
Abstract
Domestication of clonally propagated crops such as pineapple from South America was hypothesized to be a ‘one-step operation’. We sequenced the genome of Ananas comosus var. bracteatus CB5 and assembled 513 Mb into 25 chromosomes with 29,412 genes. Comparison of the genomes of CB5, F153 and MD2 elucidated the genomic basis of fiber production, color formation, sugar accumulation and fruit maturation. We also resequenced 89 Ananas genomes. Cultivars ‘Smooth Cayenne’ and ‘Queen’ exhibited ancient and recent admixture, while ‘Singapore Spanish’ supported a one-step operation of domestication. We identified 25 selective sweeps, including a strong sweep containing a pair of tandemly duplicated bromelain inhibitors. Four candidate genes for self-incompatibility were linked in F153, but were not functional in self-compatible CB5. Our findings support the coexistence of sexual recombination and a one-step operation in the domestication of clonally propagated crops. This work guides the exploration of sexual and asexual domestication trajectories in other clonally propagated crops.
Similar content being viewed by others

Main
Most grain crops, vegetables and ornamentals are produced sexually through seed propagation, whereas most fruit trees, tubers and some ornamentals are clonally propagated through grafting, tissue culture, divisions or cuttings. Sexually reproducing species undergo hundreds to thousands of generations of recombination during domestication; this recurrent selection leaves highly tractable signatures in the genome. In contrast, domestication of clonally propagated crops depends on both vegetative and sexual reproduction, the latter acting more sporadically on long-lived clones. It can even be a one-step operation, where selection is completed once a clone is selected1. Hence, clonal crops may have undergone zero to a few recombination and selection cycles postdomestication, in sharp contrast to sexually reproducing annual crops.
Pineapple (Ananas comosus (L.) Merr.) is a fruit crop originated and domesticated in South America. According to Bertoni2, the genus name Ananas means ‘excellent fruit’ in the Guaraní language of Paraguay. Pineapple was domesticated >6,000 years ago with archaeobotanical remains dated 3,500 years ago in South America and distributed to Mesoamerica >2,500 years ago3,4,5. Pineapple is clonally propagated using the leafy fruit crown, slips or suckers.
Red pineapple (Ananas comosus var. bracteatus) was anciently cultivated for fiber, fruit juice and as a living hedge, and is now a pantropical ornamental6,7. The bracteatus plant is conspicuous for its bright pink-to-red colored fruit. The name ‘bracteatus’ refers to its long bracts. The plant is vigorous with long leaves, coarse spines and abundant suckers. Plant fibers have been used in numerous applications that are beneficial to agriculture and the environment, partly because of their biodegradable nature and lack of carcinogenicity. Pineapple leaf fiber (PALF) contains 70–82% cellulose, 5–12% lignin and 1.1% ash8. PALF is a major source of natural fiber and has been used in the production of activated carbon fibers9, packaging materials10, cell scaffolds11 and apparels12.
Pineapple is in Bromeliaceae, which includes >3,000 species grouped within >50 genera13,14. Bromeliads challenge classical species concepts because of their notoriously leaky pre- and postzygotic barriers15,16,17. Ananas is unique in the family for its syncarpic fruit. Variety bracteatus is a cultigen, anciently cultivated for fiber in Southeastern Brazil, Paraguay and northern Argentina. Here, we generated a second Ananas reference genome from the bracteatus accession CB5 and resequenced numerous leading pineapple cultivars and wild Ananas species to explore the diversity and domestication history of pineapple, patterns of clonal propagation and signatures of human selection.
Results
Genome assembly and annotation of bracteatus pineapple accession CB5
The genome size of CB5 was estimated to be ~591 Mb by flow cytometry. We generated 26.9 Gb of reads from the PacBio RSII platform and ~100× Illumina short reads. Initial assembly using CANU yielded 809.6 Mb of assembled sequence. To eliminate redundant homozygous sequences, we developed a new algorithm, Pseudohaploid, that identifies and filters out heterozygous contigs based on whole-genome alignment. The resulting assembly was 513 Mb, with a contig N50 of 427 kb at 92.6% completeness and much reduced duplicated sequences (Supplementary Tables 1 and 2). Alignment of RNA sequencing (RNA-seq) assembled transcripts to the genome revealed 99.92% sequence identity (Supplementary Table 3). Additionally, 98.47% of Illumina reads were aligned to the genome, covering 99.51% of the genome (Supplementary Table 4).
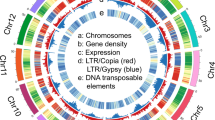
Contigs were corrected and scaffolded by high-throughput chromatin conformation capture (Hi-C) into 25 pseudo-chromosomes that anchored 456 Mb (88.8%) of the genome (Fig. 1, Supplementary Fig. 1 and Table 5). Overall, 29,412 putative protein-coding gene models were annotated (Supplementary Table 6). We identified 383.2 Mb of repetitive sequences, accounting for 74.7% of the assembled genome (Supplementary Fig. 2 and Supplementary Table 7). Kimura distances indicated a burst of long terminal repeat retrotransposon (LTR-RT) activity ~1.8 million years ago.

a–e, The rings indicate (from outermost to innermost) 25 chromosomes (a), gene density (b), transposable element abundance (c), gene copy number variation (d) and large-scale insertions compared to the F153 genome (e).
Improved assembly of pineapple F153 genome
The highest Gypsy LTR-RT content is concentrated near the centromeres in angiosperms18. The distribution of the Gypsy elements was plotted along the 25 pseudomolecules in the F153 genome (referred to as F153 v.6)19. Two peaks were observed in the linkage group (LG)01 (Supplementary Fig. 3), a chimeric pseudomolecule corresponding to two chromosomes in CB5 (Supplementary Fig. 4). There was a Gypsy peak at one end of LG24, while there was no Gypsy rich-region in LG25, which align to one chromosome in CB5. The misassembled LGs were corrected in the improved F153 genome assembly (referred to as F153 v.7), in which LG01 was separated into AccChr1 and AccChr24, while LG24 and LG25 were linked together into AccChr25.
Genomic basis of fiber production in CB5 pineapple
Both F153 and CB5 have eight CesA genes (Supplementary Table 8), grouped into those required for primary (CesA1, 3, 6 and 9) and secondary (CesA4, 7, 8 and 11) cell wall biosynthesis. The CB5 and F153 genomes share the same genes, but do not have orthologs of the CesA2, 5 and 10 genes in Arabidopsis (Supplementary Fig. 5). In F153 and CB5, genes for primary cell wall biosynthesis were all highly expressed in leaves, flowers and fruit, except for CesA9. Interestingly, the CesA4, 7 and 8, genes that are involved in secondary cell wall synthesis, were highly expressed in leaves of F153, while their expression levels were low in CB5 (Supplementary Fig. 6).
Lignin is the second major component of PALF. The full set of pineapple lignin biosynthetic genes were identified by sequence alignment to known Arabidopsis, rice and poplar lignin synthesis pathway genes20,21 (Supplementary Dataset 1). CB5 and F153 had 24 and 21 candidate genes for lignin biosynthesis, respectively. Three PAL genes in CB5 had higher expression in leaves than in F153 (Supplementary Fig. 7). Both COMT1 and CCOMT1 showed higher expression in CB5 than in F153 (Supplementary Fig. 8).
Anthocyanin biosynthetic genes
The variety bracteatus is often grown as an ornamental plant, partly because of the red color of its fruit. Anthocyanin biosynthesis shares the phenylpropanoid pathway with lignin biosynthesis in its first steps. Anthocyanin biosynthetic genes were identified in F153 and CB5 (Supplementary Table 9). The size of the CB5 gene families encoding anthocyanin biosynthetic genes was larger than in F153 (22 versus 17). Early biosynthetic genes in the pathway such as CHS, CHI, F3H and F3′H were expanded in CB5. Both F153 and CB5 did not have FLS and ANS orthologs, indicating the existence of their isozyme genes, which may take over their functions.
Sugar metabolism genes
Sweetness is a major fruit quality trait. In pineapple fruit, sucrose is the main sugar followed by glucose and fructose22. Multiple enzymes participate in their biosynthesis, transportation and metabolism with no difference in gene number between CB5 and fruit pineapple, including sucrose-phosphate synthases, sucrose-phosphate phosphatases, sucrose synthases, invertases, sucrose transporters (SUTs), sugars-will-eventually-be-exported transporters (SWEETs) and monosaccharide transporters23,24,25,26 (Supplementary Table 10). In CB5, SUTs were constantly expressed at a low level during fruit maturation (Supplementary Table 11), while two of SUT genes (AccSUT1 and AccSUT3) were highly expressed in mature fruit in MD2 (Supplementary Table 12). More SWEET genes were expressed in the late developmental stage of fruit in MD2 than in CB5 (Supplementary Tables 13 and 14). More interestingly, AccSWEET13 was located in the region of the F153 genome where a selective sweep was detected (see below). These results partially explain why MD2 accumulates more sugar in its fruit than CB5.
Bromelains
We identified 61 and 47 cysteine proteinase (CP)-type bromelains in F153 and CB5, respectively. Meanwhile, we identified 28 CPs in Amborella, 36 in Arabidopsis, 34 in papaya, 25 in grape, 50 in poplar, 47 in sorghum and 50 in rice (Supplementary Table 15). These CPs are divided into nine subfamilies (Supplementary Fig. 9). Subfamily VI had the most members, while subfamilies V, VIII and IX had fewer members, with no more than three members in each species. An expansion was observed in subfamily VI in all the selected species, especially F153. Bromelains of pineapple belong to this subfamily, and the expansion may result in a high production of bromelains. The majority of CPs showed constant expression patterns during fruit ripening (Supplementary Tables 16 and 17). Some genes such as AccCEP3 and AccPAP25 showed dynamic expression patterns at a high level during the mature stage of fruit ripening (Supplementary Table 16). In subfamily VI of CB5, only two genes displayed expression in the tissues studied. AcbPAP10 was found to be expressed in flowers, fruit and leaves, while AcbPAP17 was only expressed in flowers. More highly expressed genes in subfamily VI were detected in F153. AccPAP3 and AccPAP4 exhibited very high expression at late stages of fruit development, perhaps contributing to fruit ripening.
Patterns of genome-wide variation in pineapple
We selected 89 Ananas accessions for whole-genome resequencing, including 67 accessions of A. comosus var. comosus, nine accessions of var. bracteatus, two accessions of var. erectifolius, nine accessions of the wild var. microstachys, and two accessions from Pitcairnia gracilis and P. punicea as outgroups (Fig. 2 and Supplementary Table 18). The var. comosus samples include representatives of the three historical cultivars ‘Queen’, ‘Smooth Cayenne’, and ‘Singapore Spanish’, associated with the pantropical diffusion of the pineapple in historical times. Other important var. comosus cultivars analyzed include ‘Pérola’ and several cultivars from north-western South America and Central America, as well as admixed breeding lines and cultivars of unknown origin. We also included cultivated clones of A. comosous var. bracteatus, and the proposed wild progenitor of pineapple A. comosus var. microstachys27 (Supplementary Table 19).

Image adapted from Mapbox and OpenStreetMap
Accessions are represented by the same color code as in the admixture analysis (Fig. 3). Accessions presenting less than 10% admixture are represented by the color of their cluster. For admixed accessions, the color that comes first in importance in the admixture diagram is indicated in the wider concentric circle, and the second in importance in the smaller concentric circle: light blue, Smooth Cayenne cluster of var. comosus; dark blue, Queen cluster; violet, Singapore Spanish cluster; yellow, fourth cluster (Mordilona-related cultivars); red, var. erectifolius cluster; orange, var. bracteatus cluster; green, var. microstachys cluster. Cultivars bearing the ‘piping’ spine-suppressor gene are identified by a red P-letter.
We identified 7,428,400 high-quality SNPs and <10-base pair (bp) insertions/deletions (indels) across the 89 accessions. Cultivated pineapple yielded 3.2 million variants, including a large number of rare alleles (1.6 million with <5% minor allele frequency). This high proportion of rare alleles was probably a product of unique somatic mutations expected with clonal propagation. Nearly half (3,526,071, 47.5 %) of the SNPs were located in intergenic regions. The proportions of SNPs from genic regions assigned to exon, intron and UTR regions were 17.1%, 31.8% and 3.6% respectively (Supplementary Fig. 10). A total of 12,806 SNPs with predicted effects on gene functions, such as altering start codons, stop codons or splice sites, were discovered. Overall, 7,084 SNPs introduced stop codons, 725 SNPs disrupted stop codons, 750 SNPs disrupted start codons and 4,252 SNPs affected splicing donor or acceptor sites (Supplementary Table 20). With regards to the SNPs located in the exon regions, the number of nonsynonymous SNPs is less than synonymous SNPs for each accession (Supplementary Table 21).
The nonsynonymous and synonymous site frequency spectra were examined for cultivars Smooth Cayenne, Queen and Singapore Spanish (Supplementary Fig. 11). Smooth Cayenne has an excess of low-frequency nonsynonymous variants compared to synonymous variants, indicating purifying selection. For Queen and Singapore Spanish, there was an unusual excess of variants at an intermediate frequency for both nonsynonymous and synonymous sites. This was probably because Queen and Singapore Spanish had a higher abundance of heterozygous genotypes per SNP position (Supplementary Fig. 12).
Origin, population structure and genomic ancestry of pineapple
We used a subset of 665,162 quality-filtered SNPs to explore relationships between the genomes of divergent Ananas taxa and cultivars. Phylogenetic trees and networks estimated with RAxML28 and SplitsTree29 separated accessions of the varieties microstachys, bracteatus, erectifolius and comosus, and accessions from major cultivars within the latter. Seven mislabeled cultivars were corrected, and six cultivars were assigned to correct cultivars that could not be classified previously (Supplementary Table 18). For Singapore Spanish and Selangor Green, we confirmed and completed the history of their diffusion from Eastern Brazil to Asia. Similarly, the two var. erectifolius accessions were obtained from the same original collection through vegetative propagation (Fig. 3a, Supplementary Table 18 and Supplementary Figs. 13 and 14).

a, SplitsTree network for Ananas accessions excluding admixed samples. Green, variety microstachys; red, variety erectifolius; orange, variety bracteatus; yellow, variety comosus/Mordilona-related cultivars Cambray/Monte Lirio; purple, variety comosus/cultivar Singapore Spanish; light blue, variety comosus/cultivar Smooth Cayenne; dark blue, variety comosus/cultivar Queen; brown, genus Pitcairnia outgroups. A network of admixed samples is shown in Supplementary Fig. 14. b, MDS graphs of the studied Ananas accessions, with horizontal and vertical axes explaining 33.0% and 20.6% of the variance, respectively. Color code follows that in a and admixed A. comosus genotypes are indicated in gray. c, Ancestry results from ADMIXTURE under the K = 8 model supported by an examination of cross-validation errors (Supplementary Fig. 15).
To reduce the overrepresentation of Smooth Cayenne, Queen and Singapore Spanish/Selangor Green from population structure analyses, only five accessions were retained for each of the corresponding clusters (Fig. 3). Within variety comosus, the three groups that corresponded to the major cultivars Smooth Cayenne, Queen Singapore Spanish and a few cultivars derived from them, formed three clusters whose variation essentially originated from somatic mutations accumulated during the two to five centuries after their diffusion out of America. Smooth Cayenne and Queen dispersed from the Guianas, while Singapore Spanish and Selangor Green dispersed from the eastern coast of Brazil (south of Bahia)30. Common cultivars of A. comosus exhibit greatly reduced diversity (Fig. 3 and Supplementary Fig. 16), consistent with genetic bottlenecks from domestication. Nucleotide diversity was reduced more than 15 times in pineapple cultivars compared to their wild A. comosus var. microstachys progenitor (Supplementary Fig. 16), which is consistent with reduced diversity seen in multidimensional scaling (MDS) space and phylogenetic branch lengths (Fig. 3 and Supplementary Fig. 13) and high population differentiation (FST) among major cultivars (Supplementary Fig. 16). The typical accessions of var. bracteatus also formed a uniform group, where variation appeared to be related to somatic mutations; however, five less typical accessions showed admixture with cultivars of var. comosus. At a greater genealogical depth, composite likelihood estimation with TreeMix detected a predicted admixture event between var. bracteatus and its A. macrodontes parent (Supplementary Figs. 17,18).
The diversity and relatedness patterns were confirmed by MDS of genomic data (Fig. 3b and Supplementary Fig. 19). SplitsTree branch lengths involving the varieties microstachys, bracteatus, erectifolius and comosus were compared to those between major cultivars of comosus (Fig. 3a). Absolute genomic divergence (Dxy; Supplementary Fig. 16) was significantly greater among pairs of varieties, compared to major cultivars of comosus (P < 0.005). Dxy among Ananas varieties was on average 0.0059 (median 0.0046, s.e.m. 0.0007), which is within the range of expectations for recently derived species31,32.
Local genetic ancestry of hybrid accessions estimated with a Hidden Markov Model approach revealed a great diversity of patterns, including hybrids with large ancestry segments stemming from different modern comosus cultivars, and hybrids with small ancestry segments from different cultivars and taxa (Fig. 4). The presence of both large and small segments in hybrids indicated that admixture has affected the evolution of variety comosus over long time scales. Our most likely models of local ancestry were consistent with an average of 37 generations since the onset of admixture (range, 21–55) among the 22 var. comosus hybrids detected in our study. For the wild variety microstachys, individual estimates range from 107 to 612 generations, respectively. These numbers probably translated into several thousand years as perennial, primarily asexually propagated, plants.

Genomic regions with unusually high ancestry proportions from particular pineapple varieties are visible along chromosomes. The remaining chromosomes show similar results. For color coding labels see Fig. 3. All ancestry values sum to 1 for each genomic window.
Genomic signatures of mitotic selection and clonal propagation
Somatic mutation is a major driving force that shapes the domestication and diversification of clonally propagated plants33. One source of somatic mutation is the movement of transposable elements (TEs). We surveyed the presence/absence of variation of small DNA TEs in 89 resequenced accessions. DNA TEs were highly abundant, attaining copy numbers up to tens of thousands34 and they predominantly insert into or near gene-rich regions35. MITE-Hunter software predicted 4,614 TE junctions consisting of 2,286 Mutator, 1,156 hAT, 1,018 PIF/Harbinger, 128 CACTA and 26 unknown elements. The unique junction sites created by TE insertions were used as a reference for read mapping to assess the presence/absence of variation against the F153 reference. In total, 98,476 TE junctions were identified in the reference pineapple assembly: 46,613 Mutator, 23,634 PIF/Harbinger, 18,831 hAT, 4,091 CACTA, 254 unknowns and 12 junctions formed by two different TE superfamilies. Compared to the F153 reference genome, each accession exhibited a great number of unique TE junctions, which varied from 97% identity with F153 in Ac50 to 28% identity with F153 in Ac46c (Supplementary Table 22). The high variability of TE insertion sites in pineapple might be a driver for new traits via somatic mutation during domestication.
The process of mitotic recombination was predicted to lead to terminal homozygosity over time in tissues or organisms propagated exclusively through somatic means. This random generation of homozygosity in initially heterozygous tissues36 could provide selectable genetic variation by uncovering recessive alleles. Hence, we investigated this question by first finding all of the single-copy (SC) genes in the pineapple genome. SC genes were chosen so that identification of heterozygosity versus homozygosity for any given chromosomal location could be ascertained without the confusion generated by paralogs. The final 10,439 SC genes were distributed randomly across the genome (Supplementary Fig. 20).
Terminal runs of homozygosity at the ends of LGs were frequently detected, especially in Singapore Spanish, including LG01, 03, 04, 08, 11, 14, 15, 20, 22 and 24 (Fig. 5, and Supplementary Fig. 21). Some of this homozygosity covered the entire region, from the site of the mitotic recombination to the end of the chromosome, as expected37. The presence of such terminal runs of homozygosity indicated an early occurrence (and possible selection and fixation) of associated mitotic mutations in the domestication process. In Smooth Cayenne and Queen, short terminal homozygosity was detected sporadically in LG03 and 23, and was likely to be a product of mixed clonal and sexual reproduction. Notably, the overall level of heterozygosity (and the lack of all but a tiny number of homozygous regions) in the wild relatives of pineapple indicated that these populations were prodigious outcrossers.

Levels of heterozygosity were plotted for every 100 bp across linearly concatenated genes and across 50 accessions with low levels of admixture. A heatmap of heterozygosity is plotted where white indicates no heterozygosity and dark purple indicates high heterozygosity. The vertical dotted red line indicates predicted centromere region as determined by Gypsy LTR retrotransposon abundance. The dendrograms on the left indicate clustered heterozygosity landscapes among varieties.
Selective sweeps and selection on sexually derived forms during pineapple domestication
Genomic regions of selection during pineapple domestication were identified based on drastic reductions in nucleotide diversity (π) in cultivated accessions compared to wild lines (πc/πw) in sliding windows across the genome. Diversity within variety microstachys was used for estimating πw. Cultivars with evidence of admixture were omitted from selection scans and πc was calculated within and across each of the four cultivars. Candidate swept regions were further narrowed using an cross-population composite likelihood ratio test (XP-CLR) based approach to model the allele frequency spectrum differences between cultivated and wild accessions. This approach identified 25 putative domestication sweeps across the pineapple genome with sizes ranging from 150 kb to 1.2 Mb (Supplementary Table 23). Swept regions collectively spanned 11.9 Mb (~3.1% of the genome) which was substantially lower than patterns observed in sexually propagated crops such as tomato (186 domestication sweeps totaling 64.5 Mb, ref. 38) and soybean (121 sweeps totaling 53 Mb, ref. 39). Pineapple also had fewer putative selective sweeps than other clonally propagated crops such as cassava, which contains signatures of 224 sweeps40.
Swept regions in pineapple encompassed 392 genes with enrichment in stress response pathways (FDR = 2.1 × 10−3), but no obvious enrichment in genes previously characterized in other species as responsible for domestication-related traits. To narrow this list of candidate domestication genes, we surveyed gene expression changes in a high-resolution series of developing pineapple fruit. The strongest sweep was a 225 kb region at the beginning of LG03 with a 400-fold reduction in diversity across cultivated accessions compared to the wild var. microstachys (Fig. 6a). This sweep was in the top 5% based on XP-CLR that indicated low FST and highly negative Tajima’s D (Fig. 6b,c). Although the sweep on LG03 overlaps with a long run of terminal homozygosity (Supplementary Fig. 21.3), it was much narrower than the homozygosity run (Fig. 5). The putative sweep contains nine genes, including a pair of tandemly duplicated bromelain inhibitors (AccBI1 and AccBI2) with fruit-specific expression patterns (Fig. 6d). Bromelains coordinating with bromelain inhibitors are supposed to play an important role in pineapple fruit ripening41,42. Bromelain inhibitor is posttranslationally inactivated during fruit ripening, leading to a significant increase in bromelain activity, thus enhancing tissue proteolysis, softening and degradation41. AccBI1 and 2 are the most highly expressed genes during fruit ripening, with reads per kilobase per million mapped reads (RPKMs) as high as 443,814. Expression of AccBI1 oscillates down to 0 RPKMs in some ripening stages, suggesting strict transcriptional control. Pineapple F153 contained 61 bromelain genes, including two that have expression patterns that correlate inversely with AccBI1 and AccBI2 (Fig. 6d).

a, Top: Heat maps showing the distribution of domestication sweeps (top 5% πw/πc) for the four cultivars. Bottom: A putative swept region at the end of LG03 containing AccBI1 and AccBI2. πw/πc across all cultivars is plotted using a sliding window of 0.5 Mb with 0.1 Mb shift. b, Genetic distance (FST) between the Smooth Cayenne, Queen, Singapore Spanish and Mordilona-related clusters for the 6.5 Mb of LG03. Mean FST values are plotted in sliding windows of 50 kb with 25 kb step size. c, Tajima’s D values for the four combined cultivar clusters (var. comosus) and wild (var. microstachys). Mean Tajima’s D values are plotted in sliding windows of 50 kb with a step size of 25 kb. d, Left: Pineapple fruit at select stages from a fruit ripening series (stages 1, 2, 7 and 8). Right: Maximum likelihood phylogeny of bromelain genes with log2 transformed RPKMs of expression in fruit, flower, leaf and root tissue.
Candidate genes for self-incompatibility in pineapple
In contrast to A. macrodontes, A. comosus and its botanical varieties are self-incompatible, with exception of some clones of var. bracteatus. However, self-incompatibility tends to be stronger in var. comosus, compared to the other varieties that were not domesticated for fruit, which is probably a result of selection under domestication to reduce seed set in fruit43. Gametophytic self-incompatibility (GSI) operated in cultivated pineapple44, similar to S-RNase-based GSI, in which the S-locus encodes a single S-RNase and multiple S-locus F-box (SLFs/SFBs) proteins45. When self-pollinated in SI species, none of the SLFs/SFBs interact with their own S-RNase, which breaks down pollen tube RNA to inhibit growth; when cross-pollinated, some members of paternal SLFs/SFBs interact with maternal S-RNase, which allows pollen tube growth45. To search for genes potentially involved in pineapple GSI, we first identified S-RNase and SLF/SFB homologs in the pineapple reference genome based on sequence homology. These candidates were then tested for their selection history in diverse pineapple varieties. Twenty-five genes passed the criteria (Supplementary Table 24).
We examined the transcript levels of the 25 SI candidate genes in androecium and gynoecium, respectively (Supplementary Table 24). Two S-RNase genes (Aco001100 and Aco004758), the potential female specificity determinants in GSI, were highly expressed in both tissues but with stronger expression in androecium. For the SLFs/SFBs, two of the genes (Aco00868 and Aco011265) showed much stronger expression in androecium than gynoecium, while two of them (Aco015095 and Aco021447) showed the opposite expression bias. Expression of four genes was not detected. The remaining 13 genes showed similar expression in both tissues. Among the six genes showing differential expression in androecium and gynoecium, the ribonuclease T2 family member Aco001100 and F-box family member Aco00868 are tightly linked on LG02, only 1.8 Mb apart, and they are the most likely candidates for self-incompatibility in A. comosus var. comosus Smooth Cayenne F153. Furthermore, Aco001100 was tightly linked with two other SLF/SFB genes (Aco001170 and Aco012216), a characteristic of RNase-based GSI45. In CB5, the ribonuclease T2 family member CB5.v30014510 was the orthologous gene of Aco001100 and linked with only one SLF/SFB family gene (CB5.v30013780), which is not a functional SI system (Supplementary Table 25).
Discussion
The chromosomal-level assembly of the bracteatus pineapple CB5 genome sheds more light on the biology and evolution of Ananas. To overcome the problem of assembling a heterozygous genome, we have developed an algorithm, Pseudohaploid, that identifies and filters out heterozygous contigs by searching for redundant homologous sequences. Facilitated by long-read sequencing technology, we identified and located more repetitive sequences in the CB5 genome, providing comprehensive resources to study genome evolution driven by TEs. In addition, the misassembled pseudo-chromosomes in the F153 genome were corrected with the assistance of the CB5 genome. Comparison between these two pineapple genomes revealed genomic components associated with fiber production, color formation, sugar accumulation and fruit maturation. It also provided an additional line of evidence to verify SI candidate genes in F153.
Our genomic data indicated the presence of a continuum of divergence, ranging from low divergence among groups of modern pineapple cultivars to moderate divergence among closely related taxa, such as the cultivated botanical varieties comosus, bracteatus and erectifolius, to a much greater divergence in the wild var. microstachys, which exhibits Dxy values normally seen among recently diverged species34,35. In contrast, FST reflected low diversity in major cultivars, consistent with the domestication bottleneck. Greatly reduced diversity in cultivars relative to their wild progenitor pointed to the severe domestication bottleneck experienced by this clonally propagated crop, and an excess of intermediate frequency alleles in two major groups of modern cultivars indicated the potential for clonal propagation to mask recessive deleterious variants in heterozygotes46.
Admixture analysis of A. comosus cultivars revealed widespread admixture genotypes in 39 (44%) out of 89 accessions, detected in every cultivar and botanical variety. With regard to evolutionary processes operating during pineapple domestication, our results indicated a role for both ancient and recent admixture and thus sexual recombination and subsequent artificial selection in most cultivars. This was supported by the dearth of terminal runs of homozygosity along the chromosomes of pineapple in two out of three major cultivars. This indicated that both sexual recombination and somatic mutations have contributed to the phenotypic diversity seen in Ananas. It appears that the true degree of genomic complexity of germplasm used in 20th-century breeding programs was previously underestimated.
Early pre-Columbian pineapple cultivars were selected for low fruit fiber content and reduced seed production through lower fertility and self-incompatibility47. The pineapple genome contains 25 selective sweeps, much fewer than those in sexually reproducing crops such as the 121 in soybean39 and the 186 in tomato38, supporting the conclusion of a mixture of sexual and asexual selection for pineapple. The strongest selective sweep included a pair of tandemly duplicated bromelain inhibitors previously suggested as important regulators of pineapple fruit senescence and ripening in this nonclimacteric fruit41. Gene duplications are the drivers of evolutionary innovation and have been linked to domestication traits in tomato48 and black raspberry49. The bromelain inhibitor gene duplication event was probably selected in pre-Columbian varieties.
Our initial working hypothesis was that somatic mutations were the main source of variation for domestication in pineapple. Our efforts to identify mitotic selective sweeps were fruitful in the cultivar Singapore Spanish as shown by extensive terminal runs of homozygosity, the hallmark of mitotic selection. However, this hypothesis was rejected for two major cultivars, Smooth Cayenne and Queen, although sporadic terminal runs were detected in two chromosomes, indicating long term clonal reproduction punctuated by sexual reproductions. Meiosis in pineapple generally occurs once every 2 years, while recombination in mitotic cells is continuous but at very low rates, about 104 to 105 times less frequent than meiotic recombination50,51. At such a low frequency and the nature of clonal production, only mitotic recombination events that occurred at the single cell stage of the reproductive tissues, crowns, suckers, slips and shoots, could be transmitted to progenies and preserved to be detectable. Moreover, one sexual recombination could interrupt terminal runs of homology that had formed and been maintained over thousands of years.
The one-step operation hypothesis, wherein domestication and early improvement are an immediate outcome of a single clonal propagant might be responsible for the selection of some long-lasting clones in some lineages. Genomic analyses, particularly those searching for terminal runs of homozygosity, can be applied to other clonally propagated crops to elucidate the extent of sexual recombination versus vegetative descent in their domestication history. The coexistence of sexual recombination and the one-step operation among different cultivars might be common in clonally propagated crops. Some controversial hypotheses were rejected in the past, but later validated entirely or partly by innovative new technologies or enhanced resolution of evidence, including the ‘dominance’ versus ‘overdominance’ hypotheses for heterosis and Lamarck’s theory of the inheritance of acquired characteristics. The hypothesis of the one-step operation for the domestication of clonally propagated crops thus seems to be one of them.
Methods
Genome assembly and annotation overview
The CB5 chromosome level assembly takes advantage of PacBio Single-Molecule Real-Time technology and Hi-C based scaffolding methods, followed by Illumina short read-based polishing. Briefly, ~50× coverage of subreads were generated with the PacBio RSII platform and ~60× coverage by short reads was generated on the Illumina HiSeq X10 platform. The initial contig-level assembly was accomplished with CANU v.1.7 and heterozygous contigs were removed using our newly developed algorithm, Pseudohaploid (details in Supplementary Note). Further, Illumina short reads were recruited to polish the PacBio assembled genome using Pilon v.1.18 with parameters: —diploid —threads 6 —changes —tracks —fix bases —verbose —mindepth 4. Hi-C libraries were created from tender leaves of CB5 at BioMarker Technologies Corporation as previously described52. Chimeric fragments representing the original cross-linked fragments were then processed into paired-end sequencing libraries and sequenced on the Illumina HiSeq X10 platform. The paired-end reads were uniquely mapped onto the draft assembly and misjoined contigs were corrected by detecting abrupt long-range contact patterns using the 3D-DNA pipeline53. The Hi-C corrected contigs were further linked into 25 pseudo-chromosomes using the ALLHiC pipeline54.
We annotated the CB5 chromosomal-level assembly using a series of programs, which are fully described in the Supplementary Note. Briefly, the MAKER2 pipeline55 was used to annotate the protein-coding proteins by integrating homologous proteins, RNA-seq assembled transcripts and the results of ab initio gene predictors. In addition, repetitive sequences were predicted by RepeatMasker56 and we also predicted miRNAs by searching for candidates that matched to public miRNAs.
Identification of lignin and anthocyanin biosynthetic genes
Protein sequences for lignin biosynthetic genes of Arabidopsis thaliana, Populus trichocarpa and Oryza sativa20 were used to align with the protein sequences of F153 and CB5 using BLASTP with a cut off e value ≤ 1 × 10−10 and coverage ≥ 0.75. For anthocyanin biosynthetic genes identification, protein sequences of Arabidopsis thaliana57 were used to align with the protein sequences of F153 and CB5. Pfam was adopted to identify conserved domains for these candidate genes. Finally, we used MEGA 7 to draw a phylogenetic tree to confirm the expected relatedness of anthocyanin biosynthetic genes. The phylogenetic tree was inferred using the neighbor-joining method. The alignment was done by MUSCLE v.3.8.31 with default substitution model and 1,000 bootstraps.
Identification of CP subfamily C1 genes
Gene models of all the species used in this study were downloaded from Phytozome v.11.0 and v.12.0 (https://phytozome.jgi.doe.gov/pz/portal.html). The conserved domain of cysteine peptidase subfamily C1, peptidase_C1 domain (PF00112) was downloaded from the pfam database (http://pfam.xfam.org/). HMMER were used to search against protein databases for each species to identify proteins containing peptidase_C1 domain with threshold of e value ≤ 1 × 10−5. We further confirmed those proteins by searching their domains against Conserved Domains Database from NCBI. Full-length proteins were aligned by MUSCLE v.3.8.31 with default parameters. Phylogenetic trees were constructed by Smart Model Selection PhyML v.3.0 with statistical criteria (AIC)58 and were further edited with MEGA 7.
Variant calling and annotation
A total of 4.7 billion 150–250 bp paired-end Illumina reads yielded an average coverage of 17.5× per accession (Supplementary Table 19). This read depth is similar to other large-scale resequencing projects39,59,60. Raw reads were quality-filtered to remove adapters and low-quality bases (Q < 30). Quality-filtered reads were aligned against the unmasked F153 pineapple draft genome (v.6) using Bowtie2 (v.2.2.6) (ref. 61) with default parameters. Read mapping rates for cultivated accessions ranged from 82.3% to 94.5% with an average of 87.6% compared to 69.4–84.2% for wild Ananas and related species. Variant detection was performed using the genome analysis toolkit (GATK; v.3.5-0-g36282e4)62 following the best practices workflow for variant discovery. Resulting BAM files were locally realigned using IndelRealigner to remove erroneous mismatches around small-scale insertions and deletions. Variants were called in each accession separately using HaplotypeCaller and individual genome Variant Call Format (gVCF) files were merged using GenotypeGVCFs. This two-step approach includes quality recalibration and regenotyping in the merged vcf file, ensuring variant accuracy. The flag –output_mode EMIT_ALL_CONFIDENT_SITES was used to provide read coverage for each position in the reference genome (including invariant sites), allowing regions with no alignment to be filtered out before population genetics analysis. A total of 9,342,943 raw variants were called by GATK. These variants were filtered to remove sites with quality scores less than 100, minimum allele frequency <0.02, and missing data >10%. The final vcf file contains 7,428,400 high-quality SNPs and indels (<10 bp) across the 89 accessions. Variants were annotated using SNPEff (v.4.2) (ref. 63) with pineapple gene models19.
Nonsynonymous and synonymous site allele frequency analysis
SNPEff annotated nonsynonymous and synonymous sites were used for site allele frequency analysis. Only SNPs from Smooth Cayenne, Queen and Singapore Spanish accessions were used because of their higher sample sizes. The bracteatus botanical variety was used as an outgroup to polarize ancestral and derived variants. Allele frequency was estimated separately for each population and SNP positions in more than 70% of each population’s sample size were analyzed. Because each SNP position had different sample sizes, we used the hypergeometric distribution to down-sample the jth SNP positions’ observed sample size, Nj, to the most minimum downsampled sample size across all SNP positions, n (ref. 64). Thus, the allele frequency for a down-sample size of n was calculated as:
where pi,2n corresponds to the allele frequency of i derived alleles in a diploid 2n population, dj is the observed derived allele count for site j and k is the total number of SNP positions.
Linkage disequilibrium (LD) analysis
The final vcf file was used for genome-wide LD calculation using individuals with nonadmixed evolutionary histories (Fig. 3c). Using PLINK (v.1.90b3.46) (ref. 65), LD between SNP pairs within the same LG was calculated using a 5 Mb window and limiting to SNPs that were not more than 499,999 SNPs apart. SNPs within LGs that were at least 10 Mb in length were analyzed. SNP pairs were then grouped into 10 kb bins to average the R-squared correlation (r2) between SNPs. SNP pairs with r2 values <0.1 were omitted. The LOESS method of line of best fit was fitted using the average r2 value per bin.
RNA-seq analysis
The trimmed paired- or single-end reads of each sample were aligned to the repeat-masked F153 genome v.6 (ref. 19), using TopHat (v.2.0.9) under default settings66. The normalized RPKM value of each sample was estimated by Cufflinks v.2.2.1, followed by Cuffnorm v.2.2.1 (ref. 66) using default settings with the pineapple gene model annotation (v.6)19.
Admixture, phylogenetics and population structure analyses
SNPs from whole-genome resequencing were filtered using vcftools v.0.1.13 (ref. 67) with minimum allelic count = 2, maximum missing data = 15%, minimum coverage = 4, SNP quality > 20, retaining only biallelic variants and no indels. A maximum likelihood-based tree of Ananas accessions was built using RAxML v.8.2 (ref. 28) with 100 bootstrap replicates to determine branch support, and a phylogenetic network was constructed using the neighbor-net method implemented within SplitsTree68. Additionally, MDS was used for model-free clustering of Ananas accessions. Nucleotide diversities (π), Dxy and FST were estimated for all taxa and major cultivars. Nucleotide diversities in wild and cultivated forms were used as a simple, robust approach to document genetic bottlenecks experienced during domestication; we refrained from demographic modeling of cultivar history using diffusion- or coalescent-based approaches due to the widespread presence of clonally propagated genotypes in the sample set, which would violate basic modeling assumptions. Instead, we explored key aspects of cultivar history by analyzing genomic patterns of ancestry. Genome-wide ancestry and admixture were estimated with ADMIXTURE v.1.23 (ref. 69). For variety comosus cultivars, this analysis used only the five samples with the highest coverage for each cultivar to avoid biases due to the overrepresentation of clonal samples. Population splits and past admixture events were further explored using the TreeMix approach70. Local ancestry along confidently assembled pineapple chromosomes was estimated with a Hidden Markov Model approach modified from Price et al.71 following Wegmann et al.72, making use of the RASPberry software. The most likely number of generations since admixture was estimated for each admixed individual by this method based on likelihood ratio tests. Except where noted, statistical analyses were carried out in R.
Detecting putative selective sweeps
Regions of selection during pineapple domestication were identified based on drastic reductions in π of cultivated accessions compared to wild lines (πc/πw) in sliding windows across the genome. Variety microstachys is the likely progenitor of cultivated pineapple, so diversity within this group was used for estimating πw. Cultivars with evidence of admixture were omitted from selection scans and πc was calculated within and across each of the four cultivars. To reduce false positives due to drift, the four cultivated groups were combined into a single pool before analysis. Nucleotide diversity (π) was calculated using the –window-pi-step tool in vcftools (v.0.1.12) (ref. 67). Invariant sites were included in calculations of π to remove any inflations in estimation related to missing data. Nucleotide diversity was calculated in sliding windows of 50 kb with a 10-kb step size to identify sweeps and in sliding windows of 10 kb with a 2.5 kb step to narrow candidate genes. The top 5% of πw/πc values were considered swept regions. Adjoining swept windows were merged into blocks, producing a final set of 25 swept regions.
Candidate swept regions were further narrowed using an XP-CLR based approach to model the allele frequency spectrum differences between cultivated and wild accessions73. The following parameters were used for XP-CLR scans across each chromosome: window of 0.005 cM, window size of 1,000 bp, a maximum of 100 SNPs per grid, and a corrLevel of 0.7. The genetic distance between adjacent variants was calculated using the ultra high-density genetic map used to anchor the F153 pineapple draft genome19. Comparisons were made between var. comosus cultivars showing no evidence of recent admixture and var. microstachys. Regions with the top 10% XP-CLR scores were merged as putative swept regions and only regions overlapping with high πw/πc values were kept to remove false positives.
FST was estimated with the Weir and Cockerman approach using four-way comparisons of the cultivar clusters (Smooth Cayenne, Queen, Singapore Spanish and Mordilona-related) in the program SFselect (https://github.com/rronen/SFselect). Tajima’s D was calculated in sliding windows of 50 kb with 25 kb overlap using a suite of programs in vcftools (v.0.1.12)67.
Identification and mapping of transposable element insertion sites
MITE-Hunter74 was used with default parameters to search the pineapple genome assembly for candidate small DNA TEs. MITE-Hunter outputs were manually examined to select bona fide TEs based on their flanking sequences, TIR and TSD characteristics and classified into families following the convention used by Han et al.75. The terminal 50 bases of TEs were used as blast queries to identify TE junctions in the pineapple genome. Blast results were filtered to retain hits that have minimum alignment length of 15 bp and are within 10 bp of the TE termini. Multiple blast hits within a window of 30 bp were merged and considered as one junction. These blast hits mark unique TE junctions in the reference pineapple genome. The presence/absence of TE junctions were scored in the 89 accessions based on mapping of Illumina reads from the accessions to the reference pineapple genome. A site was marked as present in an accession when at least one read covered 20 bp upstream and downstream of the TE junction.
Identifying tracks of homozygosity
Tracks of homozygosity were identified using the 50 resequenced varieties with the highest coverage. Long tracts of homozygosity are usually genomic regions having consecutive genes without heterozygosity. We first identified all the tracts of homozygosity spanning more than three consecutive genes. In the rare cases where two tracts of homozygosity were interrupted by only one gene with only one heterozygous SNP, the three parts were still joined into longer tracts of homozygosity. Then, the number of homozygosity tracts spanning six or more consecutive genes were counted and the summation of homozygosity tract numbers among the 38 cultivars was displayed, with 100 genes as a bin size. Red dotted lines mark predicted centromere locations, based on the observation that by far the highest density of LTR retrotransposons is always found to be flanking the centromere in all studied angiosperm genomes18.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The bracteatus CB5 genome and annotation, the revised version of F153 genome, and pineapple RNA-seq data have been submitted to EBI-ENA under the study PRJEB33121. Quality filtered Illumina reads for the 89 resequenced pineapple genomes have been deposited in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA389669.
Code availability
We developed a new algorithm, Pseudohaploid, that identifies and filters out heterozygous contigs based on whole-genome alignment. This method can be run stand-alone with any assembler and is available open-source at http://github.com/schatzlab/pseudohaploid. Other public available open-source and custom software/code used to analyze the data in this study are listed in the Nature Research Reporting Summary.
References
Zohary, D. Unconscious selection and the evolution of domesticated plants. Econ. Bot. 58, 5–10 (2004).
Bertoni, M. S. Contributions à l'étude Botanique des Plantes Cultivées (Ex Sylvis. Puerto Bertoni, Alto Parana. PY, 1919).
Byers, D. S. Prehistory of the Tehuacan Valley (Univ. of Texas Press, 1967).
Coppens d’Eeckenbrugge, G. & Duval, M.-F. The domestication of pineapple: context and hypotheses. Pineapple News 16, 15–27 (2009).
Coppens d’Eeckenbrugge, G., Uriza Avila, D. E., Rebolledo Martínez, A. & Rebolledo Martínez, L. The Cascajal Block: another testimony of the antiquity of pineapple in Mexico? Pineapple News, 18, 47–48 (2011).
Baker, K. F. & Collins, J. L. Notes on the distribution and ecology of Ananas and Pseudananas. Am. J. Bot. 26, 697–702 (1939).
Duval, M. F., Coppens d’Eeckenbrugge, G., Ferreira, F. R., Bianchetti, L. D. B. & Cabral, J. R. S. First results from joint EMBRAPA-CIRAD Ananas germplasm collecting in Brazil and French Guyana. Acta Hortic. 425, 137–144 (1997).
Asim, M. et al. A review on pineapple leaves fibre and its composites. Int. J. Polym. Sci. 2015, 950567 (2015).
Beltrame, K. K. et al. Adsorption of caffeine on mesoporous activated carbon fibers prepared from pineapple plant leaves. Ecotox. Environ. Safe. 147, 64–71 (2018).
Abd Razak, S. I., Sharif, N. F. A., Nayan, N. H. M., Muhamad, I. I. & Yahya, M. Y. Impregnation of poly(lactic acid) on biologically pulped pineapple leaf fiber for packaging materials. Bioresources 10, 4350–4359 (2015).
Costa, L. M. M. et al. Bionanocomposites from electrospun PVA/pineapple nanofibers/Stryphnodendron adstringens bark extract for medical applications. Ind. Crop. Prod. 41, 198–202 (2013).
Hazarika, P. et al. Development of apparels from silk waste and pineapple leaf fiber. J. Nat. Fibers 15, 416–424 (2018).
Benzing, D. H. Bromeliaceae: Profile of an Adaptive Radiation. (Cambridge Univ. Press, 2000).
Givnish, T. J. et al. Adaptive radiation, correlated and contingent evolution, and net species diversification in Bromeliaceae. Mol. Phylogenet. Evol. 71, 55–78 (2014).
Barbará, T., Martinelli, G., Fay, M., Mayo, S. & Lexer, C. Population differentiation and species cohesion in two closely related plants adapted to neotropical high‐altitude ‘inselbergs’, Alcantarea imperialis and Alcantarea geniculata (Bromeliaceae). Mol. Ecol. 16, 1981–1992 (2007).
Wendt, T., Canela, M. B. F., de Faria, A. P. G. & Rios, R. I. Reproductive biology and natural hybridization between two endemic species of Pitcairnia (Bromeliaceae). Am. J. Bot. 88, 1760–1767 (2001).
Palma-Silva, C. et al. Sympatric bromeliad species (Pitcairnia spp.) facilitate tests of mechanisms involved in species cohesion and reproductive isolation in Neotropical inselbergs. Mol. Ecol. 20, 3185–3201 (2011).
Bennetzen, J. L. & Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530 (2014).
Ming, R. et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 47, 1435–1442 (2015).
Hamberger, B. et al. Genome-wide analyses of phenylpropanoid-related genes in Populus trichocarpa, Arabidopsis thaliana, and Oryza sativa: the Populus lignin toolbox and conservation and diversification of angiosperm gene families. Can. J. Bot. 85, 1182–1201 (2007).
Ehlting, J. et al. Global transcript profiling of primary stems from Arabidopsis thaliana identifies candidate genes for missing links in lignin biosynthesis and transcriptional regulators of fiber differentiation. Plant J. 42, 618–640 (2005).
Adisak, J. & Jintana, J. in VII International Pineapple Symposium Vol. 902 (eds Abdullah, H. et al.) 423–426 (International Society for Horticultural Science, 2011).
Jiang, S. Y. et al. Sucrose metabolism gene families and their biological functions. Sci. Rep. 5, 17583 (2015).
Ruan, Y. L. Sucrose metabolism: gateway to diverse carbon use and sugar signaling. Ann. Rev. Plant Biol. 65, 33–67 (2014).
Büttner, M. The monosaccharide transporter(-like) gene family in Arabidopsis. FEBS Letters 581, 2318–2324 (2007).
Doidy, J. et al. Sugar transporters in plants and in their interactions with fungi. Trends Plant Sci. 17, 413–422 (2012).
Coppens d’Eeckenbrugge, G., Sanewski, G. M., Smith, M. K., Duval, M.-F. & Leal, F. in Wild Crop Relatives: Genomic and Breeding Resources (ed. Kole C.) 21–41 (Springer, 2011).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Huson, D. H. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73 (1998).
Coppens d’Eeckenbrugge, G., Duval, M.-F., Leal, F. in Genetics and Genomics of Pineapple (ed. Ming, R.) 1–25 (Springer, 2018).
Chapman, M. A., Hiscock, S. J. & Filatov, D. A. Genomic divergence during speciation driven by adaptation to altitude. Mol. Biol. Evol. 30, 2553–2567 (2013).
Cruickshank, T. E. & Hahn, M. W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014).
Eckert, C. G. in Ecology and Evolutionary Biology of Clonal Plants (eds Stuefer, J.F. et al.) 279–298 (Springer, 2002).
Chen, J., Hu, Q., Zhang, Y., Lu, C. & Kuang, H. P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res. 42, D1176–D1181 (2014).
Wessler, S. R., Bureau, T. E. & White, S. E. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 5, 814–821 (1995).
Stern, C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics 21, 625 (1936).
LaFave, M. C. & Sekelsky, J. Mitotic recombination: why? when? how? where? PLoS Genet. 5, e1000411 (2009).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226 (2014).
Zhou, Z. et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotechnology 33, 408–414 (2015).
Ramu, P. et al. Cassava HapMap: Managing genetic load in a clonal crop species. Preprint at bioRxiv https://doi.org/10.1101/077123 (2016).
Neuteboom, L. W., Matsumoto, K. O. & Christopher, D. A. An extended AE-rich N-terminal trunk in secreted pineapple cystatin enhances inhibition of fruit bromelain and is posttranslationally removed during ripening. Plant Physiol. 151, 515–527 (2009).
Raimbault, A. K., Zuily-Fodil, Y., Soler, A., Mora, P. & de Carvalho, M. H. C. The expression patterns of bromelain and AcCYS1 correlate with blackheart resistance in pineapple fruits submitted to postharvest chilling stress. J. Plant Physiol. 170, 1442–1446 (2013).
Coppens d’Eeckenbrugge, G., Duval, M.-F. & Van Miegroet, F. Fertility and self-incompatibility in the genus Ananas. Acta Hortic. 334, 45–52 (1992).
Brewbaker, J. L. & Gorrez, D. D. Genetics of self-incompatibility in the monocot genera, Ananas (pineapple) and Gasteria. Am. J. Bot. 54, 611–616 (1967).
Bedinger, P. A., Broz, A. K., Tovar-Mendez, A. & McClure, B. Pollen-pistil interactions and their role in mate selection. Plant Physiol. 173, 79–90 (2017).
Gaut, B. S., Seymour, D. K., Liu, Q. P. & Zhou, Y. F. Demography and its effects on genomic variation in crop domestication. Nat. Plants 4, 512–520 (2018).
Coppens d’Eeckenbrugge, G., Leal, F. & Bartholomew, D. in The Pineapple: Botany, Production and Uses 13–32 (2003).
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E. J. & van der Knaap, E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530 (2008).
VanBuren, R. et al. The genome of black raspberry (Rubus occidentalis). Plant J. 87, 535–547 (2016).
Paques, F. & Haber, J. E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 (1999).
Petes T. D. & Symington L. S. in The Molecular and Cellular Biology of the Yeast Saccharomyces (eds Jones, E.W. et al.) 407–521 (Cold Spring Harbor Press, 1991).
Xie, T. et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol. Plant 8, 489–492 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573 (2018).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48, 11–39 (2014).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0 http://www.repeatmasker.org/ (2013–2015).
Guo, N. et al. Anthocyanin biosynthetic genes in Brassica rapa. BMC Genomics 15, 426 (2014).
Lefort, V., Longueville, J.-E. & Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424 (2017).
Hazzouri, K. M. et al. Whole genome re-sequencing of date palms yields insights into diversification of a fruit tree crop. Nat. Commun. 6, 8824 (2015).
Qi, J. et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510–1515 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strainw1118; iso-2; iso-3. Fly (Austin). 6, 80–92 (2012).
Nielsen, R. et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3, e170 (2005).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Price, A. L. et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5, e1000519 (2009).
Wegmann, D. et al. Recombination rates in admixed individuals identified by ancestry-based inference. Nat. Genet. 43, 847–853 (2011).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Han, Y., Qin, S. & Wessler, S. R. Comparison of class 2 transposable elements at superfamily resolution reveals conserved and distinct features in cereal grass genomes. BMC Genomics 14, 71 (2013).
Acknowledgements
This work was supported by the Department of Science and Technology of Fujian Province (grant no. 2016NZ0001-1), the National Natural Science Foundation of China grants (nos. U1605212 to Y.Q., 31628013 to Q.Y. and 31701874 to X.Z.), the Fuzhou Science and Technology project (grant no. 2017-N-33 to X.Z.), the Distinguished Young Scholars Fund in Fujian Agriculture and Forestry University (grant no. xjq201609 to L.Y.C.), the National Science Foundation grants (nos. DBI-1401572 to R.V. and DBI-1350041 to M.C.S.), the US National Institutes of Health (grant no. R01-HG006677 to M.C.S.), the NSF Plant Genome grants (nos. 0607123 and 043707-01 to J.L.B. and IOS-1546218 to M.D.P.), Zegar Family Foundation Grant (no. A16-0051-001 to M.D.P.) and the Swiss National Science Foundation grant (no. CRSII3_147630 to C.L.). We thank W. Till, D. Wegmann and D. Bartholomew for helpful discussions and J. Lin for assistance with data submission.
Author information
Authors and Affiliations
Contributions
R.M. conceived this genome project and coordinated research activities. R.M., L.Y.C., R.V. and J.L.B. designed the experiments. T.M., P.B., Q.W. and M.-L.W. maintained and provided Ananas germplasm. M.C.S. and X.Z. developed algorithms to resolve redundant assembly. X.Z., H.Y., S.C., M.A., S.R., M.C.S., R.V. and C.M.W. processed genome sequencing and resequencing data. L.Y.C., X.Z., Z.Liao, J.Liu, J.Lin, J.Y., M.F., Z.Lin, J.Z., L.H., J.Wu, S.Z., L.W., Y.Q., T.-Y.H., S.-M.K., C.-W.T., M.-L.W. and R.E.P. analyzed CB5 and ‘F153’ genome and RNA-seq data. R.V., M.P., H.Z., C.M.W., J.Z., L.H., H.W., J.Y.C., A.S., J.S., W.C.Y., J.C.C., J.W., Q.Y., G.C.d.E., G.M.S., M.D.P., J.L.B., C.L. and R.M. analyzed resequenced genomes. R.V., R.M., L.Y.C., X.Z., G.C.d.E., G.M.S., C.L. and J.L.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note and Supplementary Figures 1–24 and Supplementary Tables 1–28.
Supplementary Dataset 1
List of lignin biosynthesis genes in Arabidopsis thaliana, Populus trichocarpa, Oryza sativa and Ananas comosus.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, LY., VanBuren, R., Paris, M. et al. The bracteatus pineapple genome and domestication of clonally propagated crops. Nat Genet 51, 1549–1558 (2019). https://doi.org/10.1038/s41588-019-0506-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0506-8
This article is cited by
-
Identification of bromelain subfamily proteases encoded in the pineapple genome
Scientific Reports (2023)
-
Next-generation sequencing technology: a boon to agriculture
Genetic Resources and Crop Evolution (2023)
-
Molecular regulation of tomato male reproductive development
aBIOTECH (2023)
-
Loss of Heterozygosity and Its Importance in Evolution
Journal of Molecular Evolution (2023)
-
The Gastrodia menghaiensis (Orchidaceae) genome provides new insights of orchid mycorrhizal interactions
BMC Plant Biology (2022)
