
Abstract
The kidneys integrate information from continuous systemic processes related to the absorption, distribution, metabolism and excretion (ADME) of metabolites. To identify underlying molecular mechanisms, we performed genome-wide association studies of the urinary concentrations of 1,172 metabolites among 1,627 patients with reduced kidney function. The 240 unique metabolite–locus associations (metabolite quantitative trait loci, mQTLs) that were identified and replicated highlight novel candidate substrates for transport proteins. The identified genes are enriched in ADME-relevant tissues and cell types, and they reveal novel candidates for biotransformation and detoxification reactions. Fine mapping of mQTLs and integration with single-cell gene expression permitted the prioritization of causal genes, functional variants and target cell types. The combination of mQTLs with genetic and health information from 450,000 UK Biobank participants illuminated metabolic mediators, and hence, novel urinary biomarkers of disease risk. This comprehensive resource of genetic targets and their substrates is informative for ADME processes in humans and is relevant to basic science, clinical medicine and pharmaceutical research.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
Meta-analysis summary statistics for SNPs that are associated with any metabolite at P < 5 × 10−8 will be integrated into the SNiPA database with its v3.4 release. Genome-wide summary statistics will be made available publicly through the GWAS catalog (https://www.ebi.ac.uk/gwas). Raw data for Extended Data Fig. 5 are available as source data with the supplementary materials.
Code availability
Each use of software programs has been clearly indicated and information on the options that were used is provided in the Methods section. Source code to call programs is available upon request.
References
Caldwell, J., Gardner, I. & Swales, N. An introduction to drug disposition: the basic principles of absorption, distribution, metabolism, and excretion. Toxicol. Pathol. 23, 102–114 (1995).
Köttgen, A., Raffler, J., Sekula, P. & Kastenmuller, G. Genome-wide association studies of metabolite concentrations (mGWAS): Relevance for nephrology. Semin. Nephrol. 38, 151–174 (2018).
Homuth, G., Teumer, A., Volker, U. & Nauck, M. A description of large-scale metabolomics studies: increasing value by combining metabolomics with genome-wide SNP genotyping and transcriptional profiling. J. Endocrinol. 215, 17–28 (2012).
Kalim, S. & Rhee, E. P. An overview of renal metabolomics. Kidney Int. 91, 61–69 (2017).
Nigam, S. K. et al. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin. J. Am. Soc. Nephrol. 10, 2039–2049 (2015).
Suhre, K. et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 477, 54–60 (2011).
Shin, S. Y. et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014).
Long, T. et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 49, 568–578 (2017).
Suhre, K., Raffler, J. & Kastenmuller, G. Biochemical insights from population studies with genetics and metabolomics. Arch. Biochem. Biophys. 589, 168–176 (2016).
Gieger, C. et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 4, e1000282 (2008).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Titze, S. et al. Disease burden and risk profile in referred patients with moderate chronic kidney disease: composition of the German Chronic Kidney Disease (GCKD) cohort. Nephrol. Dial. Transplant. 30, 441–451 (2015).
Eckardt, K. U. et al. The german chronic kidney disease (GCKD) study: design and methods. Nephrol. Dial. Transplant. 27, 1454–1460 (2012).
Levey, A. S. et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 150, 604–612 (2009).
Evans, A. M. et al. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics 4, 132 (2014).
Raffler, J. et al. Genome-wide association study with targeted and non-targeted NMR metabolomics identifies 15 novel loci of urinary human metabolic individuality. PLoS Genet. 11, e1005487 (2015).
Suhre, K. et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 43, 565–569 (2011).
Rueedi, R. et al. Genome-wide association study of metabolic traits reveals novel gene-metabolite-disease links. PLoS Genet. 10, e1004132 (2014).
Nicholson, G. et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 7, e1002270 (2011).
Arnold, M., Raffler, J., Pfeufer, A., Suhre, K. & Kastenmuller, G. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics 31, 1334–1336 (2015).
Draisma, H. H. M. et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 6, 7208 (2015).
Pushkin, A. et al. Structural characterization, tissue distribution, and functional expression of murine aminoacylase III. Am. J. Physiol. Cell Physiol. 286, C848–C856 (2004).
Veiga-da-Cunha, M. et al. Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J. Biol. Chem. 285, 18888–18898 (2010).
Perland, E., Bagchi, S., Klaesson, A. & Fredriksson, R. Characteristics of 29 novel atypical solute carriers of major facilitator superfamily type: evolutionary conservation, predicted structure and neuronal co-expression. Open Biol. 7, 170142 (2017).
Ceder, M. M., Lekholm, E., Hellsten, S. V., Perland, E. & Fredriksson, R. The neuronal and peripheral expressed membrane-bound UNC93a respond to nutrient availability in mice. Front. Mol. Neurosci. 10, 351 (2017).
Volzke, H. et al. Cohort profile: the study of health in Pomerania. Int. J. Epidemiol. 40, 294–307 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Moe, O. W., Giebisch, G. H. & Seldin, D. W. in Genetic Diseases of the Kidney 1st edn (eds Lifton, R. P., Somlo, S., Giebisch, G. H. & Seldin, D. W.) Ch. 3 (Academic Press, 2009).
Vanholder, R. et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 63, 1934–1943 (2003).
Rhee, E. P. & Thadhani, R. New insights into uremia-induced alterations in metabolic pathways. Curr. Opin. Nephrol. Hypertens. 20, 593–598 (2011).
GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
Wu, H. et al. Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23, 869–881 e8 (2018).
Lahjouji, K. et al. Expression and functionality of the Na+/myo-inositol cotransporter SMIT2 in rabbit kidney. Biochim. Biophys. Acta. 1768, 1154–1159 (2007).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Wakefield, J. Bayes factors for genome-wide association studies: comparison with P-values. Genet. Epidemiol. 33, 79–86 (2009).
Kim, H. I. et al. Fine mapping and functional analysis reveal a role of SLC22A1 in acylcarnitine transport. Am. J. Hum. Genet. 101, 489–502 (2017).
Gillies, C. E. et al. An eQTL landscape of kidney tissue in human nephrotic syndrome. Am. J. Hum. Genet. 103, 232–244 (2018).
Tazawa, S. et al. SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-D-glucitol, and fructose. Life Sci. 76, 1039–1050 (2005).
Li, Y. et al. Genome-wide association studies of metabolites in patients with CKD identify multiple loci and illuminate tubular transport mechanisms. J. Am. Soc. Nephrol. 29, 1513–1524 (2018).
Petersen, A. K. et al. On the hypothesis-free testing of metabolite ratios in genome-wide and metabolome-wide association studies. BMC Bioinformatics 13, 120 (2012).
Schneider, M. P. et al. Blood pressure control in chronic kidney disease: a cross-sectional analysis from the German Chronic Kidney Disease (GCKD) study. PLoS One 13, e0202604 (2018).
Schlosser, P. et al. Netboost: Boosting-supported network analysis improves high-dimensional omics prediction in acute myeloid leukemia and Huntington’s disease. Preprint at https://arxiv.org/abs/1909.12551 (2019).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Fedde, K. N. & Whyte, M. P. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5’-phosphate ectophosphatase: normal and hypophosphatasia fibroblast study. Am. J. Hum. Genet. 47, 767–775 (1990).
Oddsson, A. et al. Common and rare variants associated with kidney stones and biochemical traits. Nat. Commun. 6, 7975 (2015).
Taylor, R. G., Levy, H. L. & McInnes, R. R. Histidase and histidinemia. Clinical and molecular considerations. Mol. Biol. Med. 8, 101–116 (1991).
Montoliu, I. et al. Current status on genome-metabolome-wide associations: an opportunity in nutrition research. Genes Nutr. 8, 19–27 (2013).
McMahon, G. M. et al. Urinary metabolites along with common and rare genetic variations are associated with incident chronic kidney disease. Kidney Int. 91, 1426–1435 (2017).
Nigam, S. K. What do drug transporters really do? Nat. Rev. Drug Discov. 14, 29–44 (2015).
Momper, J. D. & Nigam, S. K. Developmental regulation of kidney and liver solute carrier and ATP-binding cassette drug transporters and drug metabolizing enzymes: the role of remote organ communication. Expert Opin Drug Metab. Toxicol. 14, 561–570 (2018).
Broer, A. et al. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J. Biol. Chem. 279, 24467–24476 (2004).
Hu, Y., Ding, Q., He, Y., Xu, S. & Jin, L. Reintroduction of a homocysteine level-associated allele into east asians by neanderthal introgression. Mol. Biol. Evol. 32, 3108–3113 (2015).
Eckardt, K. U. et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382, 158–169 (2013).
Secora, A., Alexander, G. C., Ballew, S. H., Coresh, J. & Grams, M. E. Kidney function, polypharmacy, and potentially inappropriate medication use in a community-based cohort of older adults. Drugs Aging 35, 735–750 (2018).
Levin, A., Djurdjev, O., Beaulieu, M. & Er, L. Variability and risk factors for kidney disease progression and death following attainment of stage 4 CKD in a referred cohort. Am. J. Kidney Dis 52, 661–671 (2008).
Prokosch, H. U. et al. Designing and implementing a biobanking IT framework for multiple research scenarios. Stud. Health Technol. Inform. 180, 559–563 (2012).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 78, 4281–4290 (2006).
Do, K. T. et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 14, 128 (2018).
Piontek, U. et al. Sex-specific metabolic profiles of androgens and its main binding protein SHBG in a middle aged population without diabetes. Sci. Rep. 7, 2235 (2017).
Knacke, H. et al. Metabolic fingerprints of circulating IGF-1 and the IGF-1/IGFBP-3 Ratio: a multifluid metabolomics study. J. Clin. Endocrinol. Metab. 101, 4730–4742 (2016).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Fuchsberger, C., Taliun, D., Pramstaller, P. P., Pattaro, C. & CKDGen consortium. GWAtoolbox: an R package for fast quality control and handling of genome-wide association studies meta-analysis data. Bioinformatics 28, 444–445 (2012).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Loewenstein, Y., Portugaly, E., Fromer, M. & Linial, M. Efficient algorithms for accurate hierarchical clustering of huge datasets: tackling the entire protein space. Bioinformatics 24, i41–i49 (2008).
Langfelder, P., Zhang, B. & Horvath, S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24, 719–720 (2008).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Canela-Xandri, O., Rawlik, K. & Tenesa, A. An atlas of genetic associations in UK Biobank. Nat. Genet. 50, 1593–1599 (2018).
Acknowledgements
The work of A.K. was supported by grants KO 3598/3-1 and KO 3598/5-1 (A.K.), Y.L. was supported by grant KO 3598/4-1 (A.K.), the work of M.W., U.T.S., F. Kotsis and A.K. was supported by the Collaborative Research Center (CRC) 1140 project no. 246781735 (A.K.), and the work of P. Schlosser and A.K. was supported by the CRC 992 (A.K.), all German Research Foundation (DFG). U.T.S. and M.W. were also supported by the Else Kroener Fresenius Foundation (NAKSYS project, A.K.). M.K. was funded by the DFG Transregional Collaborative Research Centers (TRR) 152 project no. 239283807 (M.K.) and CRC 1140 project number 246781735 (M.K.). M.K. and G.W. were supported by the Excellence Strategy of the German Federal and State Governments (Center for Integrative Biological Signalling Studies EXC 2189). G.K. was supported by the grants from the National Institute on Aging (NIA): RF1-AG057452-01, RF1-AG059093-01, RF1-AG058942-01 and U01-AG061359-01. Genotyping was supported by Bayer Pharma AG. The GCKD study is supported by the German Ministry of Education and Research (Bundesministerium für Bildung und Forschung, FKZ 01ER 0804, 01ER 0818, 01ER 0819, 01ER 0820 and 01ER 0821) and the KfH Foundation for Preventive Medicine (Kuratorium für Heimdialyse und Nierentransplantation e.V. –Stiftung Präventiv-medizin) and corporate sponsors (www.gckd.org). We are grateful for the willingness of the patients to participate in the GCKD study. The enormous effort of the study personnel of the various regional centers is highly appreciated. We thank the large number of nephrologists who provide routine care for the patients and who collaborate with the GCKD study. The GCKD Investigators are listed in the Supplementary Note. SHIP is part of the Community Medicine Research net of the University of Greifswald, Germany, which is funded by the BMBF (grants 01ZZ9603, 01ZZ0103 and 01ZZ0403), the Ministry of Cultural Affairs as well as the Social Ministry of the Federal State of Mecklenburg-Western Pomerania, and the network ‘Greifswald Approach to Individualized Medicine (GANI_MED)’ funded by the Federal Ministry of Education and Research (grant 03IS2061A). The work of K.S. was supported by the Biomedical Research Program at Weill Cornell Medicine in Qatar, a program funded by the Qatar Foundation. We would like to thank Z. Zheng from GCTA for an update that enables selection of index SNPs based on the Χ2 statistic, and Elizaveta Freinkman for the extracted ion chromatogram and fragmentation spectra used to confirm the identity of X-13689 as α-CMBHC glucuronide.
Author information
Authors and Affiliations
Consortia
Contributions
P. Schlosser, Y.L, P. Sekula and A.K. designed the study. U.T.S., F. Kotsis, F. Kronenberg, U.V., G.W., P.J.O., K.-U.E. and A.K. carried out recruitment and management of the study. T.K., L.F. and A.B.E. carried out genotyping. M.P., B.H., M.N. and R.P.M. carried out metabolite quantification. P. Schlosser, Y.L., P. Sekula, J.R., F.G.-C., M.P., Y.C., M.W., I.S., T.K., G.K. and A.K. carried out bioinformatics and statistical analysis. P. Schlosser, Y.L., P. Sekula. and A.K. wrote the manuscript. P. Schlosser, Y.L., P. Sekula, J.R., F.G.-C., M.P., Y.C., M.W., I.S., U.T.S., F. Kotsis, T.K., L.F., B.H., A.B.E., M.N., U.V., G.W., P.J.O., F. Kronenberg, R.P.M., M.K., K.S., K.-U.E., G.K. and A.K critically read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
R.P.M. is an employee of Metabolon and, as such, has affiliations with or financial involvement with Metabolon. All other authors have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
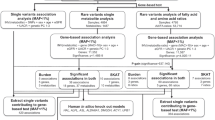
Extended Data Fig. 1 Overview of the study design.
Schematic representation of the genome-wide screens for single metabolites (a) and eigenmetabolites (b) and their follow up analyses.
Extended Data Fig. 2 Comparison of genetic effects with and without adjustment for eGFR.
Each point represents one of the 240 replicated metabolite-associated mQTLs. Genetic effect size estimates per modeled risk allele including adjustment for eGFR and UACR (x axis) by linear regression, as done in the main analysis, were plotted against those obtained after adjustment for genetic PCs, age and sex only (y axis).
Extended Data Fig. 3 Evaluation of genetic associations of replicated mQTLs from CKD patients in a healthy population sample.
(a) Each point represents the index SNP of one of 90 associations that could be matched between the Metabolon platforms of the GCKD and SHIP-Trend studies (see Supplementary Table 5). Dot size is proportional to the -log10(P value) in GCKD and error bars represent 1.96x standard errors in each study. The red line corresponds to a linear regression based on the effect estimates of the most significant index SNP in each of the 35 unique genetic regions into which the 90 associations map. (b) 81 mQTLs with -log10(P value) >12 are plotted. In subsequent panels, the color codes correspond to the detection mode of the mass spectrometer (c), metabolite super pathway (d), differences in relative standard deviation based on measurements of duplicate samples as a measure of precision (e), and the percent of imputed values in GCKD (f). For strata with at least 10 matched mQTLs, additional regression lines were added with color-coding corresponding to the respective legends.
Extended Data Fig. 4 Cell type-specific expression of associated genes in murine kidney.
(a): Resampling based enrichment testing showed that the murine homologs of the 90 associated genes are enriched for cell type-specific expression in proximal tubule in mice (see Supplementary Table 8). The vertical line indicates the statistical significance threshold after Bonferroni adjustment; the arrow indicates a P value<1e-8. (b): Heatmap illustrates the relative expression of each associated gene across the murine kidney cell types; only genes with z-score >2 in at least one cell type are plotted. The mouse gene homologs are provided in parentheses. EC: endothelial cells; PT: proximal tubule; LOH: loop of Henle; DCT: distal convoluted tubule; PC: principal cells; IC: Intercalated cells; CD-Trans: collecting duct transient cells; NK: natural killer cells.
Extended Data Fig. 5 Association between the index SNP at SLC7A9 and pair-wise metabolite ratios reveals transported substrates in vivo.
The figure uses color coding to show the strength of associations (coefficient’s t-test statistics scaled to [-1,1]) between genotype and the 83 ratios that contained information beyond the associations of their individual components (P-gain >6,728,320, Methods), based on linear regression analysis of 672,832 pair-wise metabolite ratios (1172*1171/2, excl. 13,374 ratios with <300 measurements). Coefficient’s test statistics of results that did not confer additional information (P-gain ≤ 6,728,320) are uniformly presented in gray. The metabolite on the y axis represents the numerator and on the x axis the denominator of the respective ratio. Super-pathways: 01 amino acid, 02 carbohydrate, 04 energy, 05 lipid, 06 nucleotide, 08 peptide, 09 unknown. Metabolites that are a member of more than four associated metabolite ratios with a scaled test statistic >0.5 (absolute) are marked in bold. The T allele at rs12460876 was associated with higher gene expression, in agreement with greater tubular reuptake of lysine, resulting in lower urinary levels. Source data underlying this figure including number of values per ratio (column O) can be found in SourceDataEDF5.xlxs.
Extended Data Fig. 6 Overview and examples of metabolite clustering.
(a) shows the dendrogram of the metabolite clustering. The band of color indicates membership of each of the 1,172 metabolites in one of 212 clustered metabolite modules. (b) illustrates module ME193, for which metabolites are labeled. (c) displays the distribution of the eigenmetabolite of ME193 (y axis) with genotype at rs2147896 in PYROXD2 (x axis). (d) illustrates module ME161, for which metabolites are labeled. (e) displays the distribution of the eigenmetabolite of ME161 (y axis) with genotype at rs13538 in NAT8 (x axis). In (c) and (e) horizontal lines indicate medians, violin plots are clipped to the range of the 1,627 samples.
Extended Data Fig. 7 Circular presentation of genetic associations with eigenmetabolites.
The light red band shows the –log10(P value) for genetic associations with eigenmetabolite levels, representing their respective module, by chromosomal position. Associations of all 212 eigenmetabolites in the 1627 samples are overlaid in the red band, are based on linear regressions, and association P values are capped at 1e-60. The blue line indicates genome-wide significance (P = 2.4e-10). For detail about significant associations see Supplementary Table 12. Black gene labels indicate genetic regions in which all members of a given module were also identified in the single metabolite mGWAS, orange labels indicate genetic regions where additional metabolites were implicated as members of a module. The light green band shows the maximum variance in eigenmetabolite levels explained by the index SNP at each genetic region by dark green circles, with the sizes of circles corresponding to different ranges of explained variance. The inner blue band shows a stacked representation of the number of implicated metabolites in each genetic region, is colored according to the super-pathways to which they belong, and the number of modules in the genetic region is given next to it. Color keys of metabolite super-pathways are presented in the middle.
Extended Data Fig. 8 Identification of the unknown metabolite X-13689 as the glucuronide of alpha-CMBHC.
The extracted ion chromatograms (upper right) show the same retention time for both the unknown metabolite in a reference urine matrix (“neat urine”) and the candidate molecule in a neat solution (“neat synthetic”). The MS/MS fragmentation spectra of the candidate molecule (lower left) and of the unknown metabolite (lower right) show the same fragments with equal relative intensities; consequently, the candidate molecule is verified. The m/z (observed) for X-13689 is 495.22438, and the m/z (predicted) for alpha-CMBHC glucuronide is 495.22357, representing a 1.6 ppm error. The 319.1921 fragment peak represents the loss of glucuronic acid (a loss of 176), from which a loss of CO2 (-43.9898) yields the 275.201 fragment peak.
Extended Data Fig. 9 Presence of colocalizing association signals for urinary metabolites and phenotypes and diseases in the UK Biobank.
Colocalizing associations (H4≥0.8, Methods) that showed associations at genome-wide significance (P<5e-8) with both metabolites and traits and diseases in the UK Biobank were found between 68 traits and 66 of the index SNPs. The strength of the associations based on their association P values with the UK Biobank trait are indicated by cross or asterisk as described in the legend. The traits are sorted into five groups: blood count-based parameters, anthropometry, lifestyle, medical conditions, and skin color (left to right). SNPs are sorted by gene and within gene by position. Genes where incorporation of existing biochemical and biological knowledge would have led to prioritization of another most likely causal gene are marked with a # (see Supplementary Note).
Supplementary information
Supplementary Information
Supplementary Note and Supplementary Fig. 1
Supplementary Tables
Supplementary Tables 1–16
Source data
Source Data Extended Data Fig. 5
Source Data underlying Extended Data Fig. 5: Association between the index SNP at SLC7A9 and pair-wise metabolite ratios reveals transported substrates in vivo.
Rights and permissions
About this article

Cite this article
Schlosser, P., Li, Y., Sekula, P. et al. Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans. Nat Genet 52, 167–176 (2020). https://doi.org/10.1038/s41588-019-0567-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0567-8
This article is cited by
-
Transcriptome- and proteome-wide association studies nominate determinants of kidney function and damage
Genome Biology (2023)
-
A genome-wide association study identifies 41 loci associated with eicosanoid levels
Communications Biology (2023)
-
Linkage analysis using whole exome sequencing data implicates SLC17A1, SLC17A3, TATDN2 and TMEM131L in type 1 diabetes in Kuwaiti families
Scientific Reports (2023)
-
Genetic studies of paired metabolomes reveal enzymatic and transport processes at the interface of plasma and urine
Nature Genetics (2023)
-
Integrated proteomic and metabolomic modules identified as biomarkers of mortality in the Atherosclerosis Risk in Communities study and the African American Study of Kidney Disease and Hypertension
Human Genomics (2022)
