Consortium resource21. We observed moderate inflation of the test statistics (λGC = 1.12, λ1000 = 1.003), and linkage disequilibrium (LD) score regression yielded an intercept of 1.029 (s.e. = 0.0073), indicating that the majority of inflation was due to the polygenic signal in ALS (LD score regression (LDSC): \(h_{\textrm{l}}^2\) = 0.028, s.e. = 0.003, K = 350−1, P = 5.5 × 10−21). The European ancestry analysis identified 12 loci reaching genome-wide significance (P < 5.0 × 10−8; Extended Data Fig. 1). For nine loci, the top SNP or a strong LD proxy (r2 = 0.996) was present in GWAS of ALS in Asian ancestries (2,407 patients with ALS and 11,775 control participants)15,16, and all showed a consistent direction of effects (Pbinom = 2.0 × 10−3). The three SNPs that were not present in the Asian ancestry GWAS were low-frequency variants (MAF of 0.6–1.6% in European ancestries, Table 1). The genetic overlap between ALS risk in European and Asian ancestries resulted in a trans-ancestry genetic correlation of 0.57 (s.e. = 0.28) for genetic effect and 0.58 (s.e. = 0.30) for genetic impact, which were not statistically significantly different from unity (P = 0.13 and P = 0.16, respectively). We therefore performed a cross-ancestry meta-analysis totaling 29,612 cases and 122,656 controls, which revealed three additional loci, totaling 15 genome-wide significant risk loci for ALS risk (Fig. 1, Table 1 and Supplementary Tables 4–18). Conditional and joint analysis did not identify secondary signals within these loci.

Genome-wide association statistics obtained by IVW meta-analysis of the stratified SAIGE logistic mixed model regression. The y axis corresponds to two-tailed −log10 (Pvalues); the x axis corresponds to genomic coordinates (GRCh37). The horizontal dashed line reflects the threshold for calling genome-wide significant SNPs (P = 5 × 10−8). Color coding and gene labels reflect those prioritized by the gene-prioritization analysis. Labels in bold indicate genes with known highly pathogenic mutations for ALS. SAIGE = Scalable and Accurate Implementation of Generalized mixed model software package.
Of these findings, eight loci have been reported in previous GWASs (C9orf72, UNC13A, SCFD1, MOBP–RPSA, KIF5A, CFAP410, GPX3–TNIP1 and TBK1)11,14,15. The rs80265967 variant corresponds to the p.D90A mutation in SOD1 previously identified in a Finnish ALS cohort enriched for familial ALS13. Interestingly, we observed a genome-wide significant common variant association signal within the NEK1 locus, which was previously shown to harbor rare variants associated with ALS8. The recently reported association at the ACSL5–ZDHHC6 locus16,22 did not reach the threshold for genome-wide significance (rs58854276, PEUR = 5.4 × 10−5, PASN = 4.9 × 10−7, Pcomb = 6.5 × 10−8; Supplementary Table 19), despite the fact that our analysis includes all data from the original discovery studies.
Rare variant gene-based association analyses in ALS
To assess a general pattern of underlying architectures that link associated SNPs to causal genes, we first tested for annotation-specific enrichment using stratified LDSC. This revealed that 5′ UTR regions as well as coding regions in the genome and those annotated as conserved were most enriched for ALS-associated SNPs (Extended Data Fig. 2). Subsequently, we investigated how rare, coding variants contributed to ALS risk by generating a whole-genome sequencing (WGS) dataset of patients with ALS (n = 6,538) and control participants (n = 2,415), which is a subset of the common variant GWAS cohort. The exome-wide association analysis included transcript-level rare variant burden testing for different models of allele-frequency thresholds and variant annotations (Methods). This identified NEK1 as the strongest associated gene (minimal P = 4.9 × 10−8 for disruptive and damaging variants at MAF < 0.005), which was the only gene to pass the exome-wide significance thresholds (0.05 ÷ 17,994 = 2.8 × 10−6 and 0.05 ÷ 58,058 = 8.6 × 10−7 for number of genes and protein-coding transcripts, respectively; Supplementary Table 20). This association was independent from the previously reported increased rare variant burden in selected patients with ‘familial ALS’ (ref. 8) who were not included in this study. Polygenic risk score (PRS) analyses did not illustrate a difference in PRSs in patients carrying rare variants in ALS-risk genes (SOD1, C9orf72 repeat expansion, TARDBP, FUS, NEK1, TBK1 and CFAP410) compared to all patients with ALS (Extended Data Fig. 3). Although power was limited, this is compatible with a scenario in which the genetic risk of ALS in these patients is a sum of rare variants in ALS genes and other (common) genetic variation.
Gene prioritization shows locus-specific underlying architectures
To assess whether rare variant associations could drive the common variant signals at the 15 genome-wide significant loci, we combined the common and rare variant analyses to prioritize genes within these loci. The SNP effects on gene expression were assessed by summary-based Mendelian randomization (MR) (SMR) in blood (eQTLGen23, n = 31,648) and a new brain cortex-derived eQTL dataset (MetaBrain24, n = 2,970). Finally, we analyzed methylation quantitative trait loci (mQTL) by SMR in blood-derived (n = 2,082) and brain-derived (n = 522) mQTL datasets25,26,27. Through these multi-layered gene-prioritization strategies, we classified each locus into one of four classes of most likely underlying genetic architecture to prioritize the causal gene (Supplementary Figs. 1–15).
First, in three GWAS loci, the strongest associated SNP was a low-frequency coding variant that was nominated as the causal variant. This was the case for rs80265967 (SOD1, p.D90A; Supplementary Fig. 14) and rs113247976 (KIF5A, p.P986L; Supplementary Fig. 8), which are coding variants in known ALS-risk genes. This was also the most likely causal mechanism for rs75087725 (CFAP410, formerly C21orf2, p.V58L; Supplementary Fig. 15), as the GWAS variant is a missense variant; no evidence for other mechanisms including repeat expansions or eQTL or mQTL effects was observed within this locus, and CFAP410 itself is known to directly interact with NEK1, another ALS gene6,28. These three loci illustrate the power of large-scale GWASs combined with large imputation panels to directly identify low-frequency causal variants that confer disease risk.
Second, SNPs can tag a highly pathogenic repeat expansion, as was observed for rs2453555 (C9orf72) and the known GGGGCC hexanucleotide repeat in this locus (Supplementary Fig. 7). Conditional analysis revealed no residual signal after conditioning on the repeat expansion, which was in LD with the top SNP (r2 = 0.14, |D′| = 0.99, MAFSNP = 0.25, MAFSTR = 0.047). Besides the repeat expansion, both eQTL and mQTL analyses point to C9orf72 (Supplementary Fig. 7). The HEIDI (heterogeneity in dependent instruments) outlier test, however, rejected the null hypothesis that gene expression or methylation mediated the causal effect of the associated SNP (PHEIDI,eQTL = 3.7 × 10−23 and PHEIDI,mQTL = 4.1 × 10−7). This is in line with the idea that pathogenic repeat expansion is the causal variant in this locus and that eQTL and mQTL effects do not mediate a causal effect. We found no similar pathogenic repeat expansions that fully explained the SNP association signal in the other genome-wide significant loci.
Third, in two loci (rs62333164 in NEK1 and rs4075094 in TBK1), common and rare variants converged to the same gene, which are known ALS-risk genes6,8. For both loci, the rare variant burden association was conditionally independent from the top SNP that was included in the GWAS (Supplementary Figs. 2 and 9). Here, eQTL and mQTL analyses indicated that the risk-increasing effects of the common variants were mediated through both eQTL and mQTL effects on NEK1 and TBK1. Furthermore, a polymorphic STR downstream of NEK1 was associated with increased ALS risk (motif, TTTA; threshold = 10 repeat units, expanded allele frequency = 0.51, P = 5.2 × 10−5, false discovery rate (FDR) = 4.7 × 10−4; Extended Data Fig. 4). This polymorphic repeat was in LD with the top associated SNP within this locus (r2 = 0.24, |D′| = 0.70). There was no statistically significant association for the top SNP in the WGS data to reliably determine its independent contribution to ALS risk.
Lastly, the fourth group contains seven remaining loci for which there was no direct link to a causal gene through coding variants or repeat expansions. Here, we investigated regulatory effects of the associated SNPs on target genes acting as either eQTL or mQTL. Single genes were prioritized by SMR using both mQTL and eQTL for rs2985994 (COG3; Supplementary Fig. 10), rs229243 (SCFD1; Supplementary Fig. 11) and rs517339 (ERGIC1; Supplementary Fig. 4). In other loci, both methods prioritized multiple genes, such as rs631312 (MOBP and RPSA; Supplementary Fig. 1) and rs10463311 (GPX3 and TNIP1; Supplementary Fig. 3). Aside from the prioritized genes, each of these loci harbored multiple genes that were not prioritized by any method and are therefore less likely to contribute to ALS risk.
For two loci, no gene was prioritized with these approaches. Within the UNC13A locus (rs12608932; Supplementary Fig. 12), recent studies illustrate that the genome-wide significant SNPs act as splicing quantitative trait loci conditional on dysfunction of TAR DNA-binding protein (TDP)-43, resulting in inclusion of a cryptic exon in UNC13A29,30. Furthermore, we could not prioritize a specific gene in the HLA locus (rs9275477; Supplementary Fig. 5).
Genetic modifiers of ALS disease progression
We investigated whether genetic risk factors for ALS also act as disease modifiers that affect disease onset and progression. Genotypes for the 15 genome-wide significant SNPs, PRSs and the rare variant burden for SOD1, C9orf72 (repeat expansion status), TARDBP, FUS, NEK1, TBK1 and CFAP410 were obtained for all individuals with WGS for whom the complete core clinical data (sex, age at onset, site of onset, survival, time to censoring) were available (n = 6,095). Association analyses with survival and age at onset showed that common variants had a limited effect on survival (Fig. 2a) and age at onset (Fig. 2b) but confirmed the association between faster disease progression for the UNC13A risk allele (rs12608932, hazard ratio (HR) = 1.10, 95% confidence interval (CI) = 1.05–1.15, P = 1.2 × 10−4) and slower disease progression in patients with the SOD1 p.D90A mutation (rs80265967, HR = 0.35, 95% CI = 0.16–0.77, P = 8.4 × 10−4). This limited effect of common genetic risk factors for ALS susceptibility on disease progression was reflected in the PRS analyses in which we found no effect of the full-genome PRS on survival (HR = 1.02, 95% CI = 0.98–1.06, P = 0.28) or age at onset (b = 0.10, s.e. = 0.21, P = 0.64). Analyses of rare variants confirmed faster disease progression in patients with the C9orf72 repeat expansion (HR = 1.45, 95% CI = 1.28–1.65, P = 1.2 × 10−8) with an earlier age at onset (b = −2.62, s.e. = 0.77, P = 6.4 × 10−4).

a, Cox proportional HRs for genome-wide significant SNPs (brown, n = 15), PRSs (red, n = 2) and rare variant burden in ALS-risk genes (pink, n = 7) on survival (months) tested in 6,095 patients with ALS. Estimated HRs are displayed with error bars corresponding to 95% CIs. Higher HRs correspond to shorter survival times. b, Effect estimates from a linear regression model of age at onset (years) in 6,095 patients with ALS. Lower effect estimates correspond to a younger age at onset. Effect estimates from linear regression are displayed with error bars corresponding to 95% CIs. The risk-increasing allele for ALS corresponds to the effect allele for both survival and age-at-onset analyses.
Locus-specific sharing of risk loci between ALS and neurodegenerative diseases
To investigate the pleiotropic properties of ALS-associated variants and shared genetic risk with other brain diseases, we estimated genetic correlations between neurodegenerative diseases, psychiatric traits, cerebrovascular diseases and multiple sclerosis (Extended Data Fig. 5). This showed strong genetic correlations among neurodegenerative diseases. Bivariate LDSC confirmed a statistically significant genetic correlation between ALS and PSP (rg = 0.44, s.e. = 0.11, P = 1.0 × 10−4) as previously reported20 and also revealed a significant genetic correlation between ALS and AD (rg = 0.31, s.e. = 0.12, P = 9.6 × 10−3) as well as between ALS and PD (rg = 0.16, s.e. = 0.061, P = 0.011; Fig. 3a). The point estimate for the genetic correlation between ALS and FTD was high (rg = 0.59, s.e. = 0.41, P = 0.15) but not statistically significant due to the limited size of the FTD GWAS (3,526 cases and 9,402 controls). Thus, power to detect a genetic correlation between ALS and FTD using LDSC was limited.

a, Genetic correlation analysis. Genetic correlation was estimated with LDSC between each pair of neurodegenerative diseases (ALS, AD, CBD, PD, PSP and FTD). Correlations marked with an asterisk reached nominal statistical significance (PALS,AD = 0.01, PALS,PD = 0.01, PALS,PSP = 0.0001, PPSP,PD = 0.002). b, SNP associations of ALS lead SNPs or LD proxies in neurodegenerative diseases. The association with ALS is shown at the top. Effective sample size is shown on the left. Posterior probabilities of the same causal SNP affecting two diseases were estimated through colocalization analysis and are highlighted as connections.
Patterns of sharing disease-associated genetic variants appeared to be locus specific (Fig. 3b and Supplementary Table 21). To assess whether two traits shared a common signal, indicating shared causal variants, we performed colocalization analyses for all loci meeting P < 5 × 10−5 in any of the GWASs of neurodegenerative diseases (n = 161 loci). This revealed a shared signal in the MOBP–RPSA locus between ALS, PSP and corticobasal degeneration (CBD) as well as a shared signal in the UNC13A locus between ALS and FTD (posterior probability, PPH4 > 95%; Extended Data Fig. 6). For the HLA locus, there was evidence for a shared causal variant between ALS and PD (PPH4 = 88%) but no conclusive evidence for ALS and AD (PPH4 = 51% for a shared causal variant and PPH3 = 49% for independent signals in both traits).
Furthermore, colocalization analyses identified two additional shared loci that were not genome-wide significant in the ALS GWAS: between ALS and PD at the GAK locus (rs34311866, PPH4 = 99%) and between ALS and AD at the TSPOAP1-AS1locus (rs2632516, PPH4 = 90%). Of note, the association at TSPOAP1-AS1 was not genome-wide significant in the GWAS of clinically diagnosed AD (P = 3.7 × 10−7) either but was identified in the larger AD-by-proxy GWAS31. For FTD subtypes, C9orf72 showed a colocalization signal for a shared causal variant between ALS and the motor neuron disease subtype of FTD (mndFTD, PPH4 = 93%; Extended Data Figs. 6 and 7).
Enrichment of glutamatergic neurons indicates cell-autonomous processes in ALS susceptibility
To find tissues and cell types for which gene expression profiles were enriched for genes within ALS-risk loci, we first combined gene-based association statistics calculated using MAGMA32 with gene expression patterns from the Genotype–Tissue Expression (GTEx) project (version 8) in a gene set enrichment analysis using FUMA33. We observed a significant enrichment in genes expressed in brain tissues across multiple brain regions but not in peripheral nervous tissue or muscle. Whereas this pattern roughly resembled the enrichments observed in PD and psychiatric traits, it was strikingly different from that reported31 and observed in AD in which blood, lung and spleen were mostly enriched, resembling the pattern observed in multiple sclerosis, which is a typical immune-mediated brain disease (Fig. 4a and full results in Supplementary Fig. 16 and Extended Data Fig. 8a). We subsequently queried single-cell RNA-seq datasets of human-derived brain samples to further specify brain-specific enriched cell types using the cell type analysis module in FUMA34. This showed significant enrichment for neurons but not for microglia or astrocytes (Fig. 4b). Further subtyping of these neurons illustrated that genes expressed in glutamatergic neurons were mostly enriched for genes within the ALS-associated risk loci. Again, this contrasted with AD, which showed specific enrichment of microglia, similar to multiple sclerosis (Extended Data Fig. 8b). In single-cell RNA-seq data obtained from brain tissues in mice, a similar pattern was observed showing neuron-specific enrichment in ALS and PD but microglia in AD (Extended Data Fig. 9). Together, this indicates that susceptibility to neurodegeneration in ALS is mainly driven by neuron-specific pathology and not by immune-related tissues and microglia.

a, Enrichment of tissues and brain regions included in GTEx version 8 illustrates a brain-specific enrichment pattern in ALS, similar to that in PD but contrasting with that in AD. Tissues and brain regions displayed are those significantly enriched in ALS or PD, tissues previously reported in AD and tissues of specific interest for ALS (spinal cord, tibial nerve and muscle). Color represents the enrichment coefficient, and size indicates two-sided −log10 (P-values) of enrichment obtained by the linear regression model in the MAGMA gene property analysis. b, Cell type enrichment analyses indicate neuron-specific enrichment for glutamatergic neurons. In ALS, no enrichment was found for microglia or other non-neuronal cell types, contrasting with the pattern observed in AD. Color represents the enrichment coefficient, and size indicates two-sided −log10 (P-values) of enrichment obtained by the linear regression model in the MAGMA gene property analysis. Statistically significant enrichments after correction for multiple testing over all tissues (n = 54), cell types (n = 7) and neurons (n = 3) with FDR < 0.05 are marked with an asterisk. Cx, cortex; GABA, γ-aminobutyric acid; OPCs, oligodendrocyte progenitor cells.
Brain-specific coexpression networks improve detection of ALS-relevant pathways
To determine which processes were mostly enriched in ALS, we performed enrichment analyses that combined gene-based association statistics with gene coexpression patterns obtained from either multi-tissue transcriptome datasets35 or RNA-seq data from brain cortex samples (MetaBrain24). To validate this approach, we first tested for enrichment of human phenotype ontology (HPO) terms that are linked to well-established disease genes in the Online Mendelian Inheritance in Man (OMIM) and Orphanet catalogs. Using the multi-tissue coexpression matrix, we found no enriched HPO terms after Bonferroni correction for multiple testing. Using the brain-specific coexpression matrix, however, we found a strong enrichment of HPO terms that are related to ALS or neurodegenerative diseases in general, including ‘cerebral cortical atrophy’ (P = 1.8 × 10−8), ‘abnormal nervous system electrophysiology’ (P = 4.1 × 10−7) and ‘distal amyotrophy’ (P = 8.6 × 10−7; full list in Supplementary Table 22). In general, HPO terms in the neurological branch (‘abnormality of the nervous system’) showed an increase in enrichment statistics in ALS when using the brain-specific coexpression matrix compared to the multi-tissue dataset (Extended Data Fig. 10), which illustrates the benefit of the brain-specific coexpression matrix. Subsequently, we tested for enriched biological processes using reactome and gene ontology terms. Again, using the multi-tissue expression profiles, we found that no reactome annotations were enriched. Leveraging the brain-specific coexpression networks, we identified vesicle-mediated transport (‘membrane trafficking’, P = 4.2 × 10−6, ‘intra-Golgi and retrograde Golgi-to-endoplasmic reticulum (ER) trafficking’, P = 1.4 × 10−5) and autophagy (‘macroautophagy’, P = 3.2 × 10−5) as enriched processes after Bonferroni correction for multiple testing (Supplementary Table 23). The subsequently identified enriched gene ontology terms were all related to vesicle-mediated transport or autophagy (Supplementary Tables 24 and 25).
MR analyses are in line with a causal relationship between cholesterol levels and ALS
From previous observational case–control studies and our blood-based methylome-wide study36, numerous non-genetic risk factors have been implicated in ALS. Here, we studied a selection of those putative risk factors through causal inference in an MR framework37. We selected 22 risk factors for which robust genetic predictors were available including body mass index, smoking, alcohol consumption, physical activity, cholesterol-related traits, cardiovascular diseases and inflammatory markers (Supplementary Table 26). These analyses provided the strongest evidence that cholesterol levels were causally related to ALS risk (bweighted median = 0.15, s.e. = 0.04, P = 3.2 × 10−4; Fig. 5a and full results in Supplementary Table 27). These results were robust to removal of outliers through radial MR analysis38, and we observed no evidence for reverse causality (Supplementary Tables 28 and 29). Importantly, ascertainment bias can lead to the selection of more highly educated control participants39 compared to patients with ALS who are mostly ascertained through the clinic. In line with control participants having higher education, MR analyses indicated a negative effect for years of schooling on ALS risk (inverse-variance-weighted PIVW = 2.0 × 10−4; Fig. 5b). As a result, years of schooling can act as a confounder for the observed risk-increasing effect of higher total cholesterol levels through ascertainment bias. To correct for this potential confounding, we applied multivariate MR analyses including both years of schooling and total cholesterol levels. The results for total cholesterol were robust in the multivariate analyses, suggesting a causal role for total cholesterol levels on ALS susceptibility (Supplementary Table 30).

a, MR results for ALS and total cholesterol levels. Results for the five different MR methods for two different P-value cutoffs for SNP instrument selection are presented. In total, 83 and 178 SNPs were used as instruments at cutoffs of P < 5 × 10−8 and P < 5 × 10−5, respectively. All methods show a consistent positive effect for an increased risk of ALS with higher total cholesterol levels. There is no evidence for reverse causality. Point estimates for MR are presented with error bars reflecting 95% CIs. b, MR results for ALS and years of schooling. In total, 306 and 681 SNPs were used as instruments at cutoffs of P < 5 × 10−8 and P < 5 × 10−5. Point estimates for MR are presented, with error bars reflecting 95% CIs. Statistically significant effects with a two-sided P-value passing Bonferroni correction for multiple testing over all tested traits (n = 22), instrument P-value cutoffs (n = 2) and MR methods (n = 5) are marked with an asterisk (total cholesterol, Pweighted median = 0.0003 and Pweighted median = 0.0007 for cutoffs at P < 5 × 10−8 and P < 5 × 10−5, respectively; years of schooling, PIVW = 0.0002 at the cutoff of P < 5 × 10−5). Here, SNP outliers were not removed for instrument selection. Z, genetic instrument; bxy, estimated causal effect for an increase of 1 s.d. in genetically predicted exposure.
Discussion
In summary, in the largest GWAS on ALS to date including 29,612 patients with ALS and 122,656 control participants, we identified 15 risk loci contributing to ALS risk. Through in-depth analysis of these loci incorporating rare variant burden analyses and repeat expansion screens in WGS data and blood- and brain-specific eQTL and mQTL analyses, we prioritized genes in 13 of the loci. Across the spectrum of neurodegenerative diseases, we identified a genetic correlation between ALS and AD as well as PD and PSP with locus-specific patterns of shared genetic risk across all neurodegenerative diseases. Colocalization analysis identified two additional loci, GAK and TSPOAP1-AS1, with a high posterior probability of shared causal variants between ALS and PD and between ALS and AD, respectively. We found glutamatergic neurons as the most enriched cell type in the brain, and brain-specific coexpression network enrichment analyses indicated a role for vesicle-mediated transport and autophagy in ALS. Finally, causal inference of previously described risk factors provides evidence for high total cholesterol levels as a causal risk factor for ALS.
The cross-ancestry comparison illustrated similarities in the genetic risk factors for ALS in European and East Asian ancestries, providing an argument for cross-ancestry studies and to further expand ALS GWASs in non-European populations. It is important to note that three loci including those that harbor low-frequency variants (KIF5A, SOD1 and CFAP410) were not included in the East Asian GWAS due to their low MAFs. Therefore, the shared genetic risk might not extend to rare genetic variation, for which population-specific frequencies have been observed even within Europe.
The multi-layered gene-prioritization analyses highlighted four different classes of genome-wide significant loci in ALS. First, the sample size of this GWAS combined with accurate imputation of low-frequency variants directly identified rare coding variants that increase ALS risk. These include the known p.D90A mutation in SOD1 (MAF = 0.006) as well as rare variants in KIF5A (MAF = 0.016) and CFAP410 (MAF = 0.012) for which, after their identification through GWAS, experimental work confirmed their direct role in ALS pathophysiology11,28,40. Second, we confirmed that the pathogenic C9orf72 repeat expansion is tagged by genome-wide significant GWAS SNPs and that no residual signal is left by conditioning the SNP on the repeat expansion. Although more repeat expansions are known to affect ALS risk, we found no similar loci for which the SNPs tag a highly pathogenic repeat expansion. This suggests that highly pathogenic repeat expansions on a stable haplotype are merely the exception rather than the rule in ALS. Third, common and rare variant association signals can converge on the same gene as observed for NEK1 and TBK1, consistent with observations for other traits and diseases41,42,43. We show that these signals are conditionally independent and that the common variants act on the same gene through regulatory effects as eQTL or mQTL. Fourth, we find evidence for regulatory effects of ALS-associated SNPs that act as eQTL or mQTL. These locus-specific architectures illustrate the complexity of ALS-associated GWAS loci for which not one solution fits all, but instead a multi-layered approach to prioritize genes is warranted.
In addition, we find locus-specific patterns of shared effects across neurodegenerative diseases. The MOBP locus has previously been identified in PSP and ALS, and here we show that indeed both diseases as well as CBD are likely to share the same causal variant in this locus. The same is true for UNC13A and C9orf72 with FTD and mndFTD, respectively. The colocalization analysis with PD identified a shared causal variant in the GAK locus, which was not found in the ALS GWAS alone. Furthermore, the TSPOAP1-AS1 locus harbors SNPs associated with ALS and AD risk. Although this locus was not significant in either of the GWASs, a larger GWAS including AD-by-proxy cases confirmed this as a risk locus for AD. This illustrates the power of cross-disorder analyses to leverage the shared genetic risk of neurodegenerative diseases.
We aimed to clarify the role of neuron-specific pathology in ALS susceptibility as opposed to non-cell-autonomous pathology through detailed cell type enrichment analyses. Previous experiments have illustrated multiple lines of evidence for non-cell-autonomous pathology in microglia, astrocytes and oligodendrocytes, which ultimately leads to neurodegeneration in ALS44,45,46. These experiments have shown that non-cell-autonomous processes, such as neuroinflammation, mainly act as modifiers of disease in SOD1 models of ALS45,46. Here, we show that genes within loci associated with ALS susceptibility are specifically expressed in (glutamatergic) neurons. This provides evidence for neuron-specific pathology as a driver of ALS susceptibility, which is in stark contrast to the signal of inflammation-associated tissues and cell types in AD and multiple sclerosis. It also shows that disease susceptibility and disease modification can be distinct processes, which is supported by our finding that most genetic susceptibility factors do not have a strong effect on survival. This motivates future large-scale genetic studies on modifiers of ALS progression, as these can be targets for potential new treatments for ALS as well.
The subsequent functional enrichment analyses identified that membrane trafficking, Golgi-to-ER trafficking and autophagy were enriched for genes within ALS-associated loci. These terms and their related gene ontology terms of biological processes are all related to autophagy and degradation of (misfolded) proteins. This corroborates the central hypothesis of impaired protein degradation leading to aberrant protein aggregation in neurons, which is the pathological hallmark of ALS. Our results suggest that this is a central mechanism in ALS even in the absence of rare known mutations in genes directly involved in these biological processes such as TARDBP, FUS, UBQLN2 and OPTN47.
Based on observational studies and MR analyses, conflicting evidence exists for lipid levels including cholesterol as a risk factor for ALS48,49,50. Potential selection bias, reverse causality and the subtype of cholesterol studied challenge the interpretation of these results. Here, we provided support for a causal relationship between high total cholesterol levels and ALS independent of educational attainment and ruling out reverse orientation of the MR effect. The total cholesterol effects were consistent across the different MR methods tested, indicating that this finding is robust to violation of the ‘no horizontal pleiotropy’ assumption. This is in line with our study showing methylation changes associated with increased cholesterol levels in ALS36. We do not find a clear pattern for either low-density lipoprotein (LDL) or high-density lipoprotein (HDL) cholesterol subtypes in relation to ALS risk. While cholesterol levels are closely related to cardiovascular risk, the association between cardiovascular risk and ALS risk remains controversial with conflicting reports3,48,51. Interestingly, recent work has shown that lipid metabolism and autophagy are closely related52, which brings the results of our pathway analyses and MR together. Both in vitro and in vivo experiments have shown that autophagy regulates lipid homeostasis through lipolysis and that impaired autophagy increases triglyceride and cholesterol levels. Conversely, high lipid levels were shown to impair autophagy52. Further studies on the effect of high cholesterol levels and protein degradation through autophagy illustrate that high cholesterol levels decrease the fusogenic ability of autophagic vesicles through decreased function of soluble N-ethylmaleimide-sensitive factor-attachment protein receptor (SNARE)53,54 and lead to increased protein aggregation due to impaired autophagy in mouse models of AD55. Therefore, the risk-increasing effect of cholesterol on ALS might be mediated through impaired autophagy.
In conclusion, our GWAS identifies 15 risk loci in ALS and illustrates locus-specific interplay between common and rare genetic variation that helps to prioritize genes for future follow-up studies. We show a causal role for cholesterol, which can be linked to impaired autophagy as common denominators of neuron-specific pathology that drive ALS susceptibility and serve as potential targets for therapeutic strategies.
Methods
Genome-wide association study
Data description
We obtained individual genotype-level data for all individuals in the previously published GWAS of ALS in European ancestries11,14 and publicly available control datasets including 120,971 controls genotyped on Illumina platforms. Additionally, 6,374 cases and 22,526 controls were genotyped on the Illumina OmniExpress and Illumina GSA arrays. Details for each cohort are provided in Supplementary Table 1. All patients with ALS were diagnosed and ascertained through specialized MND clinics where they were diagnosed with ALS according to the (revised) El Escorial Criteria56 by neurologists specialized in motor neuron diseases. Whole-blood samples were drawn for DNA isolation, which were specifically collected for ongoing case–control studies of ALS. Both cases with and without a family history for ALS and/or dementia were included. Cases were not pre-screened for specific ALS-related mutations. Given the late onset and relatively low lifetime risk of ALS, controls were not screened for (subclinical) signs of ALS. A detailed description of the ascertainment of newly genotyped cases and controls is provided in the Supplementary Note. All participants gave written informed consent, and the relevant local institutional review boards approved this study (Supplementary Note). Cases and controls formed cohorts when they were processed in the same laboratory and were genotyped in the same batch, resulting in 117 independent cohorts. Summary statistics were obtained for the Asian ancestry GWAS of ALS15,16 (Supplementary Note).
GWAS quality control and imputation
For each cohort, we first performed individual- and variant-level quality control, after which cohorts were merged into six strata based on genotyping platform. Subsequent stratum-wise quality control was performed, and strata were imputed up to the Haplotype Reference Consortium panel (r.1.1 2016) through the Michigan Imputation Server21. Full quality-control details are described in the Supplementary Note and Supplementary Fig. 17. Numbers of individuals and variants passing each quality-control step are described in Supplementary Table 2.
Association testing and meta-analysis
After quality control, a null logistic mixed model was fitted using SAIGE57 0.29.1 for each stratum with principal component (PC)1–PC20 as covariates. The model was fit on a set of high-quality (INFO > 0.95) SNPs pruned with PLINK 1.9 (‘–indep-pairwise 50 25 0.1’) in a leave-one-chromosome-out scheme. Subsequently, a SNP-wise logistic mixed model including the saddlepoint approximation test was performed using genotype dosages with SAIGE. Association statistics for all strata were combined in an IVW fixed-effects meta-analysis using METAL58.
Genomic inflation factors were calculated per stratum and for the full meta-analysis. To assess any residual confounding due to population stratification and artificial structure in the data, we calculated the LDSC59 intercept using SNP LD scores calculated in the HapMap3 CEU population.
Cross-ancestry analyses
GWAS summary statistics from two Asian ancestry studies were obtained15,16. These summary statistics were meta-analyzed with all European ancestry data in strata as described above. To assess genetic correlation for ALS in European and Asian ancestries, we used Popcorn60 version 0.9.9. We used population-specific LD scores for genetic impact and genetic effect provided with the Popcorn software. The regression model (‘–use_regression’) was used to estimate genetic correlation. We calculated both the correlation of genetic effects (correlation of allelic effect sizes) and genetic impact (correlation of allelic effect size adjusted for difference in allele frequencies).
Conditional SNP analysis
Conditional and joint SNP analysis (COJO, GCTA version 1.91.1b)61,62 was performed to identify potential secondary GWAS signals within a single locus. SNPs with association P ≤ 5 × 10−8 were considered. Controls of European ancestry from the Health and Retirement Study (HRS, cohort 65, Supplementary Table 1), included in stratum 4 of this study, were used as the LD reference panel.
Gene prioritization
Whole-genome sequencing
Sample selection, sequencing and data preparation
Patients with ALS and control participants from Project MinE63 were recruited for WGS. The participating cohorts were not pre-screened for ALS-associated mutations and are described in the Supplementary Note. In total, 228 patients were known to have at least one first- or second-degree relative with ALS. A full description of Project MinE and the sequencing and quality-control pipeline were described previously64. In summary, the first batch of 2,250 cases and control samples was sequenced on the Illumina HiSeq 2000 platform. All remaining 7,350 case and control samples were sequenced on the Illumina HiSeq X platform. All samples were sequenced to ~35× coverage with 100-bp reads and ~25× coverage with 150-bp reads for HiSeq 2000 and HiSeq X, respectively. Both sequencing sets used PCR-free library preparation. Samples were also genotyped on the Illumina 2.5M array. Sequencing data were then aligned to GRCh37 using the Isaac Aligner, and variants were called using the Isaac variant caller; both the aligner and caller are standard to Illumina’s aligning and calling pipeline. Full details of individual- and variant-level quality control are described in the Supplementary Note.
Genic burden association analyses
To aggregate rare variants in a genic burden test framework, we used a variety of variant filters to allow for different genetic architectures of ALS-associated variants per gene as we and others did previously64,65. In summary, variants were annotated according to allele-frequency threshold (MAF < 0.01 or MAF < 0.005) and predicted variant impact (‘missense’, ‘damaging’, ‘disruptive’). ‘Disruptive’ variants were those variants classified as frameshift, splice site, exon loss, stop gained, start loss and transcription ablation by SnpEff66. ‘Damaging’ variants were missense variants predicted to be damaging by seven prediction algorithms (SIFT67, PolyPhen-2 (ref. 68), LRT69, MutationTaster2 (ref. 70), Mutations Assessor71 and PROVEAN72). ‘Missense’ variants were those missense variants that did not meet the ‘damaging’ criteria. All combinations of allele-frequency threshold and variant annotations were used to test the genic burden on a transcript level in a Firth logistic regression framework in which burden was defined as the number of variants per individual. Sex and the first 20 PCs were included as covariates. All Ensembl protein-coding transcripts for which at least five individuals had a non-zero burden were included in the analysis.
Conditional genic burden analysis
We selected for each gene the protein-coding transcripts that were the most strongly associated with ALS across all different combinations of MAF and variant-impact thresholds. For these transcripts and variants, we applied Firth logistic regression on individuals included in both the GWAS and WGS datasets (5,158 cases and 2,167 controls). To assess whether the rare variant burden association and the signal from the GWAS were conditionally independent, we subsequently included the genotype of the top associated SNP within that locus as a covariate.
Short tandem repeat screen
For all individuals who had sequencing results in the HiSeq X dataset (5,392 cases, 1,795 controls), we screened all loci harboring SNPs associated with ALS meeting genome-wide significance for expansions of known and new STRs using ExpansionHunter73 and ExpansionHunter Denovo74.
First, we used ExpansionHunter (version 4.0) to screen for expansions of known STRs located within 1 Mb of the top ALS-associated SNP. For this, we used the STRs identified from indels in 18 high-quality genomes and the GangSTR STR catalog based on STR annotations in the reference genome75. We excluded all homopolymers from these catalogs. Repeat length was subsequently regressed on case–control status using Firth logistic regression including the first 20 PCs as covariates, recoding the STR size to a biallelic variant using a sliding window over all observed repeat lengths. To correct for multiple testing across all possible thresholds, we applied Benjamini–Hochberg correction per STR.
To screen for extremely long STR expansions (similar to the C9orf72 repeat expansion) at loci that were not included in the predefined STR catalogs, we applied ExpansionHunter Denovo74. This method aims to only find STR expansions that exceed the sequencing read length (>150 bp) by identifying reads (mapped, mismapped and unmapped) that contain STR motifs, using their mate pairs for de novo mapping to the reference genome.
For all STRs, we calculated LD statistics (r2 and |D′|) between recoded repeat genotypes at the optimal threshold and the top associated GWAS SNP. Subsequently, we conditioned the SNP association on the repeat genotype in a Firth logistic regression.
Summary-based Mendelian randomization
We used multi-SNP SMR76,77 to infer the effect of gene expression variation on ALS using eQTL (the association of a SNP with expression of a gene) on ALS risk. We chose to apply SMR because this method yielded very similar results when compared to S-PrediXcan78 and TWAS79 (Supplementary Fig. 18) when applied using GTEx version 7 eQTL, and it can be applied to the large relevant eQTL datasets (MetaBrain and eQTLGen) without access to individual-level genotype and gene expression data. MetaBrain is a harmonized set of 8,727 RNA-seq samples from seven regions of the central nervous system from 15 datasets, and we selected eQTL derived from the cortex region of the brain in samples of European ancestry (MetaBrain Cortex-EUR eQTL, n = 2,970 individuals, n = 6,601 RNA-seq samples) as our instrument variable24. European-only ALS summary statistics were used as the outcome. To supplement this analysis, we also used eQTL in blood from the eQTLGen Consortium, as this is a large available eQTL resource. Samples of European ancestry in the HRS (cohort 65 of this GWAS) were used as the LD reference panel. SNPs with MAF ≥ 1% in the HRS were included. Further SMR settings were left as default, meaning probes with at least one eQTL with P ≤ 5 × 10−8 were included.
We subsequently performed SMR using DNA mQTL data and European-only ALS summary statistics. Human prefrontal cortex and whole-blood DNA mQTL were generated as part of ongoing analyses by the Complex Disease Epigenomics Group at the University of Exeter (https://www.epigenomicslab.com/) using the Illumina EPIC HumanMethylation array that quantifies DNAm at >850,000 sites across the genome25. The prefrontal cortex mQTL dataset was generated using DNA-methylation and SNP data from 522 individuals from the Brains for Dementia Research cohort26 and includes 4,623,966 cis mQTL (distance between quantitative trait locus SNP and DNAm site ≤500 kb) between 1,744,102 SNPs and 43,337 DNA-methylation sites. The whole-blood mQTL dataset was generated using DNAm and SNP data from 2,082 individuals80 and included 30,432,023 cis mQTL between 4,030,902 SNPs and 167,854 DNA-methylation sites. mQTL reaching the significance threshold P ≤ 1 × 10−10 were taken forward for SMR analysis as described by Hannon and colleagues80. To map CpG sites to their putative target genes, we used the expression quantitative trait methylation results from a paired methylation and gene expression (RNA-seq) study in blood81. For CpG sites where no expression quantitative trait methylation was present in this dataset, we used positional mapping based on the basal regulatory domains and extended regulatory domains as defined in the Genomic Regions Enrichment of Annotations Tool (GREAT)82, which is applied in the ‘cpg_to_gene‘ function in the CpGtools toolkit83.
Polygenic risk score calculation
PRSs were constructed based on the 15 lead SNPs of genome-wide significant loci (15-SNP PRS) or a full-genome-wide model (full-genome PRS). For the 15-SNP PRS, the SNP weights were defined as the meta-analyzed effect estimates. We used the summary-BayesR framework from the Genome-wide Complex Trait Bayesian analysis (GCTB) toolkit84,85 to obtain SNP weights for the full-genome PRS based on the European ancestry meta-analysis excluding stratum 6. We used the default model parameters and the precalculated sparse LD matrix of imputed HapMap3 SNPs in 50,000 random individuals included in the UK Biobank of European ancestries. Summary-BayesR SNP effects were plotted against marginal SNP effects to rule out potential biased estimates due to non-convergence of the MCMC algorithm. Finally, the PRSs for all individuals in stratum 6 were calculated using the ‘–score’ function in PLINK and normalized to zero mean and unit variance.
Modifier analyses
For 6,095 of the patients with WGS and ALS, core clinical data were obtained including sex, site of onset (spinal or bulbar), age at onset (years), country of origin and survival, defined as time from disease onset to death, 23 h of continuous non-invasive ventilation per day or tracheostomy. Patients who were still alive were censored at the last date of follow-up.
The genetic risk factors included SNP genotypes, PRSs, C9orf72 repeat expansion status and the number of rare coding mutations in ALS-risk genes (SOD1, TARDBP, FUS, NEK1, TBK1 and CFAP410) as obtained from WGS as described above.
For survival analyses, the Cox proportional hazards mixed model from the ‘coxme‘ package in R was used, modeling country of origin as a random effect. Fixed-effect covariates included sex, age at onset, site of onset, GWAS stratum and PC1–PC5. Violation of the proportional hazards assumption for genotype on survival was assessed by inspecting Schoenfeld residuals. For age-at-onset analyses, we applied linear regression of age at onset on genotype including sex, site of onset, country, GWAS stratum and PC1–PC5 as covariates.
Cross-trait analyses
Datasets and data preparation
GWAS summary statistics for clinically diagnosed AD86, PD87, FTD88, CBD89 and PSP20 in individuals of European ancestry were obtained. For AD, we used the clinical diagnosis as the case definition to avoid spurious genetic correlations that could have been introduced through the by-proxy design31, in which by-proxy cases are defined as having a parent with AD. Although this is a powerful design for gene discovery and the genetic correlation with clinically diagnosed AD is high90, mislabeling by-proxy cases when parents suffer from other types of dementia (for example, Lewy body dementia, Parkinson’s dementia, FTD or vascular dementia) can lead to spurious genetic correlations with ALS and other neurodegenerative diseases. For FTD, we primarily used the results of the cross-subtype meta-analysis, which includes behavioral variant FTD, semantic dementia FTD, progressive non-fluent aphasia FTD and mndFTD. For CBD, allele coding was unavailable, and effect alleles were inferred by matching allele frequencies to those observed in the Haplotype Reference Consortium. SNPs with MAF > 0.4 were excluded. Because downstream methods rely on LD scores or population-specific LD patterns, the European ancestry summary statistics from the present study were used for ALS. For sample size parameters, effective sample size was calculated as described previously.
Multiple sclerosis summary statistics were obtained from the International Multiple Sclerosis Genetics Consortium91. For cerebrovascular diseases, GWAS summary statistics were obtained for ischemic stroke (any ischemic stroke)92, intracerebral hemorrhage93 and intracranial aneurysm94. For psychiatric traits, GWAS summary statistics were obtained from Psychiatric Genomics Consortium studies on anorexia nervosa95, obsessive–compulsive disorder96, anxiety disorders (anxiety score)97, post-traumatic stress disorder (all European ancestries)98, major depressive disorder99, bipolar disorder100, schizophrenia101, Tourette’s syndrome102, autism spectrum disorder103 and attention-deficit hyperactivity disorder (European ancestries)104.
Genetic correlation
Genome-wide genetic correlation between neurodegenerative traits was calculated using LDSC (version 1.0.0)59. Precomputed LD scores of European individuals in the 1000 Genomes project for high-quality HapMap3 SNPs were used (‘eur_w_ld_chr’). A free intercept was modeled to allow for potential sample overlap.
Colocalization
Before the colocalization analysis of neurodegenerative diseases, we first assessed residual confounding by estimating the LDSC intercept using LDSC (version 1.0.0) (ALS, 1.03 (s.e., 0.0073); AD, 1.03 (s.e., 0.013); PD, 0.98 (s.e., 0.0065); PSP, 1.05 (s.e., 0.0076); CBD, 0.98 (s.e., 0.0073); FTD, 1.00 (s.e., 0.0071)), showing limited inflation of test statistics due to confounding across these studies. For each locus (top SNP ±100 kb) harboring SNPs with an association with any of the neurodegenerative diseases (ALS, AD, PD, PSP, CBD, FTD) at P < 1 × 10−5, we performed colocalization analysis using the ‘coloc’ package in R105. We set the prior probabilities to π1 = 1 × 10−4, π2 = 1 × 10−4 and π12 = 1 × 10−5 for a causal variant in trait 1 or trait 2 and a shared causal variant between traits 1 and 2, respectively. Using the same parameters, we performed colocalization analysis for ALS and each of the FTD subtypes (behavioral variant FTD, semantic dementia FTD, progressive non-fluent aphasia FTD and mndFTD).
Enrichment analyses
Linkage disequilibrium score regression annotation-specific enrichment analysis
We used LDSC (version 1.0.0)59 to calculate SNP-based heritability, the LDSC intercept and SNP-based heritability enrichment for partitions of the genome. In all LDSC analyses, summary statistics excluding the HLA region of only samples of European ancestry were included. LD scores and partitioned LD scores provided by LDSC were used for genome-wide and genic region-based heritability analyses. The option ‘–overlap-annot’ was used in the partitioned heritability analysis to allow for overlapping SNPs between MAF bins. SNPs with MAF > 5% were included.
Tissue and cell type enrichment analysis
Tissue and cell type enrichment analyses were performed using the GWAS summary statistics of the European ancestry meta-analysis and FUMA33 software version 1.3.6a. FUMA performs a genic aggregation analysis of GWAS association signals to calculate gene-wise association signals using MAGMA version 1.6 and subsequently tests whether tissues and cell types are enriched for expression of these genes. For tissue enrichment analysis, we used the GTEx version 8 reference set. FDR-corrected P-values <0.05 across all tissues (n = 54) were considered statistically significant. For cell type enrichment analyses34, we used human-derived single-cell RNA-seq data on major brain cell types (GSE67835 without fetal samples106), Allen Brain Atlas cell types107 for the human-derived major neuronal subtypes and the DropViz108 dataset for mouse-derived brain cell types across all brain regions. We applied FDR correction for multiple testing within each expression dataset, and FDR-corrected P-values <0.05 were considered statistically significant.
Pathway enrichment analysis
We used Downstreamer software24 to identify enriched biological pathways and processes. First, gene-based association statistics were obtained with the Pascal method109, which aggregates SNP association statistics including SNPs up to 10 kb upstream and downstream of a gene, accounting for LD using the non-Finnish European individuals from the 1000 Genomes Project phase 3 (ref. 110) as a reference. In the Downstreamer method, putative core genes are defined as those that are coexpressed with disease-associated genes and can therefore be implicated in disease. Coexpression networks are based on either a large, multi-tissue transcriptome dataset including 56,435 genes and 31,499 individuals or brain-specific RNA-seq data obtained from the MetaBrain resource. The gene-based association statistics, coexpression matrix and gene Z scores per pathway or HPO term are then combined in a generalized least-squares regression model to obtain enrichment statistics24. Enrichment analyses were performed for reactome, gene ontology and HPO terms using multi-tissue or brain-specific transcriptome datasets to calculate the coexpression matrix.
The distribution of enrichment Z-score statistics was compared between analyses using multi-tissue or brain-specific coexpression matrices. Using the ‘pyhpo’ module in Python, all HPO terms were assigned to their parent term(s) in the ‘phenotypic abnormality’ (HP:0000118) branch, which includes phenotypic abnormalities grouped per organ system.
Mendelian randomization
Causal inference through MR analysis was performed for 22 exposures for which large-scale GWASs are available and for which there is prior evidence for an association with ALS. These include seven behavioral-related traits: body mass index (anthropometric)111, years of schooling (educational attainment)112, alcoholic drinks per week, age of smoking initiation and cigarettes per day from Liu et al.113, days per week of moderate physical activity and days per week of vigorous activity from the UK Biobank114; four blood pressure traits (coronary artery disease115, stroke92, diastolic blood pressure and systolic blood pressure116); seven immune system traits from Vuckovic et al.117 (basophil, eosinophil, lymphocyte, monocyte, neutrophil and white blood cell counts) and C-reactive protein118; and four lipid traits from Willer et al.119 (HDL cholesterol, LDL cholesterol, total cholesterol and triglyceride levels). A full description of the included studies is provided in Supplementary Table 26. From these GWASs, SNPs to serve as instruments for MR analyses were selected at two different P-value cutoffs (P < 5 × 10−8 and P < 5 × 10−5) and then LD clumped to obtain independent SNPs. SNP effect estimates on ALS risk were obtained from the European ancestry-only GWAS and, if needed, an LD proxy was selected (r2 > 0.8).
After harmonizing effect alleles and excluding palindromic SNPs, we performed a series of quality-control steps to avoid biased estimates of causal effects, checking for each exposure (1) instrument coverage (>85% overlapping SNPs; Supplementary Table 31), (2) instrument strength (F-statistic37,120,121 >10; Supplementary Table 32), (3) distribution and significance of the Wald ratios (visual inspection of volcano plots; Supplementary Table 33) and (4) heterogeneity across the instrument-exposure effects (Q-statistic at P < 0.05 indicated heterogeneity; Supplementary Table 34).
We applied five different MR methods: IVW using the random-effects model, MR-Egger and simple mode, weighted median and weighted mode methods. When only a single SNP was available, the Wald ratio test was conducted. MR analysis was conducted in R using the ‘mr()‘ function in the ‘TwoSampleMR‘ package122.
Subsequently, radial MR analysis was conducted to determine whether Wald ratio outliers needed to be removed from the IVW or MR-Egger MR estimates38. In addition, we conducted a Q-test to identify outlier SNPs (P < 0.05). These outliers were then removed from the original MR analyses (across all five MR methods). The radial MR analysis was conducted using the RadialMR R package (https://github.com/WSpiller/RadialMR). To determine whether MR effects were orientated in the correct direction (from exposure to ALS), we conducted both reverse MR123 and Steiger filtering124 on our top MR findings.
Finally, we explored whether the MR effects of our total and LDL cholesterol and systolic blood pressure exposures may be confounded by the effect we observed for years of schooling by conducting multivariate MR analysis125. Conditional F- and Q-statistics were calculated using the ‘MVMR‘ package126 in R.
Statistical analyses
All presented P-values correspond to two-sided P-values uncorrected for multiple testing unless explicitly stated otherwise.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The GWAS summary statistics generated in this study are publicly available in the NHGRI-EBI GWAS Catalog at https://www.ebi.ac.uk/gwas/ (accession IDs GCST90027163 and GCST90027164 for cross-ancestry and European ancestry meta-analyses, respectively) and through the Project MinE website (https://www.projectmine.com/research/download-data/). Summary statistics of the rare variant burden analyses and eQTL and mQTL SMR analyses are available through the Project MinE website. The following publicly available datasets were used in this project: the Wellcome Trust Case Control Consortium (https://www.wtccc.org.uk/) and dbGaP datasets (phs000101.v3.p1, NIH Genome-Wide Association Studies of Amyotrophic Lateral Sclerosis; phs000126.v1.p1, CIDR: Genome Wide Association Study in Familial Parkinson Disease (PD); phs000196.v1.p1, Genome-Wide Association Study of Parkinson Disease: Genes and Environment; phs000344.v1.p1, Genome-Wide Association Study of Amyotrophic Lateral Sclerosis in Finland; phs000336, a Genome-Wide Association Study of Lung Cancer Risk; phs000346, Genome-Wide Association Study for Bladder Cancer Risk; phs000789, Collaborative Study of Genes, Nutrients and Metabolites (CSGNM); phs000206, Whole Genome Scan for Pancreatic Cancer Risk in the Pancreatic Cancer Cohort Consortium and Pancreatic Cancer Case–Control Consortium (PanScan); phs000297, eMERGE Network Study of the Genetic Determinants of Resistant Hypertension; phs000652, Cohort-Based Genome-Wide Association Study of Glioma (GliomaScan); phs000869, Barrett’s and Esophageal Adenocarcinoma Genetic Susceptibility Study (BEAGESS); phs000812, the Breast and Prostate Cancer Cohort Consortium (BPC3) GWAS of Aggressive Prostate Cancer and ER− Breast Cancer; phs000428, Genetics Resource with the HRS; phs000360.v3, eMERGE Network Genome-Wide Association Study of Red Cell Indices, White Blood Count (WBC) Differential, Diabetic Retinopathy, Height, Serum Lipid Levels, Specifically Total Cholesterol, HDL (High Density Lipoprotein), LDL (Low Density Lipoprotein), and Triglycerides, and Autoimmune Hypothyroidism; phs000893.v1, Genome-Wide Association Study of Endometrial Cancer in the Epidemiology of Endometrial Cancer Consortium (E2C2); phs000168.v2, National Institute on Aging—Late Onset Alzheimer’s Disease Family Study: Genome-Wide Association Study for Susceptibility Loci; phs000092.v1, Study of Addiction: Genetics and Environment (SAGE); phs000864.v1, Genomic Predictors of Combat Stress Vulnerability and Resilience; phs000170.v2, a Genome-Wide Association Study on Cataract and HDL in the Personalized Medicine Research Project Cohort; phs000431.v2, IgA Nephropathy GWAS on Individuals of European Ancestry (IGANGWAS2); phs000237.v1, Northwestern NUgene Project: Type 2 Diabetes; phs000169.v1, Whole Genome Association Study of Visceral Adiposity in the Health Aging and Body Composition (Health ABC) Study; phs000982.v1, Genetic Analysis of Psoriasis and Psoriatic Arthritis: GWAS of Psoriatic Arthritis; phs000289.v2, National Human Genome Research Institute (NHGRI) GENEVA Genome-Wide Association Study of Venous Thrombosis (GWAS of VTE); phs000634.v1, National Cancer Institute (NCI) Genome Wide Association Study (GWAS) of Lung Cancer in Never Smokers; phs000274.v1, Genome-Wide Association Study of Celiac Disease; phs001172.v1, National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease; phs000389.v1, GEnetics of Nephropathy—an International Effort (GENIE) GWAS of Diabetic Nephropathy in the UK GoKinD and All-Ireland Cohorts; phs000460.v1, Genetics of 24 Hour Urine Composition; phs000138.v2, GWAS for Genetic Determinants of Bone Fragility in European–American Premenopausal Women; phs000394.v1, Autopsy-Confirmed Parkinson Disease GWAS Consortium (APDGC); phs000948.v1, Genetic Discovery and Application in a Clinical Setting: Continuing a Partnership (eMERGE Phase II); phs000630.v1, Exome Chip Study of NIMH Controls; phs000678.v1, a Family-Based Study of Genes and Environment in Young-Onset Breast Cancer; phs000351.v1, National Cancer Institute Genome-Wide Association Study of Renal Cell Carcinoma; phs000314.v1, Genetic Associations in Idiopathic Talipes Equinovarus (Clubfoot)—GAIT; phs000147.v3, Cancer Genetic Markers of Susceptibility (CGEMS) Breast Cancer Genome-wide Association Study (GWAS)—Primary Scan: Nurses’ Health Study—Additional Cases: Nurses’ Health Study 2; phs000882.v1, National Cancer Institute (NCI) Prostate Cancer Genome-Wide Association Study for Uncommon Susceptibility Loci (PEGASUS); phs000238.v1, National Eye Institute Glaucoma Human Genetics Collaboration (NEIGHBOR) Consortium Glaucoma Genome-Wide Association Study; phs000397.v1, National Institute on Aging (NIA) Long Life Family Study (LLFS); phs000421.v1, a Genome-Wide Association Study of Fuchs’ Endothelial Corneal Dystrophy (FECD); phs000142.v1, a Whole Genome Association Scan for Myopia and Glaucoma Endophenotypes using Twin Studies; phs000303.v1, Genetic Epidemiology of Refractive Error in the KORA (Kooperative Gesundheitsforschung in der Region Augsburg) Study; phs000125.v1, CIDR: Collaborative Study on the Genetics of Alcoholism Case Control Study; phs001039.v1, International Age-Related Macular Degeneration Genomics Consortium—Exome Chip Experiment; phs000187.v1, High Density SNP Association Analysis of Melanoma: Case–Control and Outcomes Investigation; phs000101.v5, Genome-Wide Association Study of Amyotrophic Lateral Sclerosis; phs002068.v1.p1, Sporadic ALS Australia Systems Genomics Consortium (SALSA-SGC)). Source data are provided with this paper.
Code availability
The following software packages were used for data analyses: R version 3.6.3 with additional packages tidyverse version 1.3.0, data.table version 1.14.0, ggplot2 version 3.3.3, MASS version 7.3.53, SNPRelate version 1.26.0, logistf version 1.24, coloc version 5.1.0, twoSampleMR version 0.5.6, RadialMR version 1.0, MVMR version 0.3, survival version 3.1.8, coxme version 2.2.16 and survminer version 0.4.9 (https://www.r-project.org/), Python version 3.7 with additional modules pandas version 1.1.3, numpy version 1.18.1, scipy version 1.4.1, CpGtools version 1.0.9, matplotlib version 3.1.3, pyliftover version 0.4 and pyhpo version 2.5.0 (https://anaconda.org/), GenomeStudio version 2.0 (https://emea.illumina.com/techniques/microarrays/array-data-analysis-experimental-design/genomestudio.html), GCTA version 1.93.2beta (https://cnsgenomics.com/software/gcta/#Overview), EIGENSOFT version 6.1.4 (https://github.com/DreichLab/EIG), SNPTEST version 2.5.4-beta3 (https://www.well.ox.ac.uk/~gav/snptest/), PLINK version 1.9 (http://www.cog-genomics.org/plink2), the Michigan Imputation Server (https://imputationserver.sph.umich.edu), EAGLE version 2.3 through the Michigan Imputation Server (https://imputationserver.sph.umich.edu), SAIGE version 0.29.1 (https://github.com/weizhouUMICH/SAIGE), METAL 2011-03-25 (https://genome.sph.umich.edu/wiki/METAL), SnpSift 4.3p (https://pcingola.github.io/SnpEff), ANNOVAR version 2017-07-17 for LRT, Polyphen-2, MutationTaster2, Mutation Assessor, PROVEAN and SIFT (https://annovar.openbioinformatics.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster2 (http://www.mutationtaster.org/), Mutation Assessor release 3 (http://mutationassessor.org/r3/), PROVEAN version 1.1 (http://provean.jcvi.org/index.php), SIFT version 6.2.1 (https://sift.bii.a-star.edu.sg/), SnpEff 4.3p (https://pcingola.github.io/SnpEff), LDSC version 1.0.1 (https://github.com/bulik/ldsc), ExpansionHunter version 4 (https://github.com/Illumina/ExpansionHunter), ExpansionHunter Denovo (https://github.com/Illumina/ExpansionHunterDenovo), SMR (https://cnsgenomics.com/software/smr/), MAGMA version 1.6 (https://ctg.cncr.nl/software/magma), FUMA (https://fuma.ctglab.nl/), FUMA Cell-type (https://fuma.ctglab.nl/celltype), summary-BayesR (https://cnsgenomics.com/software/gctb/#SummaryBayesianAlphabet), S-PrediXcan (https://github.com/hakyimlab/MetaXcan) and TWAS (http://gusevlab.org/projects/fusion/).
Change history
31 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41588-022-01020-3
References
van Es, M. A. et al. Amyotrophic lateral sclerosis. Lancet 390, 2084–2098 (2017).
Al-Chalabi, A., van den Berg, L. H. & Veldink, J. H. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat. Rev. Neurol. 13, 96–104 (2017).
Trabjerg, B. B. et al. ALS in Danish registries: heritability and links to psychiatric and cardiovascular disorders. Neurol. Genet. 6, e398 (2020).
Ryan, M., Heverin, M., McLaughlin, R. L. & Hardiman, O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 76, 1367–1374 (2019).
Byrne, S., Elamin, M., Bede, P. & Hardiman, O. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 83, 365–367 (2012).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015).
Kenna, K. P. et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042 (2016).
Brenner, D. et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28 (2016).
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012).
Nicolas, A. et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283 (2018).
van Es, M. A. et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087 (2009).
Laaksovirta, H. et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 9, 978–985 (2010).
van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Benyamin, B. et al. Cross-ethnic meta-analysis identifies association of the GPX3–TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun. 8, 611 (2017).
Nakamura, R. et al. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol. 3, 526 (2020).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS–FTD. Neuron 72, 257–268 (2011).
Diekstra, F. P. et al. C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis. Ann. Neurol. 76, 120–133 (2014).
Chen, J. A. et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener. 13, 41 (2018).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Iacoangeli, A. et al. Genome-wide meta-analysis finds the ACSL5–ZDHHC6 locus is associated with ALS and links weight loss to the disease genetics. Cell Rep. 33, 108323 (2020).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
de Klein, N. et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain-related diseases. Preprint at bioRxiv https://doi.org/10.1101/2021.03.01.433439 (2021).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208 (2016).
Shireby, G. L. et al. Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain 143, 3763–3775 (2020).
Hannon, E. et al. An integrated genetic–epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 17, 176 (2016).
Fang, X. et al. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 47, 834–841 (2015).
Brown, A.-L. et al. Common ALS/FTD risk variants in UNC13A exacerbate its cryptic splicing and loss upon TDP-43 mislocalization. Preprint at bioRxiv https://doi.org/10.1101/2021.04.02.438170 (2021).
Ma, X. R. et al. TDP-43 represses cryptic exon inclusion in FTD/ALS gene UNC13A. Preprint at bioRxiv https://doi.org/10.1101/2021.04.02.438213 (2021).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Leeuw, C. A., de Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Watanabe, K., Umićević Mirkov, M., de Leeuw, C. A., van den Heuvel, M. P. & Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 3222 (2019).
Deelen, P. et al. Improving the diagnostic yield of exome-sequencing by predicting gene–phenotype associations using large-scale gene expression analysis. Nat. Commun. 10, 2837 (2019).
Hop, P. J. et al. Genome-wide study of DNA methylation in amyotrophic lateral sclerosis identifies differentially methylated loci and implicates metabolic, inflammatory and cholesterol pathways. Preprint at medRxiv https://doi.org/10.1101/2021.03.12.21253115 (2021).
Davies, N. M., Holmes, M. V. & Smith, G. D. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362, k601 (2018).
Bowden, J. et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the radial plot and radial regression. Int. J. Epidemiol. 47, 1264–1278 (2018).
Munafò, M. R., Tilling, K., Taylor, A. E., Evans, D. M. & Davey Smith, G. Collider scope: when selection bias can substantially influence observed associations. Int. J. Epidemiol. 47, 226–235 (2018).
Watanabe, Y. et al. An amyotrophic lateral sclerosis-associated mutant of C21ORF2 is stabilized by NEK1-mediated hyperphosphorylation and the inability to bind FBXO3. iScience 23, 101491 (2020).
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
Luo, Y. et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat. Genet. 49, 186–192 (2017).
Kathiresan, S. et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40, 189–197 (2008).
Saez-Atienzar, S. et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv. 7, eabd9036 (2021).
Yamanaka, K. et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl Acad. Sci. USA 105, 7594–7599 (2008).
Ralph, G. S. et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 11, 429–433 (2005).
Blokhuis, A. M., Groen, E. J. N., Koppers, M., van den Berg, L. H. & Pasterkamp, R. J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794 (2013).
Seelen, M. et al. Prior medical conditions and the risk of amyotrophic lateral sclerosis. J. Neurol. 261, 1949–1956 (2014).
Bandres-Ciga, S. et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol. 85, 470–481 (2019).
Armon, C. Smoking is a cause of ALS. High LDL-cholesterol levels? Unsure. Ann. Neurol. 85, 465–469 (2019).
Turner, M. R., Wotton, C., Talbot, K. & Goldacre, M. J. Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: indirect evidence from record linkage study. J. Neurol. Neurosurg. Psychiatry 83, 395–398 (2012).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Koga, H., Kaushik, S. & Cuervo, A. M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 24, 3052–3065 (2010).
Fraldi, A. et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 29, 3607–3620 (2010).
Barbero-Camps, E. et al. Cholesterol impairs autophagy-mediated clearance of amyloid β while promoting its secretion. Autophagy 14, 1129–1154 (2018).
Brooks, B. R., Miller, R. G., Swash, M. & Munsat, T. L. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299 (2000).
Zhou, W. et al. Efficiently controlling for case–control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Brown, B. C. et al. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Project MinE ALS Sequencing Consortium. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 26, 1537–1546 (2018).
Spek, R. A. Avander. et al. The project MinE databrowser: bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 432–440 (2019).
Genovese, G. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, 1433–1441 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Chun, S. & Fay, J. C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561 (2009).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118 (2011).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Dolzhenko, E. et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 27, 1895–1903 (2017).
Dolzhenko, E. et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 21, 102 (2020).
Mousavi, N., Shleizer-Burko, S., Yanicky, R. & Gymrek, M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 47, e90 (2019).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Hannon, E. et al. Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am. J. Hum. Genet. 103, 654–665 (2018).
Hop, P. J. et al. Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference. Genome Biol. 21, 220 (2020).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Wei, T. et al. CpGtools: a Python package for DNA methylation analysis. Bioinformatics 37, 1598–1599 (2021).
Zeng, J. et al. Signatures of negative selection in the genetic architecture of human complex traits. Nat. Genet. 50, 746–753 (2018).
Lloyd-Jones, L. R. et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10, 5086 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Ferrari, R., Hernandez, D. G., Nalls, M. A. & Rohrer, J. D. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699 (2014).
Kouri, N. et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6, 7247 (2015).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99 (2018).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019).
Malik, R. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 50, 524–537 (2018).
Woo, D. et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am. J. Hum. Genet. 94, 511–521 (2014).
Bakker, M. K. et al. Genome-wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat. Genet. 52, 1303–1313 (2020).
Watson, H. J. et al. Genome-wide association study identifies eight risk loci and implicates metabo–psychiatric origins for anorexia nervosa. Nat. Genet. 51, 1207–1214 (2019).
International Obsessive Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC) and OCD Collaborative Genetics Association Studies (OCGAS). Revealing the complex genetic architecture of obsessive–compulsive disorder using meta-analysis. Mol. Psychiatry 23, 1181–1188 (2018).
Otowa, T. et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol. Psychiatry 21, 1391–1399 (2016).
Nievergelt, C. M. et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat. Commun. 10, 4558 (2019).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Stahl, E. A. et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803 (2019).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Yu, D. et al. Interrogating the genetic determinants of Tourette’s syndrome and other tic disorders through genome-wide association studies. Am. J. Psychiatry 176, 217–227 (2019).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Demontis, D. et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Darmanis, S. et al. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl Acad. Sci. USA 112, 7285–7290 (2015).
Hodge, R. D. et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019).
Saunders, A. et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030 (2018).
Lamparter, D., Marbach, D., Rueedi, R., Kutalik, Z. & Bergmann, S. Fast and rigorous computation of gene and pathway scores from SNP-based summary statistics. PLoS Comput. Biol. 12, e1004714 (2016).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
van der Harst, P. & Verweij, N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 122, 433–443 (2018).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231 (2020).
Ligthart, S. et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am. J. Hum. Genet. 103, 691–706 (2018).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Zeng, P., Wang, T., Zheng, J. & Zhou, X. Causal association of type 2 diabetes with amyotrophic lateral sclerosis: new evidence from Mendelian randomization using GWAS summary statistics. BMC Med. 17, 225 (2019).
Cragg, J. G. & Donald, S. G. Testing identifiability and specification in instrumental variable models. Econ. Theory 9, 222–240 (1993).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Smith, G. D., Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98 (2014).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Burgess, S. & Thompson, S. G. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 181, 251–260 (2015).
Sanderson, E., Davey Smith, G., Windmeijer, F. & Bowden, J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol. 48, 713–727 (2019).
Acknowledgements
Acknowledgements and relevant funding details are provided in the Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
Sample ascertainment: W.v.R., R.A.A.v.d.S., M.M., A.M.D., H.-J. Westeneng, G.H.P.T., N.T., J.C.-K., B.N.S., M. Gromicho, S. Chandran, S. Pal, K.E.M., P.J.S., J.H., R.W.O., M.S., T.M., N.B., A.J.v.d.K., A. Ratti, C. Gellera, G. Lauria, G.P.C., C.C., D.S., S.D.’A., G. Sorarù, G. Siciliano, M.F., A.P., A. Chiò, A. Calvo, C. Moglia, M. Brunetti, A. Canosa, M. Grassano, E.B., E.P., G. Logroscino, B.N., A.O., A.N., Y.L., M. Zabari, M. Gotkine, R.H. Baloh, S.B., P.V., P. Corcia, P. Couratier, S. Millecamps, V.M., F.S., J.S.M.P., A. Assialioui, R.R.-G., P.A.D., J.P.R., A.C.L., J.H.W., D. Brenner, A. Freischmidt., G. Bensimon, A. Brice, A.D., C.A.M.P., S.S.-D., N.W.W., S.T., R. Rademakers, A. Braun, J.K., D.C.W., C.M.O., A.G.U., A.H., M.R., S. Cichon, M.M. Nöthen, P.A., B.J.T., A.B.S., M. Mitne Neto, R.J.C., R.A.O., M.W.-P., C.L.-H., V.M.v.D., J.G., A. Roediger, N.G., A.J., T.B., E. Theele, B. Ilse., B.S., O.W.W., R.S., C.A.H., C. Graff, L.B., V.F., V. Demeshonok, A. Ataulina, B.R., B.K., J.Z., M.R.-G., D.G., Z.S., V. Drory, M.P., I.P.B., M.C.K., R.D.H., S. Mathers, P.A.M., M.N., G.A.N., R.P., D.B.R., K.A.M., P.S.S., M.d.C., S. Pinto, S. Petri, M.W., G.A.R., V.S., J.D.G., R.H. Brown, J.E.L., C.E.S., P.M.A., D. Fan, F.C.G., A.F.M., R.L.M., O.H., A.A.-C., P.V.D., L.H.v.d.B., J.H.V., SLALOM Consortium, PARALS Consortium, SLAGEN Consortium and SLAP Consortium. SNP array genotyping: W.v.R., R.A.A.v.d.S., A.M.D., A.S., I.F., G. Bensimon, A. Brice, A.D., C.A.M.P., S.S.-D., N.W.W., L. Tittmann, W.L., A. Franke, S.R., A. Braun, J.K., D.C.W., C.M.O., A.G.U., A.H., M.R., S. Cichon, M.M. Nöthen, P.A., B.J.T., A.B.S., B.B., S.F., S.T.N., F.J.S., K.L.W., A.K.H., L.W., C.J.C., G. Breen, D. Fan, F.C.G., A.F.M., N.R.W., A.A.-C., P.V.D., L.H.v.d.B. and J.H.V. GWAS quality control: W.v.R., R.A.A.v.d.S., M.K.B., R. Restuadi, R.L.M., N.R.W. and J.H.V. GWAS data analysis: W.v.R., R.A.A.v.d.S., M.K.B., R. Restuadi, R.P.B., M. Doherty, M.H., A.A.K., A.I., A.S., N.T., B.N.S., B.B., D. Fan, A.F.M., R.L.M., N.R.W. and J.H.V. WGS: W.v.R., R.A.A.v.d.S., P.J.H., R.A.J.Z., M.M., A.M.D., G.H.P.T., K.R.v.E., M.K., J.C.-K., B.N.S., K.P.K., A.A.-C., P.V.D., L.H.v.d.B. and J.H.V. WGS quality control: W.v.R., R.A.A.v.d.S., J.J.F.A.v.V., P.J.H., R.A.J.Z., M.M., K.P.K., P.V.D. and J.H.V. WGS rare variant burden analyses: W.v.R., R.A.A.v.d.S., P.J.H., R.A.J.Z., K.R.v.E., K.P.K., P.V.D. and J.H.V. WGS STR analyses: W.v.R., J.J.F.A.v.V., R.A.J.Z., E.D., M.A.E. and J.H.V. eQTL analyses: W.v.R., R.A.A.v.d.S., M.K.B., N.d.K., H.-J. Westra, O.B.B., P.A.D., J.M., L.F. and J.H.V. mQTL analyses: W.v.R., M.K.B., P.J.H., R.A.J.Z., G.S., E.H., A.M.D. and J.H.V. Cross-disorder analyses: W.v.R., R.A.A.v.d.S., M.K.B., N.d.K., H.-J. Westra, O.B.B., P.D., E.J.N.G., M.A.v.E., R.J.P., A.F.M., N.R.W., E. Tsai, H.R., L.F. and J.H.V. MR analyses: W.v.R., R.A.A.v.d.S., M.K.B., D. Baird, H.-J. Westra, G.D.S., T.R.G., E. Tsai, H.R. and J.H.V. Writing the manuscript: W.v.R., M.K.B., D. Baird, J.M., E. Tsai and J.H.V. Revising the manuscript: W.v.R., R.A.A.v.d.S., M.K.B., J.J.F.A.v.V., G.S., E.H., D. Baird, R. Restuadi, E.D., H.-J. Westra, G.H.P.T., K.R.v.E., E.J.N.G., M.A.v.E., R.J.P., G.D.S., T.R.G., R.L.M., K.P.K., N.R.W., E. Tsai, H.R., L.F., L.H.v.d.B. and J.H.V. Funding acquisition and study supervision: L.H.v.d.B. and J.H.V.
Corresponding authors
Ethics declarations
Competing interests
J.H.V. has sponsored research agreements with Biogen. L.H.v.d.B. receives personal fees from Cytokinetics outside of the submitted work. A.A.-C. has served on scientific advisory boards for Mitsubishi Tanabe Pharma, Orion Pharma, Biogen, Lilly, GSK, Apellis, Amylyx and Wave Therapeutics. A. Chiò. serves on scientific advisory boards for Mitsubishi Tanabe, Roche, Biogen, Denali and Cytokinetics. J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics, a consultant for ACI Clinical LLC sponsored by Biogen, Inc. or Ionis Pharmaceuticals, Inc. J.E.L. is also a consultant for Perkins Coie LLP and may provide expert testimony. The remaining authors declare no competing interests related to this work.
Additional information
Peer review information Nature Genetics thanks David Goldstein and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Manhattan plot in European ancestries GWAS.
Genome-wide association statistics obtained by inverse-variance weighted meta-analysis of the stratified SAIGE logistic mixed model regression in European ancestry cohorts. Y-axis corresponds to the two-tailed -log10(P-value), x-axis corresponds to the genomic coordinates (GRCh37). Loci containing a genome-wide significant SNP are highlighted in red. SNP IDs are the top associated SNPs in each locus. The dotted horizontal line reflects the threshold for genome-wide significance (P = 5 × 10−8).
Extended Data Fig. 2 Annotation specific heritability enrichment.
Enrichment of SNP-based heritability was calculated with LD-score regression. Grey dashed line represents no enrichment (enrichment = 1). Error bars denote standard error of enrichment estimate. Nominal statistically significant enrichment estimates (two-sided P < 0.05) are marked with an asterisk (Conserved_LindbladToh P = 6.5 × 10−5, SuperEnhancer_Hnisz P = 0.014, TFBS_ENCODE P = 0.017, H3K4me1_peaks_Trynka P = 0.018, Coding_UCSC P = 0.028, H3K9ac_Trynka P = 0.037). The category Conserved_LindbladToh was significant after Bonferroni correction for multiple testing across all categories (N = 28). Due to the regression framework in LDSC, enrichment estimates < 0 are possible (with large standard errors).
Extended Data Fig. 3 PRS stratified by rare variant carrier status.
Distribution of PRS in controls and ALS patients with or without one or more rare variants in ALS risk genes. There was no statistically significant difference in PRS between ALS patients with and without rare variants in ALS risk genes (labeled as gene_mut or gene_wt respectively). In total, 5,112 ALS patients and 2,132 controls from stratum 6 with whole-genome sequencing data available were included. For SOD1, TARDBP, FUS, NEK1, TBK1, and CFAP410, rare variants were included according to the model that yielded the strongest association in the rare variant burden association analyses. For C9orf72, patients with the pathogenic hexanucleotide repeat expansion were compared to those without the expansion. The ‘any ALS gene’ groups all patients together with a rare variant in any of the ALS risk genes. P-values for difference in PRS were derived by two-tailed logistic regression. The number of ALS patients carrying a rare variant per gene is denoted in the corresponding panel. Intervals for boxplots: center = median, box = lower and upper quartile, hinges = median ± 2 * IQR, IQR = interquartile range.
Extended Data Fig. 4 NEK1 repeat distribution.
The frequency of STR alleles in ALS cases and controls are shown. A repeat length of 11 and longer was used as the optimal threshold for disease-associated genotype. The P-value was calculated by Firth logistic regression and FDR correction over all possible thresholds. Y-axis shows the allele frequency of repeat lengths. Repeat position on GRCh37, and repeat motif are shown.
Extended Data Fig. 5 Genetic correlations between brain diseases.
Correlation matrix for genetic correlation estimates obtained from bivariate LD score regression. Colors correspond to genetic correlation estimates. Strongest clusters appear between neurodegenerative diseases and within the psychiatric traits. ALS = amyotrophic lateral sclerosis, FTD = frontotemporal dementia, PSP progressive supranuclear palsy, PD = Parkinson’s disease, CBD = corticobasal degeneration, AD = (clinically diagnosed) Alzheimer’s disease, MS = multiple sclerosis, IS = ischemic stroke (any), ICH = intracerebral hemorrhage, IA = intracranial aneurysm (any), AN = anorexia nervosa, OCD = obsessive compulsive disorder, Anxiety = anxiety disorder (score), PTSD = post-traumatic stress disorder, MDD = major depressive disorder, BIP = bipolar disorder, SCZ = schizophrenia, TS = Tourette’s syndrome, ASD = autism spectrum disorder, ADHD = attention-deficit hyperactivity disorder.
Extended Data Fig. 6 Colocalization signals.
Loci were selected for colocalization analysis when the top associated SNP was associated with any neurodegenerative disease at 5 × 10−5. For ALS, the European ancestries meta-analysis was used. Bayesian posterior probabilities for a shared variant driving risk of both traits (PPH4) are reported below locus names. Colors reflect LD between the variant and top associated SNP.
Extended Data Fig. 7 Colocalization analysis with FTD subtypes.
Top associated SNPs in the ALS GWAS were selected for colocalization analysis between ALS and FTD subtypes using COLOC. In the top panel, point height is the two-sided -log10(P-value) of the top-associated SNP in the ALS GWAS. In the bottom panel, association P-values of these SNPs with FTD subtypes are shown by color. The Bayesian posterior probability for a shared causal variant between traits (PPH4) is depicted by a connection between points.
Extended Data Fig. 8 Tissue and cell-type enrichment analyses for all brain diseases.
Tissue (a) and cell-type (b) enrichment for all included brain diseases obtained from two-sided MAGMA linear regression. Only brain diseases with exome-wide significant gene-based MAGMA associations (P < 2.7 × 10−6) were suitable for tissue and cell-type enrichment analyses. The color represents enrichment coefficient and size indicates two-sided -log10(P-value) of enrichment obtained by the linear regression model in the MAGMA gene-property analysis. Due to the large number of significant genes in the gene-based MAGMA analyses for schizophrenia, bipolar disorder and multiple sclerosis the enrichment P-values were truncated at P < 1.0 × 10−5. ALS = amyotrophic lateral sclerosis, PD = Parkinson’s disease, AD = Alzheimer’s disease, ADHD = attention-deficit hyperactivity disorder, ASD = autism spectrum disorder, TS = Tourette’s syndrome, SCZ = schizophrenia, BIP = bipolar disorder, MDD = major depressive disorder, PTSD = post-traumatic stress disorder, Anxiety = anxiety disorder (score), AN = anorexia nervosa, IA intracranial aneurysm (any), IS = ischemic stroke, MS = multiple sclerosis, Cx = cortex, OPC = oligodendrocyte progenitor cells.
Extended Data Fig. 9 Cell-type enrichment analysis in mice.
Cell-type enrichment analysis using the DropViz single-cell RNA sequencing dataset obtained from mice. Similar to the cell-type enrichment analyses there is neuron-specific enrichment in ALS and Parkinson’s disease. In Alzheimer’s disease microglia are the most enriched cell-types. The color represents enrichment coefficient and size indicates two-sided -log10(P-value) of enrichment obtained by the linear regression model in the MAGMA gene-property analysis. Statistically significant enrichments after correction for multiple testing with a false discovery rate (FDR) < 0.05 are marked with an asterisk. ALS = amyotrophic lateral sclerosis, PD = Parkinson’s disease, AD = Alzheimer’s disease, Cx = cortex.
Extended Data Fig. 10 Human phenotype ontology term enrichment.
Downstreamer enrichment analyses were performed using the multi-tissue and brain-specific co-expression matrix to identify co-regulated ALS-genes. The distribution of enrichment statistics (Z-scores) for all Human phenotype ontology (HPO) terms are plotted per HPO parent branch. The multi-tissue analysis indicates enrichment for the neurology parent branch ‘abnormality of the nervous system’ (dark-red), although no term passes the Bonferroni threshold for multiple testing. The brain-specific analysis illustrates stronger enrichment for the neurology parent branch. In total, 58 HPO terms pass the threshold for multiple testing of which 42 are defined within the ‘abnormality of the nervous system’ branch.
Supplementary information
Supplementary Information
Supplementary Note, Figs. 1–18 and Tables 4–25 and 27–30
Supplementary Tables
Supplementary Tables 1–3, 26 and 31–34.
Source data
Source Data Fig. 1
Link to publicly available GWAS summary statistics.
Source Data Fig. 2
Results of modifier analyses, effect estimates, 95% CIs and P values for the Cox proportional hazards mixed model (survival) and linear regression (age at onset).
Source Data Fig. 3
Bivariate LDSC results for genetic correlation estimates among neurodegenerative diseases and association statistics for genome-wide significant SNPs and two colocalization loci in neurodegenerative diseases.
Source Data Fig. 4
MAGMA enrichment statistics for GTEx tissues and cell types in ALS, PD, AD and AD by proxy.
Source Data Fig. 5
Pointer to Supplementary Table 27.
Source Data Extended Data Fig. 1
Link to publicly available GWAS summary statistics.
Source Data Extended Data Fig. 2
Results of the stratified LDSC enrichment analysis.
Source Data Extended Data Fig. 3
PRSs stratified by phenotype or rare variant burden status.
Source Data Extended Data Fig. 4
NEK1 STR alleles for patients with ALS and controls.
Source Data Extended Data Fig. 5
LDSC genetic correlation estimates for brain diseases.
Source Data Extended Data Fig. 6
Association statistics for SNPs in loci with colocalization signals.
Source Data Extended Data Fig. 7
Association statistics for genome-wide significant SNPs in ALS and FTD subtypes.
Source Data Extended Data Fig. 8
MAGMA enrichment statistics for GTEx tissues and cell types across all brain diseases.
Source Data Extended Data Fig. 9
MAGMA enrichment statistics for DropViz cell types across ALS, PD, AD and AD by proxy.
Source Data Extended Data Fig. 10
Downstreamer HPO term enrichment statistics using multi-tissue and MetaBrain coexpression matrices.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Rheenen, W., van der Spek, R.A.A., Bakker, M.K. et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 53, 1636–1648 (2021). https://doi.org/10.1038/s41588-021-00973-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00973-1
This article is cited by
-
Causal relationships between neuropsychiatric disorders and nonalcoholic fatty liver disease: A bidirectional Mendelian randomization study
BMC Gastroenterology (2024)
-
Overlaps and divergences between tauopathies and synucleinopathies: a duet of neurodegeneration
Translational Neurodegeneration (2024)
-
TBK1, a prioritized drug repurposing target for amyotrophic lateral sclerosis: evidence from druggable genome Mendelian randomization and pharmacological verification in vitro
BMC Medicine (2024)
-
Variability in SOD1-associated amyotrophic lateral sclerosis: geographic patterns, clinical heterogeneity, molecular alterations, and therapeutic implications
Translational Neurodegeneration (2024)
-
ALS-associated C21ORF2 variant disrupts DNA damage repair, mitochondrial metabolism, neuronal excitability and NEK1 levels in human motor neurons
Acta Neuropathologica Communications (2024)
